Contents lists available at ScienceDirect
Journal
of
Chromatography
A
journal homepage: www.elsevier.com/locate/chroma
Integration
of
three-phase
microelectroextraction
sample
preparation
into
capillary
electrophoresis
Amar Oedit
a, Bastiaan Duivelshof
a, Peter W. Lindenburg
a,b,∗, Thomas Hankemeier
aaDivisionofSystemsBiomedicineandPharmacology,LeidenAcademicCentreforDrugResearch,Einsteinweg55,2300RALeiden,theNetherlands bUniversityofAppliedSciencesLeiden,FacultyScience&Technology,ResearchGroupMetabolomics,Mailbox382,2300AJ,Leiden,theNetherlands
a
r
t
i
c
l
e
i
n
f
o
Articlehistory:
Received11July2019 Revised25September2019 Accepted25September2019 Availableonline26September2019
Keywords:
Electromembraneextraction Electroextraction
Samplepreparation Capillaryelectrophoresis Sampleenrichment Electrophoresis
a
b
s
t
r
a
c
t
Amajorstrengthofcapillaryelectrophoresis(CE)isitsabilitytoinjectsmallsamplevolumes.However, thereisagreatmismatchbetweeninjectionvolume(typically < 100nL)andsamplevolumes(typically 20–1500μL).Electromigration-basedsamplepreparationmethodsarebasedonsimilarprinciplesasCE. Thecombinationofthesemethodswithcapillaryelectrophoresiscouldtackleobstaclesintheanalysisof dilutesamples.
This study demonstrates coupling of three-phase microelectroextraction (3PEE) to CE for sample preparation and preconcentration of large volume samples while requiringminimal adaptation ofCE equipment.Inthisset-up,electroextractiontakesplacefromanaqueousphase,throughanorganicfilter phase,intoanaqueousdropletthatishangingatthecapillaryinlet.Thefirstvisualproof-of-conceptfor thisset-upshowedsuccessfulextractionusingthecationicdyecrystalviolet(CV).Thepotentialof3PEE forbioanalysiswasdemonstratedbysuccessfulextractionofthebiogenicaminesserotonin(5-HT), tyro-sine(Tyr)and tryptophan(Trp).Under optimizedconditionslimitsofdetection(LOD)were15nMand 33nMfor5-HTandTyrrespectively (withTrpasaninternalstandard).TheseLODsarecomparableto othersimilarpreconcentrationmethodsthathavebeenreportedinconjunctionwithCE.Goodlinearity (R2> 0.9967)wasobservedforbothmodelanalytes.RSDsforpeakareasintechnicalreplicates,interday andintraday variabilitywereallsatisfactory,i.e., below14%.5-HT,Tyrand Trpspiked tohumanurine weresuccessfullyextractedandseparated.Theseresultsunderlinethegreatpotentialof3PEEasan inte-gratedenrichmenttechniquefrombiologicalsamplesandsubsequentsensitivemetabolomicsanalysis.
© 2019TheAuthors.PublishedbyElsevierB.V. ThisisanopenaccessarticleundertheCCBY-NC-NDlicense.
(http://creativecommons.org/licenses/by-nc-nd/4.0/ )
1. Introduction
Samplepreparationisacrucialaspectofbioanalysis.Themain objectivesofsamplepreparationaretopurifyandenrichanalytes prior toseparation anddetection.Commonlyused sample prepa-ration techniquesareprotein precipitationandpartitioningbased techniques, e.g., solid phase extraction (SPE) and liquid–liquid extraction [1,2]. In the past years,electromigration-based extrac-tion methods have gained increased attention [3–5]. The main principle behind electromigration-based techniques is the use of an electric field to extract ions from a donor phase (optionally throughintermediatephases)toanacceptorphase.Themigration speed dependson theelectrophoreticmobilityoftheanalyte and theelectricfield.Theelectricfieldstrengthistypicallylowinthe
∗ Correspondingauthor.
E-mail addresses: [email protected], [email protected]
(P.W.Lindenburg).
acceptor phase and thereby leads to stacking and preconcentra-tion of analytes. Electromigration-based techniques offer several advantagesoverpartitioning-basedtechniques,suchasbeingable tohandlesmallsamplevolumes,enhancedextractionspeeds(due totheelectricfieldbeingthedrivingforceratherthanpartitioning betweenphases)andeaseofautomation[4–6].
The combination of electromigration-based sample pretreat-mentwithCEofferstwomainbenefits.First,bothapproachesare basedonelectromigration,socompoundsthatcanbeextractedare alsosuitedforCEseparation.Second,electromigration-based tech-niquescan helpovercomeone ofthedrawbacksofCE:thereis a greatmismatchbetween theinjected volume (typically<100 nL) andthe samplevolume (typically 20–1500μL). In order to over-comethismismatchminiaturizedinsertshavebeendeveloped[7]. However,whensamples aretoodiluteandcompounds fallbelow detectionlimitsthisdoesnotprovideasolution. Electromigration-based samplepreparation can help overcomethe mismatchthat is oftenpresent between injectionvolumes andsample volumes
https://doi.org/10.1016/j.chroma.2019.460570
inCE analysis and can offer significant advantages in loadability comparedtoin-linestacking methods.On-lineSPE-CE, whereSPE iscoupled tothe capillary,could also serveas asolution for en-hancingtheloadability, buthasthus faronly beendemonstrated formoreapolarcompoundsonapolarcartridges[8].Moreover,to thebestofourknowledge,nocommercialsolutionsforSPE-CEare availableandsettingthemupiscomplex.
Inelectromembraneextraction(EME)asupportedliquid mem-brane(SLM)isusedtoseparatethedonorfromtheacceptorphase. The analytes are extracted through the SLM into the acceptor phaseusing anelectrical potential. Severalset-upsinwhich EME wascoupledoff-linetoCE-UVandappliedtobioanalysishavebeen reported[6,9–15].
Off-line, at-line, as well asin-line coupling of EME to CE-UV hasbeen reported on severaloccasions. An off-line process con-sistsoftwosteps(e.g.samplepreparationfollowedbyseparation). An at-line process combines the two steps via an automated handler(e.g. a robot). An in-line process takes place within the separationsystem(e.g. SPE-CEwithsorbent inside the capillary). An on-line process takes place right before the sample stream enters the separation system, but does take place within the analytical instrument (e.g. SPE-CE with sorbent outside the cap-illary)[16]. An example of on-linecoupling of EMEto CE-UV, is nano-EME,forpreconcentrationandanalysisofdrugs.Here, basic drugswereextractedfroma samplevolumeof200μL,througha membraneover acracked capillary, intoan acceptor of∼8 nL. A singleSLM could last for morethan 200 extractions. Enrichment factors(EFs)rangingbetween25and196werereportedforbasic drugcompounds, corresponding to recoveries of 0.1% and 0.79%. Under differentconditions, higher EFs were obtained, up to 500 for loperamide [17]. Moreover, EME-CE for preconcentration and analysis of basic drugs was reported. Here, an SLM was formed between two conical polypropylene units and extraction took place from the device, which was used as a vial insert. Extrac-tion took place froma 40 μL donor compartment over the SLM into an acceptor compartment of 40 μL and therefore it only served for sample cleanup and not for sample preconcentration. Recoveriesrangingfrom37%to84% wereobtained[18].However, the device requires reassembly for each new experiment, which hampersautomatedanalysis.Chui etal.integratedthefree liquid membrane(FLM)inanelectrokineticsupercharging (EKS)method in-linetofurtherimprovedetectionlimitsinCE.Inthisapproach, a small plug of immiscible organic solvent in the capillary was usedasfilterduringtheelectrokineticsampleinjectiontoenhance stackingefficiency.Analysisofcationicherbicidesinenvironmental water samples were used to evaluate the on-line preconcentra-tion efficiency andresults showed detection limit enhancements of over 1500 times. EKS over an FLM using CE for analysis on real-world samples has only been demonstrated on river water samples,arelativelycleanmatrix.Sincethedonorphaseisdrawn partially into the capillary, protein-rich samples could cause reproducibility problems due to absorption to the fused silica capillaries[19].
EMEcan be usedwithout SLM.Thisoffers severaladvantages, suchasnotrequiringreassembly andregenerationoftheSLM. In thiscasethetechniqueisreferred toasEMEoveranFLM[20] or 3PEE[21].Off-linecouplingofaEME-FLMextractiontoCE-UVwas successfullydemonstratedforanalysisofchargedbasicdrugs (nor-triptyline,haloperidolandloperamide)inhumanurine andblood [22].
Two-phase electroextraction (EE) is a process where charged analytesaretransferredfromanorganicdonorphaseintoan aque-ousacceptor phase usingan electricfield [23–25].EE coupledto CEwasfirstreportedinconjunctionwithisotachophoresisbyVan derVlisetal.in1994[26].
Previously,wehavereported3PEEasapowerful preconcentra-tiontechnique.Herein,weproposeintegrationofthissysteminto aCEset-upforon-linesampleclean-upandpreconcentration. Ra-terink etal.demonstratedthe extractionof acylcarnitines froma 50 μL donor phase, through a filter phase into a 2 μL acceptor phase that wasformed by a conductive pipettip prior to direct analysiswithhigh-resolution massspectrometry.In3PEE,altering theorganicfilterphase compositioninfluencestheselectivityand enablesselectiveextractionofdesiredanalytes[21].
In thisarticle,we demonstrate a proof-of-principle ofa novel on-lineanalyticalsystem inwhichelectromigration-basedsample preparationtechnique,i.e., 3PEE,isdirectlyhyphenatedto CE-UV. Ourmethodoffersseveraladvantages:automation,sample precon-centration and the ability to extract analytes from salt-rich ma-trices without sample preparation. In order to enable 3PEE the electrode configuration of a commercially available CE apparatus wasmodified.Thismodificationentailsplacinganinsulatingsleeve overtheelectrodewhilstleavingthebottompartoftheelectrode exposed. 3PEE-CE-UVpermitsautomated on-lineselectiveanalyte extraction andenrichment, directly froma dilute sample. In this set-upthe analytesareextractedfromasamplevialcontaininga two-phase liquid-liquid system, i.e., the aqueous sample and an organic filter phase. Extraction takes place into a droplet of ac-ceptor phase hanging at the capillary inlet in the organic filter phase.
Ina first seriesofexperimentsthe process wasvisualized us-ingthecationicdyeCVtoassessthestabilityofthedropletof ac-ceptor phase during3PEE. Then, the proposed method was eval-uated usingselected biogenic amine model compounds to evalu-ate its potential for bioanalysis of polar metabolites. In order to enhance the CE separation, pH-mediated stacking was included inthesystem. Consecutively,importantextractionparameters (EE voltage and EE time) and selectivity of the developed 3PEE-CE-UV method were investigated to optimize extraction. Then, the analytical performance of 3PEE-CE-UV was compared to conven-tional CE-UV. Finally, the performance of 3PEE-CE-UV as a sam-ple preparation procedure for polar compounds in salt-rich bi-ological samples was investigated by analysis of spiked human urinesamples inordertoshowits applicabilityformetabolomics analyses.
2. Materials and methods
2.1. Chemicalsandreagents
Sodium chloride (NaCl), CV, ammonium hydroxide (>25%), 5-HT,TyrandTrpwereobtainedfromSigma-Aldrich(Steinheim, Ger-many).Ethylacetate(EtOAc)wasobtainedfromActu-All(Oss,The Netherlands). Formic acid(FA) wasobtainedfromAcros Organics (Geel,Belgium).Sodiumhydroxide wasobtainedfromVWR (Am-sterdam, The Netherlands). All solutions were of HPLC grade or higher.WaterwaspreparedusingaMilli-QR AdvantageA10R sys-tem(Billerica,MA,USA).
2.2. Samplesandstocksolutions
2.3. Equipmentandtechniques
2.3.1. Capillaryelectrophoresis
AnalyseswereperformedusingaBeckmanCoulterP/ACEMDQ (Fullerton,CA, USA)CEapparatususingUV diodearray detection. A fused silicacapillary of50μm I.D. and365μm O.D.witha to-tal lengthof60cmwasused (PolymicroTechnologies,USA).New capillaries were sequentiallyrinsedat1379 mbar withMeOH for 10min,1MNaOHfor10min,waterfor5minandbackground elec-trolyte(BGE)for20min.Betweenruns,thecapillarieswereflushed for5minwithBGE.
Separation wasperformed using 1M FA (pH 1.8) as the BGE buffer using a separation voltage of +17.5kV. The capillary car-tridgetemperaturewasset at20°C. Detectionwasset at195nm tomaximize thenumberandresponseofmetabolitesthat canbe detectedwithareferenceat400nm.
2.3.2. Software
32 Karat (BeckmanInstruments, Fullerton,CA, USA) wasused forcontrollingtheCE-UVsystemandfordataacquisition.Injection volumes as well as the volumes of the acceptor droplet formed by reversed pressure were calculated usingSciex CEExpert V2.2 (Framingham,MA,USA).
3. Results and discussion
3.1. ModificationoftheCEinstrumentforthree-phase electroextraction
In order to enable 3PEE using a Beckman Coulter CE appara-tustheelectrode configurationwasmodifiedbyreplacingthe ex-istingelectrodewithalongerplatinumelectrodeof4cm(Fig.1B). Fromthebottom2.8cmoftheelectrodewasisolatedusinga poly-tetrafluoroethene (PTFE)sleeve,leavingonly atip of2mmofthe electrodeexposed.Thismodificationenablesanelectricfield from
the donorphase through the FLM into the acceptor droplet.The septaoftheinletvialswereremovedtoensurethatthemodified electrode,whichhadaslightlyincreasedthicknessduetothePTFE sleeve,couldstillreliablyenterthevial.
Inorder to visually monitorthe extractionprocess a USB-pen videocamerawasmountedinsidetheCEmachineandfocusedon thecapillaryinlet.DebutVideoCapture(NCHSoftware,Greenwood Village,CO,USA)wasusedtorecordtheextractionvideos.
3.2.Three-phaseelectroextractionprocedure
PriortoplacingthesamplevialsintheCEsystem,375μLdonor solutionwaspipetted in conventional CEvials (1.5mL). Based on previous EE works the donor solution was acidified to 1M FA, whichhasproventobeagooddonorsolvent[23–25].Thiswas fol-lowedby725 μLorganicfilterphaseconsistingofwater-saturated EtOAc,whichiscrucialfortheelectricfieldandtherebythe trans-ferof ionsthrough theorganicfilterlayer [21].Fig. 1graphically depictsa typical3PEEexperimentintheCEinstrument. First,the capillarywasrinsedwithFA.Thenammoniumhydroxide solution wasinjected,followed byan injectionof BGE.After inserting the capillaryinthesamplevial,ahangingdropletof100nLiscreated intheorganicfilterphasebyapplyingapressureof−69mbarfor 1min from the BGE outlet vial. Then, electroextractionwas per-formed by applying the extraction voltage, after which the en-richeddropletwasretractedintothecapillary.Atlast,thecapillary inletwasinsertedintoaBGE vialandseparationwascarriedout. Inorder tovisualize the electroextractionprocedure, thecationic dyeCVwasaddedtothedonorphase.
3.3.Visualizationofon-linethree-phaseelectroextraction
The setup for 3PEE hyphenated to CE-UV was based on the previouslyreported 3PEE-DI-MS[21].Ina visualproof-of-concept 10μMCVwaselectroextracted at3.5kVfrom375μLdonorphase
Fig.2. Visualizationof3PEEcoupledCEusingCV.Videostillsof:(A)initial condi-tionswithsamplevialcontaining375μLcrystalviolet(10μM)and725μL water-saturatedethylacetate;(B)endofdropletformation(∼100nL,1MFA),outlineof dropletbarelyvisible;(C)start3PEEbyapplicationof3kV;(D)endof3PEEafter 10min.
into∼100nLacceptorphase(Fig.2A–C).Theliquid-liquidinterface is not visible in the sample tray(see Supporting Information S1 forvialoutsideofsampletray).Priortoapplicationofthe extrac-tionvoltage,noCVcanbe observedinthedroplet.Thisindicates thatthecontributionofpartitioningtotheprocessisminimal. Af-ter10minelectroextraction,thedropletwasenricheddramatically withCV(Fig.2D).
Theseresultsindicatethatthedevelopedset-upissuccessfully extracting CV from the donor phase into the pendant acceptor phase.
3.4.On-linethree-phaseelectroextractioncoupledtocapillary electrophoresis
3.4.1. EffectofpH-mediatedstacking
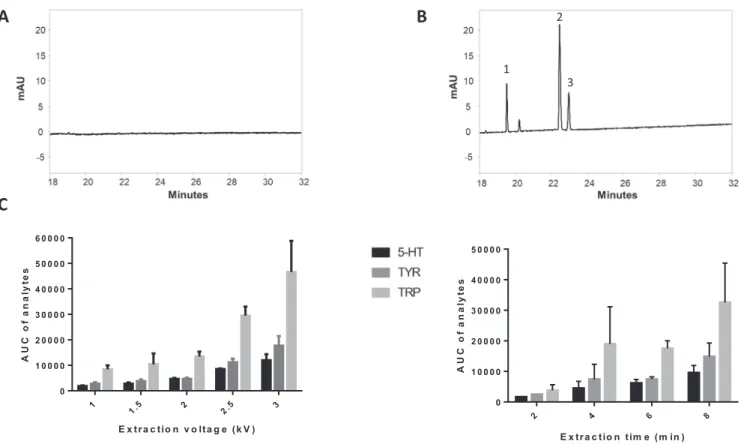
The 3PEE-CE-UV was investigated using the biogenic amines Tyr,Trpand5-HT.TheBGEconsistedof1MFA(pH1.8)toensure compounds were cationic during analysis. In a first experiment, 500nM modelanalytes were extractedfor6min at3kVand the dropletwaspartiallyretractedat34mbarfor5s, followedby CE separationat17.5kVfor30min.InFig.3Aitcanbe observedthat Trp and Tyr have poorly resolved peaks, with 5-HT overlapping. Thepeakareasarehigherdespitereducedinjectiontimecompared toFig.3B.Thisiscausedbymigrationofanalytesfromthedonor phase through the droplet into the capillary (see Supporting In-formation S2). This can possibly be explained by peak broaden-ingduring3PEEcaused byelectrophoresis andEOF, whichisstill presentto some extent, even ata low pH.In order to focus the broadsamplezoneandtherebyimproveseparation,apH-mediated stackingwasincluded.Byaddinga plugofbasicBGEafterto the acceptorphase,theacidicBGEtitratesthesamplesolutionto cre-atea neutralzone. In thiszone a higher field is presentcausing increasedmigration speed of analytes andeventually stacking at theinterfacebetweentheneutralzoneandBGE.ThispH-mediated stacking was created by injecting 15% ammonium hydroxide so-lutionfor17s at 34mbar andfollowed by 1M FA for 1.1min at 69 mbar to ensure that the droplet consisted fully of BGE. The biogenic amineswere extractedata concentration of 500nM for 8min at 3kV. The droplet could now be retracted much longer (1.5minat34mbar)whiletheseparationresolution improvedas showninFig.3B.
3.4.2. Optimizationofextractionvoltageandextractiontime The methodwasoptimizedinordertoobtainthehighest pos-sibleareaunder thecurve (AUC)forthe analytes. Inthe first se-riesofexperiments (n=3) the extractiontime waskeptconstant at5min andthe extractionvoltagewasvaried (1,1.5, 2,2.5, 3,4 and5kV). In theseexperiments 250nM Trp, Tyr and 5-HT were used. When 0kV was used no analytes were detected (Fig. 4A). Thisindicates thatanalytemigrationfromthedonorphasetothe acceptorphase issolelydriven byelectricpotential. Moreover, in-creasingthevoltage up to3kV significantlyenhanced signalsfor
Tyr,Trpand5-HTcomparedtolowervoltages(Fig.4BandC). Volt-agesbeyond3kVresultedinlossofcurrentanddropletinstability (datanotshown).
Subsequently,theextractiontimewasoptimizedwhilekeeping theextractionvoltageconstantattheoptimalvalueof3kV. Extrac-tionswereperformedfor2,4,6,8and10min.Itwasshownthat increasing the extraction time increasedenrichment and thereby peakareasoftheanalytes. Beyond8minofextractioncaused fre-quentcurrentlossesduringCE.
In summary, the optimized 3PEE procedure was as fol-lows. First, the capillary was flushed with 1M FA for 5min at 1378 mbar, followed by a 17s 34 mbar injection of 15% ammonium hydroxide and subsequent 1.1min 69 mbar injec-tion of 1M FA. Then, a droplet was formed using 1min 69mbar,afterwhichelectroextractionwascarriedoutat3kVfor 8min. Finally, the enriched droplet was retracted using 1.5min 34 mbar and CE-UV separation was performed for 35min at 17.5kV.
3.4.3. Analyticalfiguresofmerit
Table 1 shows the analytical performance of the optimized methodforthebiogenicamines5-HTandTyrincomparisonwith conventionalCE-UV.Trpwasusedasinternalstandard.
The aforementioned conditions were used to evaluatethe ex-tractionof5-HTandTyrusingdifferentconcentrations(0,0.05,0.1, 0.5,1,5μM;n=3)resultinginalinearrangeof0.01–5μM, yield-ing regressioncoefficients(R2)of 0.9967and0.9995, respectively
(Table 1). LODs were estimated usingsignal-to-noise (S/N) ratios of triplicatemeasurements at50nM and extrapolatedto S/N=3. Detectionlimitsof15nM and33nM were observedfor5-HTand Tyr,respectively.
For comparison, calibration curves were constructed with identical electrophoresis conditionsusing hydrodynamicinjection without pH-mediated stacking injecting 80nL (similarto the re-tracted volume using the optimized 3PEE-CE-UV method) using different concentrations (0, 1, 5, 10, 25, 50μM; n=3) of 5-HT andTyr with25μM Trp as internal standard. Since CE-UV could not reach the nM range of 3PEE-CE-UV the examined rangewas adjusted to a micromolar range to be able to construct a cali-bration curve (Table 1). Regression analysis yielded high R² val-ues (exceeding >0.999) for 5-HT and Tyr with conventional CE-UVandobservedLODsfor5-HTandTyrwere5μMand1μM, re-spectively. The LODsfor the 3PEE-CE-UV method were improved ∼333×and ∼30×for 5-HT andTyr, respectively. Moreover, com-pared to conventional CE-UV, linear range of 3PEE-CE-UV was extended an order of magnitude downwards to the 50–100nM range.
3.4.4. Repeatabilityandtechnicalreplicates
Intra-andinter-dayvariabilityofthemethodweredetermined usingoptimizedconditionsat500nM.AsshowninTable2, intra-dayvariabilityanalysisshowedgoodrepeatability,asRSDsforAUC valuesrangedfrom4.7%forTyrupto6.9%for5-HT.Forinter-day variability,RSD valuesrangedbetween7.9%forTyr and13.8%for 5-HT, indicating goodrepeatabilityof thedeveloped method.The increased RSD values obtainedcompared to conventional CE can bepartiallyexplainedbytheaddedsteps,includingdroplet forma-tionandextractionvs.hydrodynamicinjection.
Fig.3. Stackingeffectsin3PEEcoupledtoCE-UV.Topfiguresshowthecurrentprofileofextractionof500nM5-HT(1),Trp(2)andTyr(3)from375μldonorphaseusing 3PEE-CE-UVaswellasthecorrespondingelectropherograms.(A)retractingfor0.5minat6.9mbarand(B)retractingfor1.5minat34mbarwithpH-mediatedstacking.
2 4 6 8
0 1 0 0 0 0 2 0 0 0 0 3 0 0 0 0 4 0 0 0 0 5 0 0 0 0
E x tr a c tio n tim e (m in )
A
U
C
o
f
a
n
al
yt
es
A
B
C
1
1 .5 2 2 .5 3
0 1 0 0 0 0 2 0 0 0 0 3 0 0 0 0 4 0 0 0 0 5 0 0 0 0 6 0 0 0 0
E x t r a c tio n v o lta g e ( k V )
AUC
o
f
a
n
al
yt
es
1
2
3
Table1
Comparisontheanalyticalperformanceof3PEE-CE-UVandconventionalCE-UVinneatsolutions.AUCswerecorrectedusingTrpasinternalstandard.
Analyte Linearrange(μM) Sensitivity±SD(×10−2AU/μM) Intercept±95%CIa(×10−2AU/μM) Linearity(R²) LODb(μM)
CE 3PEE CE 3PEE CE 3PEE CE 3PEE CE 3PEE
5-HT 5–50 0.05–5 2.54±0.01 102.2±1.3 −0.40(−2.5–2.5) −2.10(−11.6–7.4) > 0.9999 0.9995 5 0.015 Tyr 1–50 0.1–5 9.65±0.06 76.8±2.6 1.67(−3.5–6.9) 5.98(−12.8–24.8) 0.9999 0.9967 1 0.033
Note:forrepeatabilityseeTable2.
a Nosignificantinterceptvalueswereobserved(p< 0.05).
bExtrapolationtowardsS/Nof3fromlowestmeasuredconcentration.
Table2
Intra-andinterdayrepeatabilityandtechnicalreplicatesof3PEE-CE-UVanalysisoftargetcompounds.AUCswerecorrectedusingTrpasinternalstandard.
Analyte 3PEE-CE-UVintraday(n=3) CE-UVintraday(n=3) 3PEE-CE-UVinterday(n=6) 3PEE-CE-UVtechnicalreplicatesa(n=5)
MeanAUCratio RSDarearatio(%) MeanAUCratio RSDarearatio(%) MeanAUCratio RSDarearatio(%) MeanAUCratio RSDarearatio(%)
5-HT 0.34 6.9 0.12 1.85 0.35 13.8 0.41 6.6
Tyr 0.44 4.7 0.50 0.87 0.43 7.9 0.43 10.0
a Obtainedfrom5consecutiveextractionsfromasinglesamplevial.
3.4.5. Enrichmentandrecovery
Inordertoassesstheperformanceof3PEE-CE-UVcorrectly,the extractionrecovery(ER)andEFwerecalculated[6].
TheseresultsshowthateventhoughtheEFmaxismuchgreater,
enrichmentwaslimited. Tyr inthe acceptorphase wasaround 8 times more concentrated than the donor phase after extraction (SupportinginformationS3). Itwasobservedthattheenrichment factorof 5-HT was 41.4and therefore fivetimes higher than for Tyr. A possible explanation for this is the lack of the carboxylic acidmoiety(pKa = 2.38)in5-HT, therebyenablingmoreefficient
transferfromtheFAcontainingdonorphase.Likewise,alower re-coverywasobserved forTyr(0.2%) thanfor5-HT (1.1%) after ex-tractionwhichcorrelateswiththeEFvalues.Theimprovementsin LOD in Table 1 differ fromthe obtained EF values asthe devel-opedmethodincludedbothstackingthroughon-line electroextrac-tion (increasing loading) and in-line stacking through a dynamic pH-mediated stacking (improving peak shapes). Both techniques are essential to thefinal methodand thereforethe final method wascompared to a simplehydrodynamic injectionmethod (thus withoutdynamicpH-mediated stacking).These resultsshow that the extraction process is not exhaustive and is a soft extraction method,which offers severaladvantagessuch asopeningup the possibilityofstudying (bio)chemical reactions and concentration-timemonitoringwithoutdisturbingtheoverall system.EFandER can be further improved by reducing the volume (and thereby height)of the organic phase to enhance the electric field distri-butiontobe morefavorabletowardsanalyteextraction.Moreover, thecomposition oftheorganicphasecanbe modifiedtoenhance EFandERaswell[21].
3.4.6. Comparisontootherset-ups
A comparison of3PEE-CE-UV method to other sample extrac-tiontechniquesthat werehyphenateddirectlytoCEandreported inliteratureisshowninTable3andSupportingInformationS5.
Singledropmicro-extraction(SDME)techniqueshavebeen cou-pledtoCE[27,28]withEFsrangingbetween130–150andEMEhas been coupled on-line to capillaries [17] with reported EFs rang-ingbetween25–196,withloperamidereachinganEFofupto500 underoptimal conditions.On-line back extraction field amplified sample injection relies on both partitioning between an organic chloroformdonorphaseandanaqueousacceptorphaseand simul-taneousdepletionoftheacceptorphaseviaelectrokineticinjection intothecapillary [29].3PEE-CE-UV bears similaritiestothe elec-trokineticsuperchargingoverFLMset-up,butdiffersintwoways: (1)preconcentrationtakesplaceatthe capillaryinletintoa pen-dantdropletratherthaninsidethecapillary,and(2)the
electroki-netic supercharging over FLM set-up incorporatesa t-ITP step to furtherenhancestacking oftheanalytesratherthanpH-mediated stacking.Mostpreconcentrationset-upsinTable3are coupledto CEarereportedforanalysisofapolarbasicdrugcompounds,with theexceptionelectrokineticsuperchargingoveranFLM[19],which wasusedpolarherbicides.Unlikeelectrokineticsuperchargingover an FLM, 3PEE-CE-UV does not require removal of FLM from the capillary, which can be a convoluted procedure and requires re-optimizationforeachneworganicFLMphase.
3PEE-CE-UV hasarelatively longtotalanalysistime compared to thediscussed set-ups. However, asthis isa proof-of-principle, separation parameters such as separation voltage and capillary lengthcouldstillbeoptimizedtoyieldshorteranalysistimes.The obtainedEFsof3PEE-CE-UV were1–2orders ofmagnitudelower than other reported methods. However, due to the combination withdynamicpH-mediated stacking similar LODswere obtained. Aprobableexplanationforthisisthefactthatwestudiedthe po-tentialof our methodfor polarmetabolites, while inother work apolardrugs,whicharemoreeasilyextractedarestudied.Transfer ofpolarmolecules,such asthebiogenicaminesinthiswork,has always beena challengingendeavor in EME andtuning of com-position and size of the organic phase remains at the forefront of interest [10]. The developed 3PEE-CE-UV has LODsin the low nMrange,whichissimilar toothertechniquesinTable2,despite having lower EFs likely due to its higher injection volume com-binedwithin-capillarystacking.Thelowrecoveriesof3PEE-CE-UV make itsuitable asa softextractiontechnique. Moreover,on-line methods such asdescribedinTable 3 aremore suitedfor analy-sisoflargedilutesamplesasthesecanhandlelargersamplesizes comparedtohighnLrangesamplesinin-linemethods.Finally,as 3PEE-CE-UVisnotexhaustiveitcanbeusedtomeasuretechnical replicates(i.e.repeatanalysesofthesamesamplevial).
3.4.7. Proofofconcept:urinebioanalysis
extrac-Table3
Comparisonofthenewlydeveloped3PEE-CE-UVmethodtoothermethodsthatwerehyphenatedtoCE.
Set-up Ref. Compounds EF(range) ER(range) LOD(range)
Total analysis time(min)
3PEE-CE-UV Thispaper Serotonin,Tyrosine, (Phenylalanine)
7.8–42 0.2–1.1% 15–33nM 52
InlineSDME-CE-MS [27] Methoxyphenamine, Methamphetamine, Amphetamine, Phenetylamine
130–150 Not
reported
2–5nM 62
Nano-EMEcoupledto CE-UV
[17] Pethidine, Nortiptyline, Methadone, Haloperidol,
Loperamide
25–196 0.1%−0.79% 8–31nM(0.2–
15ngmL−1)
> 29d
FLM– electrokinetic superchargingcoupled toCE-UV
[19] Paraquat,Diquat Not reported
(Relative recoverya:
97.0– 97.5%b)
58– 58nM (0.15– 0.20ngmL−1)
> 20d
On-lineback extractionfield amplifiedsample injection(coupledto CE-UV)
[28] Cocaine,Thebaine, (Metamphetamine)
Not reported
(Relative recoverya:
94.71– 98.65%c):
16– 16nM (0.005– 0.005μgmL−1)
18
UnderlinedcompoundsandvaluesarethosewiththelowestLOD,compoundsinitalicsarethosewiththehighestLOD.
aRelativerecoverieswerereportedbymeasuringaspikedsampleandcomparingtothecalibrationcurve. bSpikedat20ngmL−1.
cSpikedat0.5μgmL−1.
dDurationofinitialflushpriortoeachanalysisnotspecified.
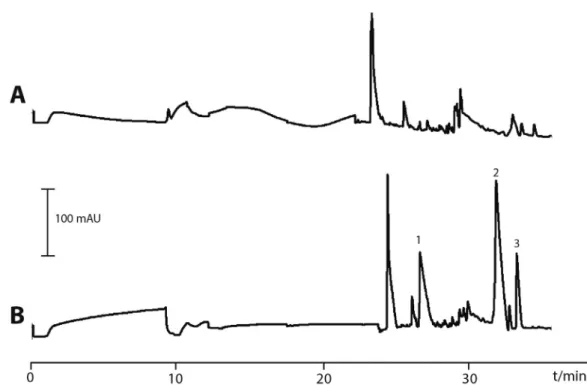
Fig.5. Proofofconceptshowingurinebioanalysisusing3PEE-CE-UV.Electropherogramsobtainedfrom(A)non-spikedurineand(B)spikedurinesamplesextractedby3PEE priortoCE-UVdetection.Urinewasspikedwith5-HT(1),Tyr(3)andTrp(2;50μM).ExtractionandanalyticalconditionscanbefoundinSection3.6.
tion and separation current profiles to extractions performed in neatsolutions(datanotshown).Beforeanalysisofthetarget ana-lytes,3PEE-CE-UVwasfirstappliedtoanon-spiked urinesample. In the corresponding electropherogramat thenon-specific wave-length 195nm, many unidentified endogenous compounds were observedaftertheextractionprocedureshowingitsabilityto ana-lyzeaurinesamplewithoutrequiringpriordilution(Fig.5A).Then, 3PEE-CE-UV wasemployed for the analysisof 5-HT, Trp andTyr (50μM), spikedtourineofthesameorigin. Theelectropherogram inFig.5B,showsdetectionof5-HT,TrpandTyr.Inordertoconfirm theidentitiesoftheendogenouscompoundsinurineamore
selec-tivedetectorisrequired.Thesepreliminaryresultsshowthe poten-tialof3PEE-CE-UV asan easyon-linesamplepreparationmethod that could be appliedforthe analysisof smallpolarmetabolites, e.g.,inametabolomicssetting.
4. Conclusion
commer-cially available CE instrument. By placing an immiscible organic filterphase on top ofan aqueous sample, cationic analyteswere extracted into an aqueous acceptor droplet formed at the capil-laryinlet,whenanelectricalfield wasapplied.Inordertoenable full droplet retraction without loss of resolution after extraction, pH-mediatedstackingwasintroducedtoefficientlystackanalytes. Theperformanceofon-line3PEE-CE-UVwasevaluatedby extract-ing5-HT,Trp andTyrfroma375μLneatsolutionintoapendant dropletof 100 nL BGE. Low extraction recoveries were obtained, demonstratingthatthetechniqueisasoftextractiontechnique.To thebestofourknowledge,EMEoveran FLMofpolarmetabolites wasneverreported.Itwasdemonstratedthatdetectionlimits im-provedto15nM5-HTand33nMTyrwith3PEE,comparedto5μM 5-HTand1μMTyr inCEwithhydrodynamicsampleinjection.As proof-of-concept,theon-line3PEE-CE-UVprocedurewasevaluated forthe analysis ofhuman urine. It was demonstrated that 5-HT, Trp and Tyr were successfully extracted from spiked urine, thus signifyingthepotential ofthe developedprocedureforurine bio-analysisandmetabolomics.
Declaration of Competing Interest
None.
Acknowledgements
The research leading to these results has received funding from the European Community’s Seventh Framework Program (FP7/2007-2013) under Grant Agreement No. 306000 (STATegra) andGrantAgreement No. 602783 (CAM-PaC).The authors would like to acknowledge Raphaël Zwier for his help in adjusting the electrodeconfigurationoftheCEinstrument.
Supplementary materials
Supplementary material associated with this article can be found,intheonlineversion,atdoi:10.1016/j.chroma.2019.460570.
References
[1] I.Kohler, J.Schappler,S. Rudaz,Microextraction techniquescombinedwith capillary electrophoresis in bioanalysis, Anal. Bioanal. Chem. 405 (2013) 125–141.
[2] R.-J.Raterink,P.W.Lindenburg,R.J.Vreeken,R.Ramautar,T.Hankemeier, Re-centdevelopmentsinsample-pretreatment techniquesfor mass spectrome-try-basedmetabolomics,TrACTrendsinAnal.Chem.61(2014)157–167.
[3] A.Wuethrich,P.R.Haddad,J.P.Quirino,Theelectricfield– Anemergingdriver insamplepreparation,TrACTrendsinAnal.Chem.80(2016)604–611.
[4] P.W.Lindenburg,R.Ramautar,T.Hankemeier,Thepotentialofelectrophoretic sample pretreatment techniques and new instrumentation for bioanalysis, withafocusonpeptidomicsandmetabolomics,Bioanalysis5(2013).
[5] S.Pedersen-Bjergaard,C.Huang,A.Gjelstad,Electromembrane extraction-Re-centtrendsandwheretogo,J.Pharm.Anal.7(2017)141–147.
[6] A.Oedit,R.Ramautar,T.Hankemeier,P.W.Lindenburg,Electroextractionand electromembraneextraction:advancesinhyphenationtoanalyticaltechniques, Electrophoresis37(2016)1170–1186.
[7] A.Šlampová,P.Kubá ˇn,Injectionsfromsub-μLsamplevolumesincommercial capillaryelectrophoresis,JournalofChromatographyA1497(2017)164–171.
[8] R.Ramautar,G.W.Somsen, G.J.deJong, CE-MSformetabolomics: develop-mentsand applicationsinthe period2014–2016,Electrophoresis38(2017) 190–202.
[9]A.Šlampová,P.Kubá ˇn,Two-phasemicro-electromembraneextractionacross freeliquidmembranefordeterminationofacidicdrugsincomplexsamples, Anal.Chim.Acta1048(2019)58–65.
[10]N.Drouin,S.Rudaz,J.Schappler,Newsupportedliquidmembranefor elec-tromembrane extraction of polar basic endogenous metabolites, J. Pharm. Biomed.Anal.159(2018)53–59.
[11]M.Silva,C.Mendiguchía,C.Moreno,P.Kubá ˇn,Electromembraneextractionand capillaryelectrophoresiswithcapacitivelycoupledcontactlessconductivity de-tection:multi-extractioncapabilitiestoanalysestracemetalsfromsaline sam-ples,Electrophoresis39(2018)2152–2159.
[12] M.Dvoˇrák,K.F.Seip,S.Pedersen-Bjergaard,P.Kubá ˇn,Semi-automatedset-up forexhaustivemicro-electromembraneextractionsofbasicdrugsfrom biolog-icalfluids,Anal.Chim.Acta1005(2018)34–42.
[13]A.Šlampová,P.Kubá ˇn,Directanalysisoffreeaqueousandorganicoperational solutionsasatoolforunderstandingfundamentalprinciplesof electromem-braneextraction,Anal.Chem.89(2017)12960–12967.
[14]M.Forough,K.Farhadi,A.Eyshi,R.Molaei,H. Khalili,V.Javan Kouzegaran, A.A. Matin, Rapid ionicliquid-supported nano-hybridcomposite reinforced hollow-fiberelectromembraneextractionfollowedbyfield-amplifiedsample injection-capillary electrophoresis:aneffective approachfor extractionand quantificationofimatinibmesylateinhumanplasma,J.Chromatogr.A1516 (2017)21–34.
[15]A.R. Fakhari, H. Mohammadi Kosalar, S. Asadi, K.S. Hasheminasab, Surfac-tant-assistedelectromembraneextractioncombinedwith cyclodextrin-modi-fiedcapillaryelectrophoresisfor theseparationandquantificationof tranyl-cypromineenantiomersinbiologicalsamples,J.Sep.Sci.41(2018)475–482.
[16]R. Ramautar, G.W.Somsen, G.J. de Jong, Recent developments in coupled SPE-CE,Electrophoresis31(2010)44–54.
[17]M.D.Payan,B.Li,N.J.Petersen,H.Jensen,S.H.Hansen,S.Pedersen-Bjergaard, Nano-electromembraneextraction,Anal.Chim.Acta785(2013)60–66.
[18]P.Pant˚uˇcková,P.Kubá ˇn,P.Boˇcek,Asimplesamplepretreatmentdevicewith supportedliquidmembranefordirectinjectionofuntreatedbodyfluidsand in-line coupling toa commercialCE instrument,Electrophoresis 34 (2013) 289–296.
[19]M.Q.Chui,L.Y.Thang,H.H.See,Integrationofthefreeliquidmembraneinto electrokineticsupercharging-capillaryelectrophoresisforthedetermination ofcationicherbicidesinenvironmentalwatersamples,J.Chromatogr.A1481 (2017)145–151.
[20]P.Kubá ˇn,P.Boˇcek,Micro-electromembraneextractionacrossfreeliquid mem-branes.instrumentation and basic principles,J. Chromatogr. A 1346(2014) 25–33.
[21]R.J.Raterink,P.W.Lindenburg,R.J.Vreeken,T.Hankemeier,Three-phase elec-troextraction,anew(online)samplepurificationandenrichmentmethodfor bioanalysis,Anal.Chem.85(2013)7762–7768.
[22]P.Kuban,P.Bocek,Micro-electromembraneextractionacrossfreeliquid mem-branes.extractionsofbasicdrugsfromundilutedbiologicalsamples,J. Chro-matogr.A1337(2014)32–39.
[23]P.W.Lindenburg,R.Seitzinger,F.W.A.Tempels,U.R.Tjaden,J.vanderGreef, T.Hankemeier,Onlinecapillaryliquid–liquidelectroextractionofpeptidesas fastpre-concentrationpriortoLC–MS,Electrophoresis31(2010)3903–3912.
[24]P.W.Lindenburg,F.W.Tempels,U.R.Tjaden,J.vanderGreef,T.Hankemeier, On-linelarge-volumeelectroextractioncoupledtoliquidchromatography-mass spectrometrytoimprovedetectionofpeptides,J.Chromatogr.A1249(2012) 17–24.
[25]P.W. Lindenburg,U.R.Tjaden,J. vanderGreef,T.Hankemeier, Feasibility of electroextractionas versatilesamplepreconcentrationfor fastandsensitive analysisofurinemetabolites,demonstratedonacylcarnitines,Electrophoresis 33(2012)2987–2995.
[26]E.VanderVlis,M.Mazereeuw,U.R.Tjaden,H.Irth,J.vanderGreef,Combined liquid-liquidelectroextractionandisotachophoresisasafaston-linefocusing stepincapillaryelectrophoresis,J.Chromatogr.A(1994)333–341.
[27]Z.A.AlOthman,M.Dawod,J.Kim,D.S.Chung,Single-dropmicroextractionasa powerfulpretreatmenttoolforcapillaryelectrophoresis:areview,Anal.Chim. Acta739(2012)14–24.
[28]J.Kim,K.Choi,D.S.Chung,In-linecouplingofsingle-dropmicroextractionwith capillaryelectrophoresis-massspectrometry,Anal.Bioanal.Chem.407(2015) 8745–8752.
[29]H.Fang,Z.Zeng,L.Liu,D.Pang,On-lineback-extractionfield-amplifiedsample injectionmethodfordirectlyanalyzingcocaineandthebaineintheextractants bysolventmicroextraction,Anal.Chem.78(2006)1257–1263.