Abstract
Investigation of the reduction-protonation pathway from ruthenium polypyridyl hydroxymethyl to free methanol is conducted. Metal-coordinated acetonitrile ylide (-CH2NCCH3) complexes are formed following protonation of [Ru(5,5’-Me2bpy)2(CO)(CH2OH)][PF6] and
(π-C5H5)Fe(CO)2(CH2OCH3) by triflic acid (CF3SO3H) in the presence of excess acetonitrile indicating H+ attack at the oxygen atom. DFT computational methods are utilized to optimize gas-phase structures of Ru(tpy)(bpy)(X)+ and Ru(5,5’-Me2bpy)2(CO)(X)+ (where X = H, OCH3, CH2OH) to construct a reaction coordinate diagram describing hydride transfer to formaldehyde to yield the isomer adducts, hydroxymethyl and methoxy.
Introduction
The growing global energy demand and consequential increase in greenhouse gas
emissions are two of the most prominent issues facing present-day society. The past few decades have seen advances in efficiency for numerous sources of renewable energy; however, the world as a whole is still largely dependent on extracting energy from fossil fuels. For these reasons, continued research on photochemical and photoelectrochemical reduction of CO2 is essential to balancing the global carbon footprint and producing liquid fuels to replace coal and petroleum. There are many aspects of photoelectrochemical CO2 reduction that make it an attractive alternative energy source: the abundance and stability of CO2; the limitless, free supply of solar radiation; and the proof of concept provided by nature in photosynthesis. Recycling carbon dioxide into a useable energy source would radically alter the world’s energy production landscape; however, significant challenges make difficult the production of an economically viable fuel. Severe overpotentials make electrochemical CO2 reduction energetically expensive. Additionally, the production of commercially-important energy sources like formic acid and methanol require coupled multi-electron and multi-proton addition steps. Fortunately, solar energy utilization continues to be a major research focus, resulting in major advances in overcoming the obstacles associated with photoelectrochemical CO2 reduction1.
As previously mentioned, photoelectrochemical reduction is the process by which nearly all plants convert CO2 into metabolic fuel (in the form of glucose). Thus, using this idea to produce liquid fuels is often called “artificial photosynthesis” with the commercial device being a solar-driven, water oxidation-CO2 reduction-coupled fuel cell whereby the chlorophyll, enzymes, and biomolecules of photosynthesis are replaced with chromophore dye, molecular catalysts, and nanomaterials. Figure 1 shows the design of the dye-sensitized
Figure 1. Dye-sensitized photoelectrosynthesis cell (DSPEC) schematic showing the proposed conversion of H2O and CO2 into O2 gas and fuel (in this case CO)10.
However, for a majority of catalysts, reduction of CO2 results in the formation of toxic, corrosive, or otherwise undesirable fuel sources such as CO and HCOOH11,12,13. Methanol, CH3OH, has also been produced from the reduction of CO2 and can serve as a medium for the storage of hydrogen, another promising energy source14,15.The high energy density of methanol (twice that of liquid hydrogen) and ability to drive conventional combustion engines without modification make it an attractive fuel source as well.16
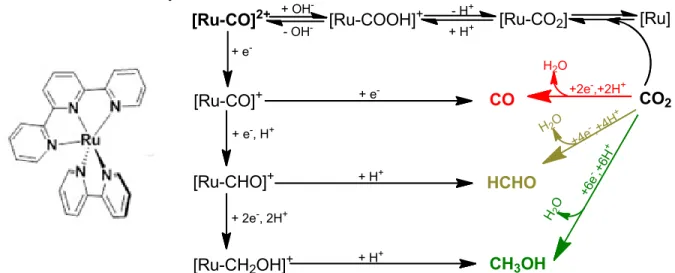
Building on nearly 100 years of industrial catalytic CO2 reduction to methanol, Tanaka, et al.9 investigated the hydride reduction pathways of a reduced Ru(tpy)(bpy) catalyst (tpy = 2,2’:6,2’’-terpyridine, bpy = 2,2’-bipyridine). The study reported a quantitative conversion of CO2 to methanol proposed to occur via formyl (-CHO) and hydroxymethyl (-CH2OH) group formation on the Ru catalyst.
[Ru-CO]2+ [Ru-COOH]+ [Ru-CO2] [Ru]
[Ru-CO]+
[Ru-CHO]+
[Ru-CH2OH]+ CH3OH
HCHO
CO2 CO
+ OH -- OH
-- H+ + H+ + e
-+ e-, H+
+ 2e-, 2H+
+ H+ + H+
+ e- +2e-,+2H+
H2O
+4e -,+4H
+
+6e
-,+6H +
H2O
H2O
As seen in the bottom step, the conversion of [Ru-CH2OH]+ to CH3OH (Figure 2), Tanaka et al. proposes that protonation occurs at carbon.9 However, research by the Schauer group indicates this may not be the case, launching an investigation into the products of various reactions along the protonation/reduction pathway using Ru(tpy)(bpy) and analogous ruthenium polypyridyl complexes.
An exploration of the protonation reactions of d6 ruthenium and iron analogs to Ru(tpy) (bpy)(CH2OH)+ is presented. From there, conversion between hydrido, hydroxymethyl, and methoxy adducts of ruthenium polypyridyl complexes is investigated though synthetic and DFT methods.
Experimental Section
Materials. All manipulations were performed under atmosphere of dry nitrogen using either standard Schlenk-line or glove box techniques, except where noted. Optima grade
dichloromethane and acetonitrile solvents were purchased from Fisher Scientific and dried through two columns of alumina and dispensed into Teflon-sealed schlenk flasks for storage. Solvents acetone, diethyl ether, tetrahydrofuran, ethanol (200 proof), and hexanes were purchased from Fisher Scientific then purged with nitrogen gas and dried over 4Å molecular sieves. Ruthenium(II) trichloride hydrate and cyclopentadienyl iron(II) dicarbonyl dimer were purchased from Sigma-Aldrich. Sodium borohydride and trimethylamine-N-oxide were also purchased from Fisher Scientific and stored under nitrogen. Infrared spectra was taken on a Bruker ALPHA Fourier Transform IR spectrometer. NMR spectra were obtained on a Bruker 400 MHz Nanobay. Variable temperature spectra were obtained on a Bruker AVANCE III 500 MHz spectrometer with BBI (inverse broadband).
Computational Methods. All calculations were carried out with the Gaussian 09 program package17 with LanL2DZ ECP basis18–20 for Ru. For all carbon, hydrogen, oxygen, and nitrogen atoms, the 6-31G(d,p) basis set was used21. All calculations employed the hybrid DFT B3LYP method22–24 in the gas phase. Transition state structures and energies were obtained from calculations using the Synchronous Transit-Guided Quasi-Newton (STQN) Method25 requested by the QST3 option in Gaussian.
Syntheses. cis-[Ru(bpy’)2(CO)2][PF6]226 (bpy’ = 5,5’-dimethyl-2,2’-bipyridine),
(π-C5H5)Fe(CO)2(CH2OCH3)27 ([Fp-CH2OCH3]+), and [Ru(tpy)(bpy)(H)][PF6]28 were prepared by using modifications of literature procedures.
[Ru(bpy’)2(CO)(CH2OH)][PF6] ([Ru-CH2OH]+). Preparation was adapted from Nagao, et al.9A sample of [Ru(bpy’)2(CO)2][PF6]2 (206.8 mg, 0.307 mmol) was dissolved in MeCN in a schlenk flask. An 11 mL aqueous solution of NaBH4 (208.0 mg, 5.50 mmol) was prepared in a separate schlenk flask. Excess NaBH4 (1.8 equivalents = 1.1 mL of stock solution) was added to MeCN solution of ruthenium complex cooled to approximately -30°C using asalt water/ice bath. The slightly yellow-tinted solution became bright yellow, then orange and finally deep red in color. The crude sample mixture was allowed to gradually warm to room temperature, stirring
and diethyl ether. The resulting bright orange-red solid powder was stored in drybox. Average isolated yield, 175.5 mg, 0.215 mmol, 70%.
1H NMR (25 °C, CD2Cl2, δ, ppm): 0.85 (t, 1H, OH in CH2OH), 2.16, 2.19, 2.53, 2.57 (s, 3H, Me on Me2bpy), 4.40, 4.52 (dd, 1H, CH2 in CH2OH, 2JHH = 7.0 Hz, 3JHH = 5.0 Hz), 7.03 (s, 1H), 7.43 (s, 1H), 7.74, 7.77, 7.83, 7.97 (d, 1H, 3JHH = 8.5 Hz), 8.10 (m, 3H), 8.19, (d, 1H), 9.02 (s, 1H), 9.30 (s, 1H). ) [7 – 10 ppm: bpy on Me2bpy].
FTIR (CH2Cl2): v(CO) at 1934 cm-1. ESI-MS m/z (CH3CN): 529.16
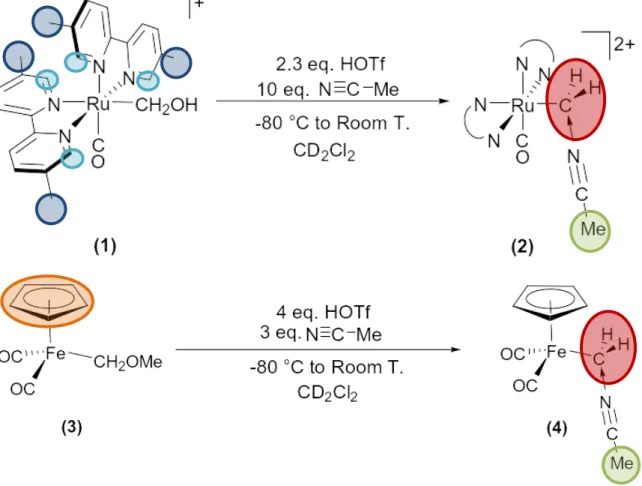
Low Temperature NMR Protonation of [Ru(bpy’)2(CO)(CH2OH)][PF6]. A sample of [Ru-CH2OH]+ (3.2 mg, 0.0048 mmol) was added to an NMR tube and dissolved using 600 μL CD2Cl2 via cannula transfer of the solvent at room temperature. The sample was cooled to -78°C using a dry ice/acetone cold bath in a tall dewar fitted with foam lid used to keep NMR tube in place. A 113 mM stock solution of HOTf in CD2Cl2 was prepared by addition of 5 µL of neat HOTf to 495 µL of CD2Cl2. For [Ru-CH2OH]+, an estimated 10.0 equivalents of dry acetonitrile (2.5μL, 0.048 mmol) and an estimated 2.3 equiv. of HOTf stock solution (99.3 μL, 0.0112 mmol) were added via syringe to the sample. The sample was stirred with a 2 mm glass rod slowly to mix at low temperature, and then transferred to an NMR probe. 1H NMR spectra were collected starting at -70 °C, and in 10°C increments until reaching room temperature.
1H NMR for initial product ([Ru-CH2NMe]2+) (20 °C, CD2Cl2, δ, ppm): 2.20 (s, 6H, Me on Me2bpy), 2.33 (br t, 3H, Me on MeCN-ylide, 5JHH = 3.0 Hz), 2.61 (d, 6H, Me on Me2bpy, 2JHH = 5.0 Hz), 3.90 (dq, 1H, CH2 on MeCN-ylide, 2JHH = 13.5 Hz, 5JHH = 3.0 Hz), 3.30 (dq, 1H, CH2 on MeCN-ylide, 2JHH = 13.5 Hz, 5JHH = 3.0 Hz), 7.03 (s, 1H), 7.27 (s, 1H), 7.82, 7.91, 8.05 (d, 1H, 3JHH = 8.5 Hz), 8.10 (d, 2H, 3JHH = 8.0 Hz), 8.20, 8.23, 8.26 (d, 1H, 3JHH = 8.0 Hz), 8.37 (s, 1H), 8.99 (s, 1H) [7 – 10 ppm: bpy on Me2bpy].
Low Temperature NMR Protonation of (π-C5H5)Fe(CO)2(CH2OCH3) ([Fp-CH2OMe]). A sample of [Fp-CH2OMe] (42 mg, 0.189 mmol) was added to an NMR tube and dissolved using 600 μL CD2Cl2 via cannula transfer of the solvent at room temperature. The sample was cooled to -78°C using a dry ice/acetone cold bath in a tall dewar fitted with foam lid used to keep NMR tube in place. A 113 mM stock solution of HOTf in CD2Cl2 was prepared by addition of 5 µL of neat HOTf to 495 µL of CD2Cl2. For [Fp-CH2OMe]+, an estimated 3.0 equiv. of dry acetonitrile (29.7 μL, 0.567 mmol) and an estimated 4.0 equiv. of HOTf stock solution (66.8 μL, 0.756 mmol) were added via syringe to sample. The sample was stirred with a 2 mm glass rod slowly to mix at low temperature, and then transferred to an NMR probe. 1H NMR spectra were collected starting at -70°C and in 10°C increments until reaching room temperature.
1H NMR for initial product ([Fp-CH2NMe]+) (20°C, CD2Cl2, δ, ppm): 2.70 (t, 3H, Me on MeCN-ylide, 5JHH = 3.0 Hz) 3.53 (q, 2H CH2 on MeCN-ylide, 5JHH = 3.0 Hz) 3.77 (s, 3H, MeOH by product) 5.06 (s, 5H, Hs of Cp ring).
NaBH4 addition to [Ru(bpy’)2(CO)(CH2OH)][PF6]. A sample of [Ru-CH2OH)+] (5 mg, 0.0074 mmol) was added to an NMR tube, dissolved in 600 μL CD3CN, and cooled in ice water (-10°C). A 1.2 M NaBH4 solution was prepared by dissolving 32.3 mg NaBH4 in 700 μL D2O. To the NMR sample, 4 equiv. of NaBH4 stock solution (25 μL, 0.030 mmol) was added via syringe. The sample was analyzed by 400 MHz 1H NMR at room temperature.
3JHH = 3.6 Hz), 7.07 (s, 1H), 7.50 (s, 1H), 7.75, 7.78, 7.84, 8.00 (d, 1H, 3JHH = 8.5 Hz), 8.21 (m, 3H), 8.28 (d, 1H), 8.98 (s, 1H), 9.08 (s, 1H). [7 – 10 ppm: bpy on Me2bpy]. [Ru(bpy’)2(CO)(NCCH3)][PF6]2 ([Ru-NCMe]2+). A solution of dry trimethylamine-N-oxide (0.486 mmol, 1.2 eq) (TMNO) in dry acetonitrile was added dropwise to a solution of [Ru(5,5’-Me2bpy)2(CO)2][PF6]2 (0.330 g, 0.405 mol) dissolved in 30 mL dry acetonitrile and stirred overnight. Addition of TMNO resulted in a color change from pale yellow to bright yellow. Addition of diethyl ether precipitated a bright yellow solid which was washed with 3 x 10 mL diethyl ether, and dried in vacuo; yield, 268.5 mg, 0.324 mmol, 80 %.
1H NMR (25°C, CD3CN, δ, ppm): 2.16, 2.27, 2.60, 2.62 (s, 3H, Me of Me2bpy), 7.20, 7.33 (s, 1H), 7.88 (d, 1H, 3JHH = 8.4 Hz), 7.990 (d, 1H, 3JHH = 8.4 Hz), 8.13 (d, 1H, 3JHH = 8.4 Hz), 8.21 (m, 2H), 8.32-8.35 (m, 4H), 8.81, 9.06 (s, 1H) [7 – 10 ppm: bpy on
Me2bpy].
FTIR (NCMe): v(CO) at 2011 cm-1.
Ru(bpy’)2(CO)(H)][PF6]. Synthesis was adapted from Konno, et al28. An aqueous solution of NaBH4 (0.637 mol, 2.3 eq.) was added dropwise to [Ru(bpy’)2(CO)(NCCH3)][PF6]2 (0.278 g, 0.335 mmol) dissolved in 2:1 ethanol (200 proof) – degassed H2O and the mixture was stirred for 5 hours under N2(g) atmosphere. Over course of reaction, the yellow solid turned to a deep red. After stirring, the solution was concentrated in vacuo to one-quarter of the original volume precipitating a fine red-brown solid, which was filtered, washed with 3 x 5 mL H2O, and dried in vacuo;Average isolated crude yield, 70 %. FTIR (NCMe): v(CO) at 1933 cm-1. The product was further purified on an alumina column using 2:1 toluene-acetonitrile as eluent. Red and yellow bands elute first and both give the purified complex. All eluent was evaporated off and the product was recrystallized with acetonitrile and water; Isolated pure yield, 43.1 mg, 0.067 mmol, 20 %.
1H NMR (25°C, CD3CN, δ, ppm)): –11.57 (s, 1H, Ru-H), 2.17, 2.21, 2.50, 2.51 (s, 3H, Me on Me2bpy), 7.08 (s, 1H), 7.42 (s, 1H), 7.78 (t, 3H, 3JHH = 6.4 Hz), 7.93 (d, 1H, 3JHH = 8.4 Hz), 8.10 (t, 3H, 3JHH = 8.0 Hz), 8.18 (d, 1H, 3JHH = 8.4 Hz), 9.02 (s, 2H) [7 – 10 ppm: bpy on Me2bpy].
FTIR (NCMe): v(CO) at 1933 cm-1
ESI MS (NCMe): m/z 499 (main peak, M+ where M = [Ru(bpy’)2(CO)(H)].
Results and Discussion
Protonation of [Ru(bpy’)2(CO)(CH2OH)][PF6] and (π-C5H5)Fe(CO)2(CH2OCH3) leading to acetonitrile ylide formation. Tanaka et al. provides evidence for the formation of formyl and hydroxymethyl adducts of Ru(tpy)(bpy) and liberation of methanol following NaBH4 addition in protic solvents. The NaBH4 addition experiment was repeated by the Schauer group with the less reactive Ru(bpy’)2(CO)(CH2OH)+ analog and although methanol was present in the 1H NMR spectrum, it was revealed the NaBH4 sample was the source of methanol. Tanaka’s claim of protonation in the conversion of the hydroxymethyl adduct to free methanol should elicit a red flag. Chemical intuition assigns H+ as the electrophile and following nucleophilic attack, would form a bond with the electronegative –OH group, not tetravalent carbon. In response to this disparity between experimental and theory, protonation reactions were conducted on
(CH2OH)]+ allowing protonation reactions to be studied by NMR spectroscopy. The iron
complex was chosen based on the work of Nelson29, who observed carbene formation following the protonation of the (π-C5H5)Fe(CO)2(CHPhOCH3). Carbene formation, we hypothesize, is part of the protonation mechanism and will be discussed later. Trifluoromethanesulfonic acid, or triflic acid (HOTf), was chosen as the reagent for protonation and acetonitrile (NCMe) was chosen as the Lewis base to coordinate to either the carbene or ruthenium, depending on the site of protonation. NMR scale protonation reactions were conducted under the conditions described in Figure 3 and the subsequent NMR spectra are shown in Figure 4 for the ruthenium and iron systems.
Figure 4. 500 MHz 1H NMR (273 K, CD2Cl2) spectra A and B following protonation reaction on (1) and (2).
For 1H NMR spectrum A (Figure 4), the resonances indicated by the red spheres show two doublets of quartets consistent with what would be expected for the diastereotopic
methylene hydrogens of the acetonitrile ylide. The quartets and lone triplet splitting patterns result from the 5-bond J-coupling between the hydrogens of the methylene and nitrile methyl group. As proof, both sets of resonances display 5JHH values of 3.0 Hz. NMR spectrum B (Figure 4) also shows evidence for the formation of the acetonitrile ylide. Again, coupling between a quartet and triplet is observed with an identical 5JHH value of 3.0 Hz. A single quartet exists in place of the doublet of quartet pair due to the σv symmetry of the iron system resulting in a loss of coupling between the methylene hydrogens of the ylide. Although they are not shown in Figure 4, appropriate integration values were found for the respective resonances.
Figure 5. Mechanism for protonation and Lewis base attack that yields the acetonitrile ylide products mentioned previously. Protonation occurs on the oxygen, followed by loss of water, formation a carbene intermediate, and coordination of a Lewis base to methylene.
These NMR results provide experimental evidence contradictory to the Tanaka-proposed reduction pathway to methanol, specifically the last step in which protonation on the
hydroxymethyl carbon affords free methanol. Formation of the acetonitrile ylide following protonation of [Ru-CH2OH]+ and [Fp-CH2OCH3]+ provides direct evidence for protonation occurring at oxygen. Additionally, it is hypothesized that the pathway to the acetonitrile ylide
A
B
goes through a carbene intermediate suggesting an SN1-type mechanism for Lewis base coordination; however, future experiments are needed to give irrefutable confirmation. Numerous experiments have been undertaken by the Schauer group to probe the reactivity of Ru(bpy’)2(CO)(CH2OH)+ under varying Lewis acid and base conditions with the underlying goal being to better understand the true pathway to methanol (if one even exists). However, the results of these experiments are the subject of a future paper.
Investigation of C1 reduction via hydride transfer by Ru polypyridyl complexes. The information learned in the previous study is valuable because free methanol could,
hypothetically, be produced via protonation by forming the oxygen-coordinated isomer of hydroxymethyl, more commonly referred to as methoxy (Figure 6).
Figure 6 . Mechanism for methanol liberation following protonation of the methoxy adduct.
Fortunately, methoxy adducts have been observed in ruthenium polypyridyl complexes. A study by Creutz et al.30 is of particular interest because it investigates the hydride transfer ability of [Ru(tpy)(bpy)(H)]+ (tpy = 2,2’,6’,2’’-terpyridine; bpy = 2,2’-bipyridine) to C1 species CO2, CO, and CH2O using water as a solvent. Results of this investigation warrant attention because, for one, the metal-coordinated products of hydride transfer are all oxygen-bonded. Secondly, rate constants suggest a direct dependence on Gutmann solvent acceptor number31 and indicate extremely fast rates for the reactions of CO2 and formaldehyde with [Ru-H]+ in water (k = 8.5x105 M-1 s-1 and k ~ 1x106 M-1 s-1, respectfully). Finally, characterization of hydride transfer products was conducted using mass spectrometry. This approach is somewhat problematic, however, as mass spec is unable to differentiate between structural isomers. An NMR analysis would help confirm the product identities and provide greater insight as to the hydride transfer mechanism. To develop a basic understanding of the mechanism for this class of hydride transfer reactions, both synthetic and computational studies were attempted. The synthetic study will be discussed first.
The scope of the synthetic investigation includes preparation of ruthenium polypyridyl hydride complexes [Ru(tpy)(bpy)(H)][PF6] and [Ru(bpy’)2(CO)(H)][PF6]and monitoring of their reaction with CH2O by NMR. Again, the bis-bpy’-carbonyl analog was chosen for the
investigation in case the tpy-bpy system showed reactivity too rapid for NMR analysis. Synthesis and characterization of [Ru(tpy)(bpy)(H)][PF6] has been well documented28 and involves
Figure 7. Developed synthetic pathway to [Ru(bpy’)2(CO)(H)]+ with accompanying isolated solid colors and CO stretching frequencies for each complex. Colored spheres are used for NMR identification in Figure 8.
At each successive step in the pathway, the CO stretching decreases in frequency in agreement with the decreasing electron withdrawing ability of the substituted ligand, and thus increasing π-backbonding to CO from left to right. The 1H NMR spectrum of alumina column-purified Ru(bpy’)2(CO)(H)+ (Figure 8) gives characteristic upfield bpy’-CH3 singlet resonances from 2.1-2.5 ppm (dark blue circles). Downfield bpy’-H singlet resonances are present (light blue circles), two of which are coincident at a shift of ~9.0 ppm. Finally, the far upfield
resonance at -11.56 ppm is indicative of a metal hydride, in this case Ru-H of Ru(bpy’)2(CO)(H) +.
Figure 8. 400 MHz 1H NMR (298 K, CD3CN) spectrum of column-purified Ru(bpy’)2(CO)(H)+ sample.
Naturally, the next step in the investigation is to react both [Ru-H]+ complexes with formaldehyde; however, isolation of pure formaldehyde proved to be exceedingly difficult. Acid-and base-catalyzed decompositions Acid-and thermal “cracking” of paraformaldehyde were attempted; however, they were not yet successful in producing pure formaldehyde.
Figure 9. Calculated reaction coordinate diagram for Ru(tpy)(bpy)(X)+ system where X = H, OCH3, or CH2OH.
hydroxymethyl complexes is slightly larger for the Ru(tpy)(bpy)+ system than the Ru(bpy’)2(CO) + system (7.50 kcal/mol versus 6.71 kcal/mol, respectively). The oxygen-coordinated, methoxy complex being the lower energy isomer is a seemingly unexpected outcome when considering the resulting electron repulsion between the filled dπ (t2g) orbitals of the d6 ruthenium center and filled p orbitals of oxygen. However, it may be that the σ-donating ability of the methoxy is able to overcome this repulsion and bring the complex to lower energy. In each system, the Ru-O bond distance for methoxy is ~0.07 Å smaller than the Ru-C bond distance of hydroxymethyl (see Figures 11 and 12). Transition state energies were also calculated
to assess the kinetics of hydride transfer. In each case, the transition from methoxy and
hydroxymethyl complexes to the hydrido complex and free formaldehyde was predicted to go by a β-hydride elimination-like reaction whereby hydride coordination to the metal occurs
Ru—O Ru—C Ru—H --C—O-- --C—H
Ru(tpy)(bpy)(H)+ --- --- 1.609 ---
---Ru(tpy)(bpy)‡ 2.689 (Å) 2.706 2.068 1.351 1.161
Ru(tpy)(bpy)(OCH3)+ 2.037 3.034 3.307 1.396 1.106
Ru(bpy’)2(CO)(H)+ --- --- 1.602 ---
---Ru(bpy’)2(CO)‡ 2.712 2.761 2.156 1.346 1.174
Ru(bpy’)2(CO)(OCH3)+ 2.042 3.019 3.270 1.399 1.105
Figure 11. Calculated bond distances (in angstroms) for states along reaction pathway between Ru-OCH3+ and Ru-H+ + CH2O for both polypyridyl systems. ‡Indicates a transition state.
Ru—O Ru—C Ru—H --C—O-- --O—H
Ru(tpy)(bpy)(H)+ --- --- 1.609 ---
---Ru(tpy)(bpy)‡ 3.216 2.410 1.900 1.373 1.351
Ru(tpy)(bpy)(CH2OH)+ 2.937 2.104 3.808 1.442 0.968
Ru(bpy’)2(CO)(H)+ --- --- 1.602 ---
---Ru(bpy’)2(CO)‡ 3.144 2.440 1.828 1.383 1.338
Ru(bpy’)2(CO)(CH2OH)+ 2.935 2.125 3.811 1.441 0.968
simultaneously with carbon/oxygen dissociation liberating free formaldehyde. All calculated transition state free energies (ΔG‡) are exceedingly large (see Figures 9 and 10). Applying the Eyring equation, which relates ΔG‡ to the reaction rate constant, to the lowest energy barrier reaction at room temperature ([Ru(tpy)(bpy)(H)]+ + CH2O → Ru(tpy)(bpy)(OCH3)]+; ΔG‡ = 42.89 kcal/mol) gives a rate constant of 1.70x10-19 s-1 and a corresponding t1/2 of 130 billion years. Unfortunately, reducing the rate with high reaction temperatures is not practical as both hydride systems are rather heat sensitive and decompose above ~313K. DFT calculations have managed to predict the correct product in hydride transfer to formaldehyde but fail to confirm the low energy barrier necessary for rapid hydride transfer as observed by Creutz. It is important though to realize that the calculations discussed involve molecules in the gas-phase, and it is possible that inclusion of an appropriate solvent model could stabilize the transition state and significantly lower ΔG‡. Additionally, the disparity between experimental and computational results might be a consequence of calculating the wrong mechanistic pathway. There may exist a lower energy pathway nullifying the assumption that the transition state follows from a concerted hydride transfer-O/C coordination reaction (or β-hydride elimination reaction, depending on the direction). Thus, future work will study the effect of including a solvent model and will also test alternative mechanistic pathways. One idea to be explored involves dechelation of a pyridine ring of terpyridine or bipyridine followed by β-hydride elimination to the vacant coordination site, accompanied by loss of CH2O.
Figure 12. Overlaid calculated reaction coordinate diagrams for Ru(tpy)(bpy)(X)+ and Ru(bpy’)-2(CO)(X)+ systems where X = H, OCH3, or CH2OH.
Conclusion
Protonation of [Ru(bpy’)2(CO)(CH2OH)]+ and (π-C5H5)Fe(CO)2 (CH2OCH3) followed by acetonitrile coordination yields the acetonitrile ylide complex in each case suggesting
proposed. A new pathway for the synthesis of [Ru(bpy’)2(CO)(H)]+ is reported. This complex and its tpy-bpy analog await reaction with CH2O once it can be isolated in its monomeric form. Lastly, DFT-calculated ΔG‡ values for the reaction of [Ru-H]+ complexes with CH2O to form hydroxymethyl or methoxy adducts could indicate the importance of solvent in stabilization or the likelihood of a more complex hydride transfer mechanism than originally proposed.
Acknowledgment. This research was supported by the University of North Carolina at Chapel Hill Energy Research Frontier Center, a grant from the US Dept. of Energy. A special thanks goes out to Dr. Cynthia Schauer and Marsha Massey for their keen advice and constant encouragement.
References
(1) Kumar, B.; Llorente, M.; Froehlich, J.; Dang, T.; Sathrum, A.; Kubiak, C. P. Annu. Rev. Phys. Chem. 2012, 63, 541.
(2) Fisher, B. J.; Eisenberg, R. J. Am. Chem. Soc. 1980, 102 (24), 7361.
(3) Bolinger, C. M.; Story, N.; Sullivan, B. P.; Meyer, T. J. Inorg. Chem. 1988, 27 (25), 4582.
(4) Grodkowski, J.; Neta, P.; Fujita, E.; Mahammed, A.; Simkhovich, L.; Gross, Z. J. Phys. Chem. A 2002, 106 (18), 4772.
(5) Raebiger, J. W.; Turner, J. W.; Noll, B. C.; Curtis, C. J.; Miedaner, A.; Cox, B.; DuBois, D. L. Organometallics 2006, 25 (14), 3345.
(6) Sahara, G.; Ishitani, O. Inorg. Chem. 2015.
(7) Bruce, M. R. M.; Megehee, E.; Sullivan, B. P.; Thorp, H. H.; O’Toole, T. R.; Downard, A.; Pugh, J. R.; Meyer, T. J. Inorg. Chem. 1992, 31 (23), 4864.
(8) Norris, M. R.; Concepcion, J. J.; Glasson, C. R. K.; Fang, Z.; Lapides, A. M.; Ashford, D. L.; Templeton, J. L.; Meyer, T. J. Inorg. Chem. 2013, 52 (21), 12492.
(9) Nagao, H.; Mizukawa, T.; Tanaka, K. Inorg. Chem. 1994, 33 (15), 3415.
(10) Alibabaei, L.; Luo, H.; House, R. L.; Hoertz, P. G.; Lopez, R.; Meyer, T. J. J. Mater. Chem. A 2013, 1 (13), 4133.
(11) Grice, K. A.; Kubiak, C. P. Adv. Inorg. Chem. 2014, 66, 163.
(12) Kang, P.; Cheng, C.; Chen, Z.; Schauer, C. K.; Meyer, T. J.; Brookhart, M. J. Am. Chem. Soc. 2012, 134 (12), 5500.
(14) Barton, E. E.; Rampulla, D. M.; Bocarsly, A. B. J. Am. Chem. Soc. 2008, 130 (20), 6342.
(15) Magdesieva, T. V; Zhukov, I. V; Kravchuk, D. N.; Semenikhin, O. A.; Tomilova, L. G.; Butin, K. P. Russ. Chem. Bull. 2002, 51 (5), 805.
(16) Bill, A. Carbon dioxide hydrogenation to methanol at low pressure and temperature, EPFL, 1998.
(17) Frisch, M. J. et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, 2009.
(18) Hay, P. J.; Wadt, W. R. J. Chem. Phys. 1985, 82 (1), 270.
(19) Wadt, W. R.; Hay, P. J. J. Chem. Phys. 1985, 82 (1), 284.
(20) Hay, P. J.; Wadt, W. R. J. Chem. Phys. 1985, 82 (1), 299.
(21) Hariharan, P. C.; Pople, J. A. Theor. Chim. Acta 1973, 28 (3), 213.
(22) Becke, A. D. Phys. Rev. A 1988, 38 (6), 3098.
(23) Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37 (2), 785.
(24) Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Chem. Phys. Lett. 1989, 157 (3), 200.
(25) Peng, C.; Bernhard Schlegel, H. Isr. J. Chem. 1993, 33 (4), 449.
(26) Anderson, P. A.; Deacon, G. B.; Haarmann, K. H.; Keene, F. R.; Meyer, T. J.; Reitsma, D. A.; Skelton, B. W.; Strouse, G. F.; Thomas, N. C. Inorg. Chem. 1995, 34 (24), 6145.
(27) Green, M. L. H.; Istaq, M.; Whiteley, R. N. J. Am. Chem. Soc. 1967, No. 1508.
(28) Konno, H.; Kobayashi, A.; Sakamoto, K.; Fagalde, F. Inorganica Chim. Acta 2000, 299, 155.
(29) Brookhart, M.; Nelson, G. O. J. Am. Chem. Soc. 1977, 99, 6099.
(30) Creutz, C.; Chou, M. H. J. Am. Chem. Soc. 2007, 129 (33), 10108.


![Figure 7. Developed synthetic pathway to [Ru(bpy’) 2 (CO)(H)] + with accompanying isolated solid colors and CO stretching frequencies for each complex](https://thumb-us.123doks.com/thumbv2/123dok_us/8331561.2210318/9.918.120.803.105.288/figure-developed-synthetic-pathway-accompanying-isolated-stretching-frequencies.webp)


