Comparison of advancing methodologies for diatom
composition analysis within a coastal upwelling community
By: Emily Pierce
Senior Honors Thesis
Environment, Ecology, and Energy Program
University of North Carolina at Chapel Hill
05/01/2020
Approved:
Adrian Marchetti, Thesis Advisor
Comparison of advancing methodologies for diatom composition analysis within a coastal upwelling community
Abstract
Diatom community composition has an important influence on global oceans and ecological processes. Developing accurate and efficient methods for characterizing diatom taxonomy under different environmental conditions is critical to understanding the implications of changes in the diatom community. Two developing methods for identifying and enumerating diatoms, cell imaging and molecular sequencing, are experiencing recent rapid advancements. This project aims to compare diatom taxonomic composition results within natural assemblages derived from rapidly advancing methods, FlowCam imaging and 18S rDNA sequencing techniques with traditional light microscopy cell counting techniques. FlowCam imaging allows taxonomic identification via microscopy through semi-automated morphological characterization. Whereas, the molecular sequencing method used in this study is dependent on similarity of sequences obtained from the environment to those known sequences within a reference sequence database. Both methods were implemented in tandem along with traditional light microscopy cell counts to analyze phytoplankton samples collected throughout the California Upwelling Zone. Overall, our study provides a comparison across two rapidly evolving taxonomic approaches and a traditional approach used to determine plankton abundance and distributions in the ocean.
Introduction
Marine phytoplankton play key roles in supporting ecological and human health. Diatoms are an especially important group of marine phytoplankton because of their role as components of the global carbon cycle (Falkowski et al., 1998)8), bases for the marine food web (Sarthou et al., 200520), and potential harmful algal bloom formers (Bates & Trainer, 20062). Diatom
community composition, which describes the abundance of specific taxa within an assemblage, is critical for several ecological processes and is also experiencing rapid transformations due to the impacts of climate change (Marinov et al., 201314). These compositional changes are likely to have huge implications on global carbon cycling, marine food webs, and water quality; thus, it is imperative to be able to accurately and efficiently analyze diatom communities.
The traditional approach to phytoplankton community composition analysis has consisted of counting and identifying cells via light microscopy (Utermöhl, 195822). This traditional approach is associated with several practical drawbacks including subjective identification and lengthy analysis time. As technology has advanced, many new methods have emerged and evolved over time. These new methodologies often produce more rapid results than traditional approaches and can offer additional data insights. While advancing methodologies present exciting new information and opportunities, it is critical to understand these new methods in context of one another (Karlusich et al., 202011). Advancing methodologies offer different qualitative and quantitative capabilities that must be validated to ensure the most efficient and accurate phytoplankton community analysis as well as comparability and continuity with previous methodologies.
metadata on each particle’s shape, size, and other parameters. A Fluid Imaging software package can be applied in order to taxonomically classify and sort particle images. The FlowCam
provides several practical advantages because it can be easily transported and employed in the field and its real-time data output allows for rapid sampling of a large number of cells (Fluid Imaging Technologies, 20119).
Amplicon sequencing is another evolving method for phytoplankton community analysis. All eukaryotic phytoplankton, including diatoms, possess at least one copy of the 18S ribosomal RNA gene which contains variable regions unique to each taxon. Because the 18S gene is highly conserved and unique to each taxon, it is a prime candidate for sequencing in order to derive community composition data. Gene sequences are classified using peer reviewed reference databases, removing the subjectivity used in other user dependent classification methods. However, the number of 18S gene copies in an organism is variable and there is not yet an accepted method for converting copy number to cell number; therefore, sequencing methods do not yield the same quantitative results that other methods allow (Gong & Marchetti, 20196).
There has been substantial research comparing results from advancing methods for
phytoplankton community composition analysis to results from traditional light microscopy techniques. Studies comparing phytoplankton cell abundance results from FlowCam and light microscopy concluded that the methods yield comparable results for quantitative abundance. The FlowCam has been determined to be ideal for rapid analysis in which coarse taxonomic
resolution at the genus level and semi-quantitative abundance analysis are acceptable (Álvarez et al., 20141, Hrycik et al., 201910). Studies comparing 18S sequencing results with light
microscopy results conclude that the methods have different quantitative capabilities and do not show widely comparable results for semi-quantitative abundance (Gong et al., 20205, Kittelmann et al., 201512). There has been a limited number of studies comparing phytoplankton community composition across different methods such as FlowCam and 18S sequencing, creating a gap in our understanding of how new methods intercompare to each other.
This study aims to compare diatom community composition analysis results derived from three different taxonomic identification approaches – FlowCam imaging, 18S rDNA amplicon sequencing, and traditional light microscopy counts. Samples analyzed in this study were collected from a coastal upwelling community that have been exposed to experimental manipulation by simulating upwelling combined with iron additions. Therefore, diatom community composition methods are being compared across a set of samples with drastically varying diatom abundance and diversity. Additionally, analyzing samples exposed to
Methods
Sampling Locations and Experimental Design
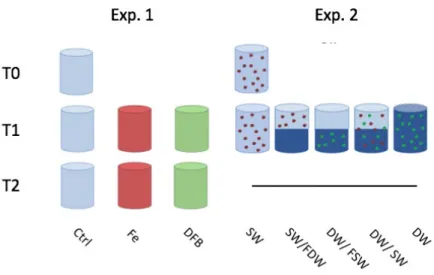
Iron response (Exp1) and seed population (Exp 2) experiments were conducted onboard the R/V Oceanus from May 27th, 2019 through June 6th, 2019. Both experiments were conducted at a site overlying a wide continental shelf location, 4100.896’N 12425.165’W, and a narrow continental shelf location, 3555.329N 12132.439W, (Fig. 1) during non-upwelling, relaxation conditions. In Exp. 1, water was collected at the 10C isotherm (90m at wide shelf and 80m at narrow shelf). Cubitainers (10L) were filled in triplicates with one of the following inoculation treatments: iron, desferrioxamine B(DFB), or no treatment (Fig. 2). Iron stock was added to a concentration of 5nM. DFB, an iron chelating agent that limits the uptake of iron by phytoplankton was added to a concentration of 200nM. Exp. 1 controls were filled with deep water and no additives. In Exp. 2, water was collected at the 10C isotherm (90m at wide shelf and 80m at narrow shelf) and at the surface (15m at both sites). Cubitainers were filled in triplicate with deep water, surface water, or a mixture of the two (Fig. 2). Exp. 2 controls were filled with either deep water or surface water with no additives. All filled cubitainers were incubated in on-deck tanks circulated with 10C sea water and shaded to achieve 30% of the incident irradiance (Io). Cubitainers were
harvested at two time points for experiment 1 and one time point
for experiment 2 in addition to the initial communities (T0).
Subsampling for each of the following parameters relevant to
this study were collected- size-fractionated chlorophyll a, DNA,
preservations for light microscopy, and FlowCam
measurements.
Figure 2- Experimental setup for Exp. 1 and Exp. 2 (SW= Surface water, DW= Deep water, FSW= Filtered Surface Water, FDW = Filtered Deep Water).
Chlorophyll a extractions and quantification
Approximately 400 mL from each cubitainer was collected in a dark bottle and filtered onto a Millipore isopore filters (5m pore size , 47mm) and subsequently onto a Whattman GF/F filter (0.7m nominal pore size, 25mm) using a series filter cascade and a vacuum pump. Filters were collected and stored at -20C until onshore analysis. Filters were placed in 6mL of 90% acetone and stored at -20C for 24 hours before analysis. Samples were analyzed using the acidification method on a Turner 10AU fluorometer blanked with a 90% acetone solution (Parsons et al., 198416).
FlowCam Diatom Enumeration
At the various time points, approximately 25mL of seawater was collected from each cubitainer in a 50mL centrifuge tube. The whole water samples were analyzed using a Portable Series FlowCam set to auto trigger mode using a 10x objective with a 100 m glass flow cell installed. FlowCam analysis yielded a set of images associated with each sample. Auto trigger mode captures images of a subarea of the flow cell at a set shutter speed and then saves the particles captured in each image. The number of particles is normalized by the volume imaged to produce a sample density (particles mL-1). Any images not containing particles (i.e., bubbles or smears on the flow cell caused by a disruption of the calibration setting) were deleted from the dataset and the particle density was corrected. Remaining images were categorized to diatom genus using Fluid Imaging software combined with manual taxonomic identification (Fluid Imaging Technologies, 20119).
The image clarity of particle images captured by the FlowCam (Fig. 3) is variable and thus limits taxonomic classification to the genus level. Particles are imaged by the FlowCam at a set
chain, a chain oriented in such a way that it is impossible to count individual cells, or a chain with indistinguishable segments. The variability in the number of cells captured in a particle image presents a barrier to cell enumeration that is unique to chain forming phytoplankton. Underestimation could be especially problematic considering the deleterious toxin production from the chain-forming genus, Pseudo-nitzschia.
Figure 3. Selected images from FlowCam sample (Exp. 2, Wide Shelf, SWDW-1). From left to right genera – Chaetoceros, Navicula, Asterionellopsis,and Thalassiosira.
DNA Extractions and 18S rDNA Sequencing
Microscopy Counts
Approximately 100mL of seawater from a single replicate cubitainer of each treatment type was collected in glass bottles and inoculated with Lugol’s to achieve a final 2% Lugol’s
concentration. Samples were then stored in the dark at 4 °C until analysis onshore. A subsample was prepared for analysis by settling a fixed volume using Utermöhl settling chambers
(Utermöhl, 195822). Slides were analyzed on an Olympus CKX-31 invertedmicroscope under a 200x magnification. Diatoms cells were enumerated and classified in 10 randomly selected fields of view per sample.
Results and Discussion
Analysis for this project remains underway as datasets for DNA and light microscopy are not yet complete. All results presented here are preliminary. While results are preliminary, several observations can be made and discussed.
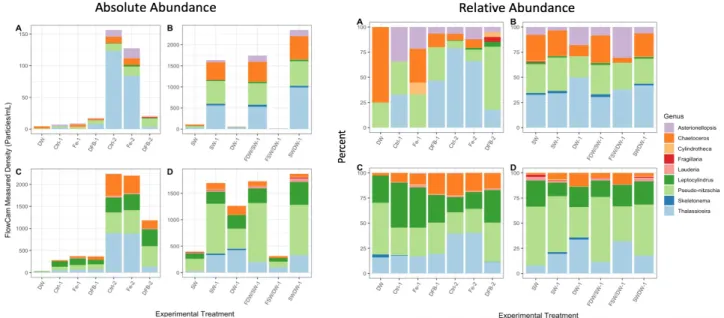
Figure 4- Absolute abundance andpercent relative abundance of identified diatom genera from the FlowCam methodology (A= Exp1, Wide Shelf, B= Exp2, Wide Shelf, C= Exp1, Narrow Shelf, D=Exp2, Narrow Shelf).
absolute and relative abundance results from each method, DNA, FlowCam, and microscopy, should be included in the comparison. Discrepancies in comparison of abundance datasets from these methods may further inform method choices. For example, complete results may indicate that all of the methods included here detect similar relative abundance results but dissimilar absolute abundance results. This potential disagreement may indicate that not all of the included methods are appropriate for quantification but can be used to detect dominant genera.
As datasets are completed, more work will be needed in order to achieve the same unit of quantification across all absolute abundance datasets. Light microscopy methods measure absolute abundance in cells mL-1; whereas, FlowCam measures particles mL -1 and DNA
sequencing measures 18S gene copies mL -1. Several assumptions will need to be made in order to convert FlowCam and DNA sequencing datasets into units of cells mL-1. These assumptions may impact variability across absolute abundance results.
Figure 5- Standard error for three dominant genera from FlowCam absolute abundance measurements. (A= Exp1, Wide Shelf, B= Exp2, Wide Shelf, C= Exp1, Narrow Shelf, D=Exp2, Narrow Shelf)
errors for the FlowCam abundances of Pseudo-nitzschia, especially for experiment 2 measurements. It will be important to compare standard errors across methods to further
investigate the cause of large standard error values. It will also be important to compare standard errors for absolute abundances from the dataset with initial units of quantification, particles mL -1, and converted units, cells mL -1, to understand error associated with the conversion factor.
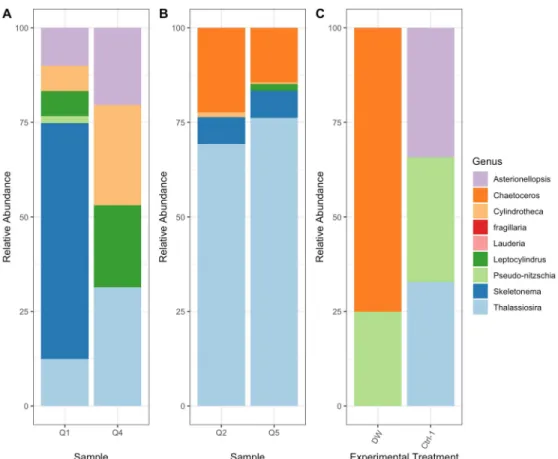
Figure 6- Relative abundance data derived from three different methods (A= Light Microscopy Cell Counts, B= Amplicon Sequencing, C= FlowCam Analysis). Preliminary results from the FlowCam method are an average of three samples corresponding to the experimental treatment shown. Preliminary results from microscopy
and sequencing methods are limited to individual samples corresponding to the experimental treatment shown in FlowCam results. For comparison purposes, analysis was limited to diatom genera detected by each method.
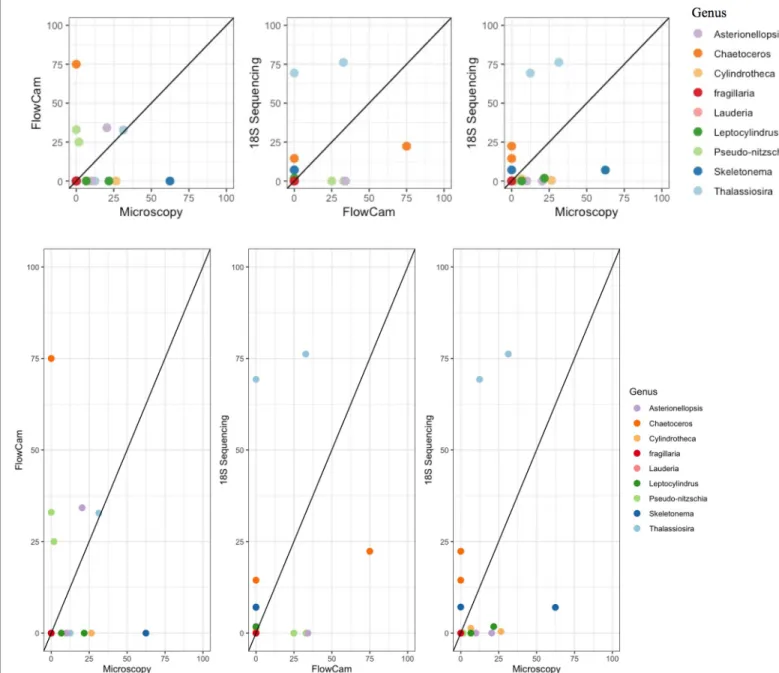
Figure 7-Comparison of relative abundance between methods including a 1:1 reference line.
method. Upon completion of the DNA and light microscopy datasets, a full comparison of the number and diversity of genera detected will allow for further evaluation of the detection capabilities of these methods.
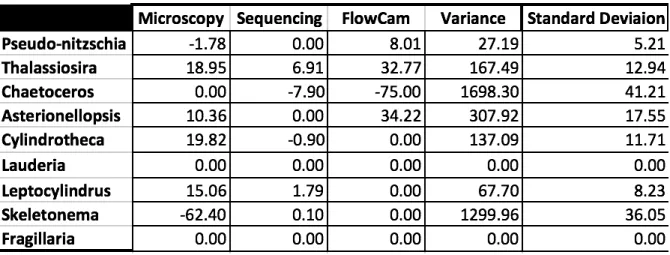
Table 1- The change in relative percent abundance across initial time points is shown for each genus using each method. The Variance and standard deviation for each genus is included.
As a consequence of not detecting certain taxa dependent on the method, the preliminary data showed strong disagreement across methods is the detection of community composition changes in response to the treatments. The high standard deviation values for most genera may also indicate that the different methods do not detect similar changes across the experimental treatments included in the preliminary results. This potential discrepancy in results across methods could have large implications for researchers interested in measuring community composition change. The completion of the DNA and sequencing datasets will allow for further comparison of each method’s results across experimentally stimulated shifts in the diatom community composition.
Preliminary results are limited to a small number of samples with comparatively low biomass. Because early samples being compared are low in diatom biomass, the number of cells in each sample is low and may therefore be contributing to higher measurements of variation. It will be important to determine if high variation is specific to low abundance samples, in which case there may be implications for researchers hoping to compare results across a variety of studies for low abundance communities. As datasets are completed, it will not only be important to further explore the amount of variation present, but also the source of that variation. As highlighted in these preliminary results, there may be variation across methods due to certain taxa going undetected or inaccurately enumerated. The factors resulting in high standard error associated with specific taxa needs to be further explored. In addition, preliminary results are limited to comparison of relative abundance, which, as noted provides different information than absolute abundance. Therefore, preliminary results cannot be taken as a complete assessment of the methods in question; however, they provide initial analysis of some of the factors researchers should consider when choosing methodologies or comparing results from studies that utilized different approaches for taxonomic identification and enumeration of phytoplankton.
Preliminary results indicate disagreement across FlowCam imaging, 18S rDNA sequencing, and light microscopy methods in detection of diatom genera, the relative proportions of those genera, and the changes in diatom community composition in response to experimental treatments. These preliminary results are limited to low abundance samples and semi-quantitative relative abundance; therefore, it is important to further explore the disagreement across methods as datasets are completed.
Combining preliminary results with knowledge of the practical consequences of the methods investigated here, we can begin to highlight the implications of method choice for different researchers. The FlowCam will provide the most rapid results which may be ideal for field monitoring programs; however, it is limited in taxonomic depth, appears not to detect rarer taxa, and may skew abundance data on chain forming diatoms, presenting hurdles for researchers interested in detecting specific species. Light microscopy requires more time and expertise, but it appears to detect the greatest number of taxa for those that can be morphologically differentiated, potentially making it the best option for researchers aiming to detect specific ecologically
important species (e.g., HAB species). Sequencing of rDNA removes the subjectivity that FlowCam and microscopy rely on, but there are significant hurdles to cell enumeration and clear quantification.
Future work on comparing advancing methodologies should consider evaluating methods using a mock community. This study allows for insights on how the results of these methods employed in the field compare, providing valuable information for researchers making method choices or comparing cross study results; however, there is no way to determine which results are the most accurate. The use of mock communities with known cell abundance would allow for an
References
1. Álvarez, E., Moyano, M., López-Urrutia, A., Nogueira, E., Scharek, R. (2014). Routine determination of plankton community composition and size structure: a comparison between FlowCAM and light microscopy, Journal of Plankton Research, 36(1),170–184, https://doi
org.libproxy.lib.unc.edu/10.1093/plankt/fbt069
2. Bates, S. S. & Trainer, V. L. in Ecology of Harmful Algae (eds Graneli, E. & Turner, J. T.) Ch. 7 (Springer, 2006)
3. Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, Bai Y, Bisanz JE, Bittinger K, Brejnrod A, Brislawn CJ, Brown CT, Callahan BJ, Caraballo-Rodríguez AM, Chase J, Cope EK, Da Silva R, Diener C, Dorrestein PC, Douglas GM, Durall DM, Duvallet C, Edwardson CF, Ernst M, Estaki M, Fouquier J, Gauglitz JM, Gibbons SM, Gibson DL, Gonzalez A, Gorlick K, Guo J, Hillmann B, Holmes S, Holste H, Huttenhower C, Huttley GA, Janssen S, Jarmusch AK, Jiang L, Kaehler BD, Kang KB, Keefe CR, Keim P, Kelley ST, Knights D, Koester I, Kosciolek T, Kreps J, Langille MGI, Lee J, Ley R, Liu YX, Loftfield E, Lozupone C, Maher M, Marotz C, Martin BD, McDonald D, McIver LJ, Melnik AV, Metcalf JL, Morgan SC, Morton JT, Naimey AT, Navas-Molina JA, Nothias LF, Orchanian SB, Pearson T, Peoples SL, Petras D, Preuss ML, Pruesse E, Rasmussen LB, Rivers A, Robeson MS, Rosenthal P, Segata N, Shaffer M, Shiffer A, Sinha R, Song SJ, Spear JR, Swafford AD, Thompson LR, Torres PJ, Trinh P, Tripathi A, Turnbaugh PJ, Ul-Hasan S, van der Hooft JJJ, Vargas F, Vázquez-Baeza Y, Vogtmann E, von Hippel M, Walters W, Wan Y, Wang M, Warren J, Weber KC, Williamson CHD, Willis AD, Xu ZZ, Zaneveld JR, Zhang Y, Zhu Q, Knight R, and
Caporaso JG. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nature Biotechnology 37: 852–857. https://doi.org/10.1038/s41587-019-0209-9
4. Callahan, B.J., McMurdie, P.J., Rosen, M.J., Han, A.W., Johnson, A.J. Holmes, S.P. (2016). Dada2: high-resolution sample inference from illumina amplicon data. Nature methods, 13(7):581.
doi:10.1038/nmeth.3869.
5. Gong, W., Hall, N., Hans, P., Marchetti, A. (2020). Phytoplankton composition in a eutrophic estuary: Comparison of multiple taxonomic approaches and influence of environmental factors. Env. Microbiol, 6. Gong W and Marchetti A. (2019) Estimation of 18S gene copy number in marine eukaryotic plankton using
7. Fadrosh, D. W. et al. (2014). An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome 2, 1–7
8. Falkowski, P. G., R. T. Barber, and V. Smetacek (1998), Biogeochemical controls and feedbacks on ocean primary production, Science, 281(5374), 200–206.
9. Fluid Imaging Technologies. (2011). FlowCam Manual. Version 3.0.
10. Hrycik, A., Shambaugh, A., Stockwell, J.D. (2019). Comparison of FlowCAM and microscope biovolume measurements for a diverse freshwater phytoplankton community, Journal of Plankton Research,
41(6),849–864, https://doi.org/10.1093/plankt/fbz056
11. Karlusich, J.P.P., Ibarbalz, F.M., Bowler, C. (2020). Phytoplankton in the TARA ocean. Annual Review of Marine Science, 12(1), 233-265. https://doi.org/10.1146/annurev-marine-010419-010706
12. Kittelmann, S., Devente, S.R., Kirk, M.R., Seedorf, H., Dehority, B.A., Janssen, P.H. (2015). Phylogeny of intestinal ciliates, including Charonina ventriculi, and comparison of microscopy and 18S rRNA gene pyrosequencing for rumen ciliate community structure analysis. Appl Environ Microbiol, 81(7),2433–2444. doi:10.1128/AEM.03697-14.
13. Kozich, J. J., Westcott, S. L., Baxter, N. T., Highlander, S. K. & Schloss, P. D. (2013). Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 79, 5112–5120.
14. Marinov, I., Doney, S. C., Lima, I. D., Lindsay, K., Moore, J. K., and Mahowald, N. ( 2013), North‐South asymmetry in the modeled phytoplankton community response to climate change over the 21st century,
Global Biogeochem. Cycles, 27(4),1274– 1290, doi:10.1002/2013GB004599.
15. Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. journal, 17(1):10. doi:10.14806/ej.17.1.200.
16. Parsons, T. R., Y. Maita, And C. M. Lalli. (1984). A manual of chemical and biological methods for seawater analysis. Pergamon Press.
17. Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. (2012). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res
41:D590–D596. doi:10.1093/nar/gks1219
18. Rognes T, Flouri T, Nichols B, Quince C, Mahé F. (2016). VSEARCH: a versatile open source tool for metagenomics. PeerJ 4:e2584. doi:10.7717/peerj.2584.
19. Sieracki C. K., Sieracki M. E., Yentsch C. S. (1998). An imaging-in-flow system for automated analysis of marine microplankton, Mar. Ecol. Prog. Ser., 168,285-29610.3354/meps168285
20. Sarthou, G., Timmermans, K. R., Blain, S. & Treguer, P. (2005) Growth physiology and fate of diatoms in the ocean: a review. J. Sea Res. 53, 25–42
21. Tkacz, A., Hortala, M. & Poole, P.S. (2018). Absolute quantitation of microbiota abundance in environmental samples. Microbiome6, 110. https://doi.org/10.1186/s40168-018-0491-7 22. Utermöhl, H. (1958) Methods of collecting plankton for various purposes are discussed., SIL