re
v
ie
w
s
re
ports
de
p
o
si
te
d r
e
se
a
rch
refer
e
e
d
re
sear
ch
interacti
o
ns
inf
o
rmation
Dmitri M Krylov
*
, Kira S Makarova
*
, Raja Mazumder
*†
,
Sergei L Mekhedov
*
, Anastasia N Nikolskaya
*
, B Sridhar Rao
*
,
Igor B Rogozin
*
, Sergei Smirnov
*
, Alexander V Sorokin
*
,
Alexander V Sverdlov
*
, Sona Vasudevan
*
, Yuri I Wolf
*
, Jodie J Yin
*
and
Darren A Natale
*†
Addresses: *National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, MD 20894, USA. †Current address: Protein Identification Resource, Georgetown University Medical Center, 3900 Reservoir Road, NW, Washington, DC 20007, USA.
Correspondence: Eugene V Koonin. E-mail: [email protected]
© 2004 Koonin et al; licensee BioMed Central Ltd. This is an Open Access article: verbatim copying and redistribution of this article are permitted in all media for any purpose, provided this notice is preserved along with the article's original URL.
A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes
Sequencing the genomes of multiple, taxonomically diverse eukaryotes enables in-depth comparative-genomic analysis which is expected to help in reconstructing ancestral eukaryotic genomes and major events in eukaryotic evolution and in making functional predictions for currently uncharacterized conserved genes.
Abstract
Background: Sequencing the genomes of multiple, taxonomically diverse eukaryotes enables in-depth comparative-genomic analysis which is expected to help in reconstructing ancestral eukaryotic genomes and major events in eukaryotic evolution and in making functional predictions for currently uncharacterized conserved genes.
Results: We examined functional and evolutionary patterns in the recently constructed set of 5,873 clusters of predicted orthologs (eukaryotic orthologous groups or KOGs) from seven eukaryotic genomes: Caenorhabditis elegans, Drosophila melanogaster, Homo sapiens, Arabidopsis thaliana, Saccharomyces cerevisiae, Schizosaccharomyces pombe and Encephalitozoon cuniculi. Conservation of KOGs through the phyletic range of eukaryotes strongly correlates with their functions and with the effect of gene knockout on the organism's viability. The approximately 40% of KOGs that are represented in six or seven species are enriched in proteins responsible for housekeeping functions, particularly translation and RNA processing. These conserved KOGs are often essential for survival and might approximate the minimal set of essential eukaryotic genes. The 131 single-member, pan-eukaryotic KOGs we identified were examined in detail. For around 20 that remained uncharacterized, functions were predicted by in-depth sequence analysis and examination of genomic context. Nearly all these proteins are subunits of known or predicted multiprotein complexes, in agreement with the balance hypothesis of evolution of gene copy number. Other KOGs show a variety of phyletic patterns, which points to major contributions of lineage-specific gene loss and the 'invention' of genes new to eukaryotic evolution. Examination of the sets of KOGs lost in individual lineages reveals co-elimination of functionally connected genes. Parsimonious scenarios of eukaryotic genome evolution and gene sets for ancestral eukaryotic forms were reconstructed. The gene set of the last common ancestor of the crown group consists of 3,413 KOGs and largely includes proteins involved in genome replication and expression, and central metabolism. Only 44% of the KOGs, mostly from the reconstructed gene set of the last common ancestor of the crown group, have detectable homologs in prokaryotes; the remainder apparently evolved via duplication with divergence and invention of new genes.
Conclusions: The KOG analysis reveals a conserved core of largely essential eukaryotic genes as well as major diversification and innovation associated with evolution of eukaryotic genomes. The results provide quantitative support for major trends of eukaryotic evolution noticed previously at the qualitative level and a basis for detailed reconstruction of evolution of eukaryotic genomes and biology of ancestral forms.
Published: 15 January 2004
Genome Biology 2004, 5:R7
Received: 23 October 2003 Revised: 1 December 2003 Accepted: 4 December 2003 The electronic version of this article is the complete one and can be
phylogeny. In particular, the comparative genomics of prokaryotes has revealed previously underappreciated major trends in genome evolution, namely, extensive lineage-spe-cific gene loss and horizontal gene transfer (HGT) [1-7]. To efficiently extract functional and evolutionary information from multiple genomes, rational classification of genes based on homologous relationships is indispensable. The two prin-cipal classes of homologs are orthologs and paralogs [8-11]. Orthologs are defined as homologous genes that evolved via vertical descent from a single ancestral gene in the last com-mon ancestor of the compared species. Paralogs are homolo-gous genes, which, at some stage of evolution, have evolved by duplication of an ancestral gene. Orthology and paralogy are intimately linked because, if a duplication (or a series of duplications) occurs after the speciation event that separated the compared species, orthology becomes a relationship between sets of paralogs, rather than individual genes (in which case, such genes are called co-orthologs).
Correct identification of orthologs and paralogs is of central importance for both the functional and evolutionary aspects of comparative genomics [12,13]. Orthologs typically occupy the same functional niche in different organisms; in contrast, paralogs evolve to functional diversification as they diverge after the duplication [14-16]. Therefore, robustness of genome annotation depends on accurate identification of orthologs. A clear demarcation of orthologs and paralogs is also required for constructing evolutionary scenarios, which include, along with vertical inheritance, lineage-specific gene loss and HGT [5,7].
In principle, orthologs, including co-orthologs, should be identified by means of phylogenetic analysis of entire families of homologous proteins, which is expected to define ortholo-gous protein sets as clades [17-19]. However, for genome-wide protein sets, such analysis remains extremely labor-intensive, and error-prone as well. Accordingly, procedures have been developed for identifying sets of likely orthologs without explicit referral to phylogenetic analysis. These pro-cedures are based on the notion of a genome-specific best hit (BeT), that is, the protein from a target genome that is most similar (typically in terms of similarity scores computed using BLAST or another sequence-comparison method) to a given protein from the query genome [20,21]. The assumption cen-tral to this approach is that orthologs have a greater similarity to each other than to any other protein from the respective genomes. When multiple genomes are analyzed, pairs of probable orthologs detected on the basis of BeTs are com-bined into orthologous clusters represented in all or a subset of the analyzed genomes [20,22]. This approach, amended with additional procedures for detecting co-orthologous pro-tein sets and for treating multidomain propro-teins, was imple-mented in the database of Clusters of Orthologous Groups
been used for functional annotation of new genomes [26-29], target selection in structural genomics [30-32], identification of potential drug targets [33,34] and genome-wide evolution-ary studies [4,13,35-38]. Sonnhammer and co-workers inde-pendently developed a similar methodology for identification of co-orthologous protein sets from pairwise genome com-parisons and applied it to the sequenced eukaryotic genomes [39].
A central notion introduced in the context of the COG analysis is that of a phyletic pattern, that is, the pattern of representa-tion (presence-absence) of analyzed species in each COG [13,20]. Similar concepts have been independently developed and applied by others [40,41]. The COGs show a remarkable scatter of phyletic patterns, with only a small minority repre-sented in all sequenced genomes. A recent quantitative study showed that parsimonious evolutionary scenarios for most COGs involve multiple events of gene loss and HGT [7]. Both similarity and complementarity among the phyletic patterns of COGs, in conjunction with other information, such as con-servation of gene order, have been successfully employed to predict gene functions [13,42,43]. The comparison of phyletic pattern has been formalized in set-theoretical algorithms and systematically applied to the computational and experimen-tal analysis of bacterial flagellar systems, which demonstrated the considerable robustness of this approach [44].
We recently extended the system of orthologous protein clus-ters to complex, multicellular eukaryotes [25]. Here, we examine the phyletic patterns of KOGs in connection with known and predicted protein functions. In-depth analysis of some of these KOGs resulted in prediction of previously uncharacterized, but apparently essential, conserved eukary-otic protein functions. We also reconstruct the parsimonious scenario of evolution of the crown-group eukaryotes by assigning the loss of genes (KOGs) and emergence of new genes to the branches of the phylogenetic tree and explicitly delineate the minimal gene sets for various ancestral forms. To our knowledge, this is the first systematic, genome-wide examination of the sets of orthologous genes in eukaryotes.
Results and discussion
KOGs for seven sequenced eukaryotic genomes: functional and evolutionary implications of phyletic patterns
en
t
re
v
ie
w
s
re
ports
refer
e
e
d
re
sear
ch
de
p
o
si
te
d r
e
se
a
rch
interacti
o
ns
inf
o
rmation
detail elsewhere ([25]; see also Materials and methods). An important difference stems from the fact that complex eukaryotes encode many more multidomain proteins than prokaryotes and, furthermore, orthologous eukaryotic pro-teins often differ in domain composition, with additional domains accrued in more complex forms [3,45]. Accordingly, and unlike the original COG construction procedure, proba-ble orthologs with different domain architectures were assigned to one KOG and were not split if they shared a com-mon core of domains. In addition to the KOGs, which con-sisted of at least three species, clusters of putative orthologs from two species (TWOGs) and lineage-specific expansions (LSEs) of paralogs from each of the analyzed genomes were identified ([25,52]; see also Materials and methods). In most of the analyses discussed below, KOGs and TWOGs are treated together, unless otherwise specified.
[image:3.612.311.554.85.288.2]Figure 1 shows the assignment of the proteins from each of the analyzed eukaryotes to KOGs with different numbers of species, TWOGs and LSEs. The fraction of proteins assigned to KOGs tends to decrease with the increasing genome size, from 81% for S. pombe to 51% for the largest, the human genome. (For reasons that remain unclear, but might be related to its intracellular parasitic lifestyle, E. cuniculi has a relatively small fraction of conserved proteins that belonged to KOGs: approximately 60%.) The contribution of LSEs shows the opposite trend, being the greatest in the largest genomes, that is, human and Arabidopsis, and minimal in the microsporidian (Figure 1). A notable difference was observed between eukaryotes in terms of their representation in KOGs found in different numbers of species. While the three unicel-lular organisms are represented mainly in the highly con-served seven- or six-species KOGs, a much larger fraction of the gene set in animals and Arabidopsis is accounted for by LSEs, and by KOGs found in three or four genomes. These include animal-specific genes and genes that are shared by plants and animals but not by fungi and the microsporidian (Figure 1). The large number of KOGs in the latter group (700 KOGs represented in Arabidopsis and at least two animal species) is notable and probably results from massive, line-age-specific loss of genes during eukaryotic evolution (see below).
The phyletic patterns of KOGs reveal both the existence of a conserved eukaryotic gene core and substantial diversity. The 'pan-eukaryotic' genes, which are represented in each of the seven analyzed genomes, account for around 20% of the KOGs, and approximately the same number of KOGs include all species except for the microsporidian, an intracellular par-asite with a highly degraded genome [51]. Among the remain-ing KOGs, a large group includes representatives of the three analyzed animal species (worm, fly and humans) but a sub-stantial fraction (approximately 30%) are KOGs with
unexpected patterns, for example, one animal, one plant and one fungal species (see [53] and examples in Table 1).
During the manual curation of the KOG set, the KOGs with unexpected patterns were scrutinized in an effort to detect potential highly diverged members from one or more of the analyzed genomes. Some of these unexpected patterns might indicate that a gene is still missing in the analyzed set of pro-tein sequences from one or more of the species included; reports of newly discovered genes have appeared since the release of the initial reports on genome sequences of complex eukaryotes, for example, as a result of massive sequencing of human cDNAs [54], exhaustive annotation of the Drosophila genome [55] and comparative analysis of closely related yeast genomes [56]. The unexpected phyletic patterns seem, how-ever, largely to reflect the extensive, lineage-specific gene loss that is characteristic of eukaryotic evolution [57]; on many occasions, this scenario is supported by the presence of orthologs in other eukaryotic lineages and/or in prokaryotes (Table 1). However, interesting exceptions to the multiple loss explanation might exist as exemplified by the ATP/ADP-translocase, which is present in Arabidopsis and Encepha-litozoon and could have evolved via independent HGT from intracellular bacterial parasites ([58] and Table 2).
Common phyletic patterns of genes that otherwise were not suspected to be functionally linked might suggest the exist-ence of such connections and prompt additional analysis
Assignment of proteins from each of the seven analyzed eukaryotic genomes to KOGs of with different numbers of species and to LSEs
Figure 1
Assignment of proteins from each of the seven analyzed eukaryotic genomes to KOGs with different numbers of species and to LSEs. 0, Proteins without detectable homologs (singletons); 1, LSEs. Species abbreviations: Ath, Arabidopsis thaliana; Cel, Caenorhabditis elegans; Dme, Drosophila melanogaster; Ecu, Encephalitozoon cuniculi; Hsa, Homo sapiens; Sce, Saccharomyces cerevisisae; Spo, Schizosaccharomyces pombe.
0 1
2 3
4 5
6 7 Ecu Sce
SpoAth CelDme
Hsa
Number of prote
ins
Number of species in KOGs 0
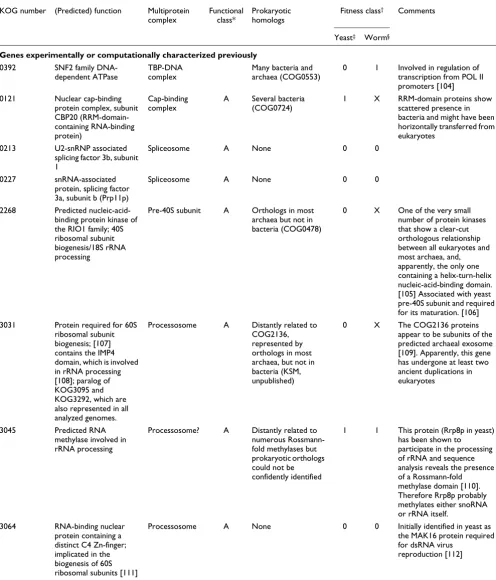
KOG/TWOG number
Phyletic pattern* (Predicted) structure and function
Prokaryotic homologs Comments
TWOG0892 ---H--E Discoidin domain protein, potential regulator of proteasome activity
Detected in a few phylogenetically scattered bacteria, no COG so far [69]
TWOG0263 A---E ATP/ADP translocase ATP/ADP translocases of chlamydia, rickettsia, Xylella fastidiosa
ATP/ADP translocase is a hallmark of intracellular parasites and symbionts, which allows them to scavenge ATP from the host cell; chloroplast protein in plants. Could be acquired by plants and microsporidia via independent HGT from bacteria. [58]
TWOG0689 ---HY-- Uncharacterized protein essential for propionate metabolism
PrpD protein of several bacteria and archaea (COG2079)
The yeast and human (and the orthologs from other vertebrates) proteins show the greatest similarity to different subsets of bacterial orthologs, which might suggest independent HGT events.
TWOG0871 ---H-P- Uncharacterized conserved protein, probably enzyme
COG4336, sporadic representation in several bacterial lineages
The human (and mouse) protein has an additional domain conserved in the archaeon Pyrococcus. Human and S. pombe proteins are most similar to different subsets of bacterial homologs, which suggests the possibility of independent HGT events. TWOG0788 A----P- Urease Ureases of many bacterial
species
Highly conserved enzyme present in plants and many fungi but not S. cerevisiae. Plant and fungal ureases have a common domain architecture distinct from that of bacterial orthologs, which suggests monophyletic origin. Might have evolved via early HGT from bacteria (proto-mitochondria?) with subsequent loss in animals and some fungi.
4751 A--H--E Recombination repair protein BRCA2, contains varying number of BRCA2 repeats
None Although sequence conservation is limited to the BRC repeats [101] the number of which varies substantially, statistical significance of the observed sequence similarity and the absence of other homologs suggests that the proteins in this KOG are true orthologs. Apparent orthologs of BRCA2 are detectable also in other species from the taxa represented in the KOGs (mosquito Anopheles gambiae, fungus Ustilago maydis) [102] and in early-branching eukaryotes (Leishmania, Trypanosoma; E.V.K., unpublished work), suggesting that evolution of BRCA2 involved multiple gene losses
4597 A--H--E TATA-binding protein 1-interacting protein
None Probable multiple gene losses
4486 A--H--E 3-methyl-adenine DNA glycosylase
Orthologs in many bacteria (COG2094)
The plant protein and those from mammals and microsporidia show the greatest similarity to different subsets of bacterial orthologs. Evolution might have included a combination of gene loss and independent HGT events
1594 A-D-Y-- Predicted epimerase related to aldose 1-epimerase
Bacterial orthologs, primarily proteobacteria (COG0676)
Eukaryotic proteins are more closely related to each other than to bacterial orthologs, indicating monophyletic origin. Function remains unknown; might be involved in a distinct and still
uncharacterized pathway of polysaccharide biosynthesis. LSE in Arabidopsis (seven paralogs). 4141 ---HYPE Rad52/22, protein involved
in double-strand break repair
[image:4.612.57.563.117.692.2]en
t
re
v
ie
w
s
re
ports
refer
e
e
d
re
sear
ch
de
p
o
si
te
d r
e
se
a
rch
interacti
o
ns
inf
o
rmation
leading to concrete functional predictions [42,59-61]. The pair of KOG5324 and KOG4246 is a case in point that has not been described previously. The initial observation that these KOGs share the same unusual pattern of presence-absence in eukaryotes, and have similar phyletic patterns in prokaryotes, with a ubiquitous presence in archaea, prompted a more detailed examination of the multiple alignments of the respective proteins and the conservation of the (predicted) operon organization in archaea and bacteria (Table 2 and data not shown). The combination of clues from these analy-ses suggests that the two proteins interact in a still uncharac-terized pathway of RNA processing, which also includes RNA 3'-phosphate cyclase (KOG3980)) [62] and cytosine-C5-methylase (NOL1/NOP2 in eukaryotes; KOG1122). The pro-teins in KOG3833 and KOG4528 are likely to represent novel enzyme families, possibly a kinase-phosphatase pair (E.V.K. and L. Aravind, unpublished data). Notably, these predicted new enzymes are present in animals and E. cuniculi but not in Arabidopsis or yeasts. In contrast, KOG3980 is present in all analyzed eukaryotic genomes except for Arabidopsis, whereas KOG1122 is pan-eukaryotic. These differences in the phyletic patterns of the components of the predicted pathway are concordant with the patterns in eukaryotes in that.
Figure 2 shows the distribution of known and predicted func-tions of eukaryotic proteins among 20 functional categories for the entire set of KOGs and, separately, for KOGs repre-sented in six or seven species and the animal-specific KOGs. Compared to the functional breakdown of prokaryotic COGs [25], the prevalence of signal transduction is notable among eukaryotes. This feature is particularly prominent in animal-specific KOGs, whereas the highly conserved set is compara-tively enriched in proteins that are involved in translation, transcription, chaperone-like functions, cell cycle control and chromatin dynamics (Figure 2). The large number of KOGs
for which only general functional prediction was feasible, and those whose functions remain unknown, even among the sub-set that is represented in six or seven eukaryotic species, emphasizes that our current understanding of eukaryotic biology is seriously lacking with even in respect of the func-tions of highly conserved genes.
The distribution of KOGs by the number of paralogs in each genome is shown in Figure 3. The preponderance of lineage-specific duplication of conserved genes, that is, intra-KOG LSEs, in multicellular eukaryotes is obvious. Cases when a single gene in yeast or, particularly, Encephalitozoon, has two or more co-orthologs in animals and/or plants are most com-mon in KOGs, whereas the reverse situation is rare. These observations support the notion of the major contribution of LSE to the evolution of eukaryotic complexity [52]. However, 131 KOGs are represented by a single ortholog in all genomes compared (Table 2) and a substantial number of KOGs have one member from a majority of the genomes (data not shown). Recent theoretical modeling of the evolution of par-alogous families has suggested that, in general, ancient pro-tein families tend to have multiple paralogs [5,63]. Therefore, whenever a KOG has a single member in all or most species, this should be attributed to selection against duplication of this particular gene. A prominent cause of such selection could be the involvement of the respective gene products in essential multisubunit complexes, such that imbalance between subunits leads to deleterious effects [64].
Known and new functions of single-member, pan-eukaryotic KOGs
We examined in greater detail the 131 KOGs that are repre-sented by a single gene in each of the seven genomes (Table 2). As can be envisaged from their presence in diverse eukary-otic taxa, including the 'minimal' genome of 4528 -CDH--E Uncharacterized predicted
enzyme, possibly a polynucleotide kinase (structure of the ortholog from the bacterium Thermotoga maritima has been determined - pdb code 1j5u)
Conserved in all archaea and several bacteria (COG1371)
Context analysis of archaeal and bacterial genomes suggests functional interaction between proteins of KOG5324 and KOG4246, RNA 3'-terminal phosphate cyclase (KOG4398, COG0430), and tRNA/rRNA cytosine C5-methylase (KOG1299/ COG0144) ([103] and E.V.K., unpublished observations). Taken together, the observations appear to implicate KOG5324 and KOG4246 in a still uncharacterized pathway of rRNA and/or tRNA processing and modification. Conservation of these proteins in archaea and early-branching eukaryotes suggests lineage-specific gene loss in plants and fungi.
3833 -CDH--E Uncharacterized predicted enzyme, possibly a polynuclotide phosphatase
Conserved in all archaea and several bacteria (COG1690)
See comment for KOG5324
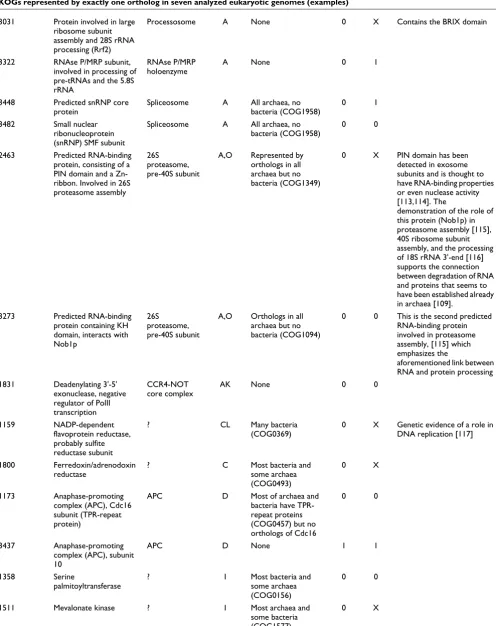
[image:5.612.51.563.117.269.2]KOG number (Predicted) function Multiprotein complex
Functional class*
Prokaryotic homologs
Fitness class† Comments
Yeast‡ Worm§
Genes experimentally or computationally characterized previously 0392 SNF2 family
DNA-dependent ATPase
TBP-DNA complex
Many bacteria and archaea (COG0553)
0 1 Involved in regulation of transcription from POL II promoters [104]
0121 Nuclear cap-binding protein complex, subunit CBP20 (RRM-domain-containing RNA-binding protein)
Cap-binding complex
A Several bacteria (COG0724)
1 X RRM-domain proteins show scattered presence in bacteria and might have been horizontally transferred from eukaryotes
0213 U2-snRNP associated splicing factor 3b, subunit 1
Spliceosome A None 0 0
0227 snRNA-associated protein, splicing factor 3a, subunit b (Prp11p)
Spliceosome A None 0 0
2268 Predicted nucleic-acid-binding protein kinase of the RIO1 family; 40S ribosomal subunit biogenesis/18S rRNA processing
Pre-40S subunit A Orthologs in most archaea but not in bacteria (COG0478)
0 X One of the very small number of protein kinases that show a clear-cut orthologous relationship between all eukaryotes and most archaea, and, apparently, the only one containing a helix-turn-helix nucleic-acid-binding domain. [105] Associated with yeast pre-40S subunit and required for its maturation. [106]
3031 Protein required for 60S ribosomal subunit biogenesis; [107] contains the IMP4 domain, which is involved in rRNA processing [108]; paralog of KOG3095 and KOG3292, which are also represented in all analyzed genomes.
Processosome A Distantly related to COG2136, represented by orthologs in most archaea, but not in bacteria (KSM, unpublished)
0 X The COG2136 proteins appear to be subunits of the predicted archaeal exosome [109]. Apparently, this gene has undergone at least two ancient duplications in eukaryotes
3045 Predicted RNA methylase involved in rRNA processing
Processosome? A Distantly related to numerous Rossmann-fold methylases but prokaryotic orthologs could not be confidently identified
1 1 This protein (Rrp8p in yeast) has been shown to participate in the processing of rRNA and sequence analysis reveals the presence of a Rossmann-fold methylase domain [110]. Therefore Rrp8p probably methylates either snoRNA or rRNA itself.
3064 RNA-binding nuclear protein containing a distinct C4 Zn-finger; implicated in the biogenesis of 60S ribosomal subunits [111]
Processosome A None 0 0 Initially identified in yeast as the MAK16 protein required for dsRNA virus
[image:6.612.58.554.121.701.2]en
t
re
v
ie
w
s
re
ports
refer
e
e
d
re
sear
ch
de
p
o
si
te
d r
e
se
a
rch
interacti
o
ns
inf
o
rmation
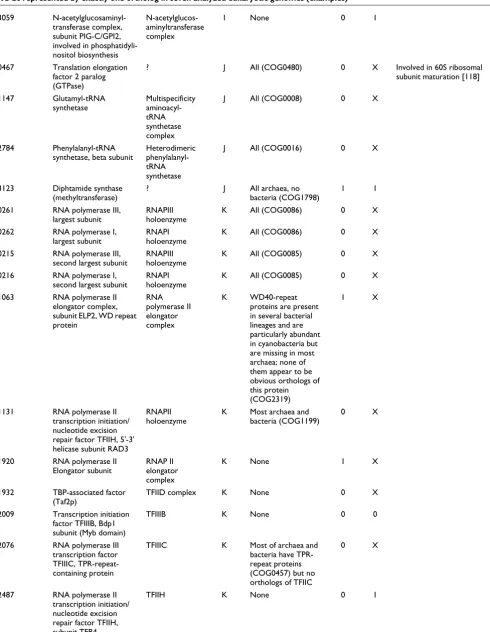
0291, 0302, 0306, 310, 0319, 0650, 1272
WD40-repeat proteins, subunits of rRNA processing complexes [69,70]
Processosome A WD40-repeat proteins are present in several bacterial lineages and are particularly abundant in cyanobacteria but are missing in most archaea; none of them appear to be obvious orthologs of this protein (COG2319)
all 0 X,X,1, X,1,1,1
0284 Polyadenylation factor I complex, subunit PFS2, WD40-repeat protein
Poly-adenylation complex
A Same as above (COG2319)
0 X
0337 RNA helicase involved in 28S rRNA processing
Processosome A Most of the archaea and bacteria (COG0513)
0 X
0343 RNA helicase involved in 28S rRNA processing
Processosome A Most of the archaea and bacteria (COG0513)
0 X
1069 3'-5' exoribonuclease (RNAse PH), exosome subunit Rrp46
Exosome A Most bacteria and archaea (COG0689)
0 1
1070 Exosome subunit Rrp5 (RNA-binding S1 domain fused to TPR repeats)
Exosome A Most bacteria (COG0539, COG0457)
0 1
1135 mRNA cleavage and polyadenylation complex subunit CFT2 (CPSF)
Cleavage and polyadenylation complex
A Most archaea and some bacteria (COG1236)
0 0
1914 mRNA cleavage and polyadenylation factor I complex, subunit RNA14
Cleavage and polyadenylation complex
A None 0 X
1975 RNA (guanine-7-) methyltransferase (capping enzyme subunit)
Capping enzyme
A Numerous methyltrans-ferases (COG0500) but no ortholog
0 1
2051 Nonsense-mediated mRNA decay complex, subunit 2
NMD complex A None 1 X
2554 Pseudouridylate synthase ? A Most archaea and bacteria (COG0101)
1 1
2613 Upf1p-interacting protein, NMD complex subunit Nmd3p
NMD complex A All archaea, no bacteria (COG1499)
0 X
2771 tRNA-specific adenosine-34 deaminase subunit Tad3p
Heterodimeric RNA-specific deaminase
A Most bacteria and some archaea (COG0590)
0 X
2780 Protein involved in ribosomal large subunit assembly (RPF1), contains IMP4 domain
Processosome A Most archaea, no bacteria (COG2136)
0 1
2781 Subunit of the small (ribosomal) subunit (SSU) processosome (snoRNP), IMP4
Processosome A Most archaea, no bacteria (COG2136)
0 1
2874 Protein involved in rRNA processing and ribosomal assembly
? A All archaea, no bacteria (COG1094)
0 1 Predicted RNA-binding protein containing KH domain
3013 Exosome subunit Rrp4 Exosome A Most archaea, on bacteria (COG1097)
[image:7.612.55.557.112.737.2]3031 Protein involved in large ribosome subunit assembly and 28S rRNA processing (Rrf2)
Processosome A None 0 X Contains the BRIX domain
3322 RNAse P/MRP subunit, involved in processing of pre-tRNAs and the 5.8S rRNA
RNAse P/MRP holoenzyme
A None 0 1
3448 Predicted snRNP core protein
Spliceosome A All archaea, no bacteria (COG1958)
0 1
3482 Small nuclear ribonucleoprotein (snRNP) SMF subunit
Spliceosome A All archaea, no bacteria (COG1958)
0 0
2463 Predicted RNA-binding protein, consisting of a PIN domain and a Zn-ribbon. Involved in 26S proteasome assembly
26S proteasome, pre-40S subunit
A,O Represented by orthologs in all archaea but no bacteria (COG1349)
0 X PIN domain has been detected in exosome subunits and is thought to have RNA-binding properties or even nuclease activity [113,114]. The
demonstration of the role of this protein (Nob1p) in proteasome assembly [115], 40S ribosome subunit assembly, and the processing of 18S rRNA 3'-end [116] supports the connection between degradation of RNA and proteins that seems to have been established already in archaea [109].
3273 Predicted RNA-binding protein containing KH domain, interacts with Nob1p
26S proteasome, pre-40S subunit
A,O Orthologs in all archaea but no bacteria (COG1094)
0 0 This is the second predicted RNA-binding protein involved in proteasome assembly, [115] which emphasizes the
aforementioned link between RNA and protein processing
1831 Deadenylating 3'-5' exonuclease, negative regulator of PolII transcription
CCR4-NOT core complex
AK None 0 0
1159 NADP-dependent flavoprotein reductase, probably sulfite reductase subunit
? CL Many bacteria (COG0369)
0 X Genetic evidence of a role in DNA replication [117]
1800 Ferredoxin/adrenodoxin reductase
? C Most bacteria and some archaea (COG0493)
0 X
1173 Anaphase-promoting complex (APC), Cdc16 subunit (TPR-repeat protein)
APC D Most of archaea and bacteria have TPR-repeat proteins (COG0457) but no orthologs of Cdc16
0 0
3437 Anaphase-promoting complex (APC), subunit 10
APC D None 1 1
1358 Serine
palmitoyltransferase
? I Most bacteria and some archaea (COG0156)
0 0
1511 Mevalonate kinase ? I Most archaea and some bacteria (COG1577)
[image:8.612.59.555.107.733.2]en
t
re
v
ie
w
s
re
ports
refer
e
e
d
re
sear
ch
de
p
o
si
te
d r
e
sea
rch
interacti
o
ns
inf
ormation
3059 N-acetylglucosaminyl-transferase complex, subunit PIG-C/GPI2, involved in phosphatidyli-nositol biosynthesis
N-acetylglucos-aminyltransferase complex
I None 0 1
0467 Translation elongation factor 2 paralog (GTPase)
? J All (COG0480) 0 X Involved in 60S ribosomal subunit maturation [118]
1147 Glutamyl-tRNA synthetase
Multispecificity aminoacyl-tRNA synthetase complex
J All (COG0008) 0 X
2784 Phenylalanyl-tRNA synthetase, beta subunit
Heterodimeric phenylalanyl-tRNA synthetase
J All (COG0016) 0 X
3123 Diphtamide synthase (methyltransferase)
? J All archaea, no bacteria (COG1798)
1 1
0261 RNA polymerase III, largest subunit
RNAPIII holoenzyme
K All (COG0086) 0 X
0262 RNA polymerase I, largest subunit
RNAPI holoenzyme
K All (COG0086) 0 X
0215 RNA polymerase III, second largest subunit
RNAPIII holoenzyme
K All (COG0085) 0 X
0216 RNA polymerase I, second largest subunit
RNAPI holoenzyme
K All (COG0085) 0 X
1063 RNA polymerase II elongator complex, subunit ELP2, WD repeat protein
RNA polymerase II elongator complex
K WD40-repeat proteins are present in several bacterial lineages and are particularly abundant in cyanobacteria but are missing in most archaea; none of them appear to be obvious orthologs of this protein (COG2319)
1 X
1131 RNA polymerase II transcription initiation/ nucleotide excision repair factor TFIIH, 5'-3' helicase subunit RAD3
RNAPII holoenzyme
K Most archaea and bacteria (COG1199)
0 X
1920 RNA polymerase II Elongator subunit
RNAP II elongator complex
K None 1 X
1932 TBP-associated factor (Taf2p)
TFIID complex K None 0 X
2009 Transcription initiation factor TFIIIB, Bdp1 subunit (Myb domain)
TFIIIB K None 0 0
2076 RNA polymerase III transcription factor TFIIIC, TPR-repeat-containing protein
TFIIIC K Most of archaea and bacteria have TPR-repeat proteins (COG0457) but no orthologs of TFIIC
0 X
2487 RNA polymerase II transcription initiation/ nucleotide excision repair factor TFIIH, subunit TFB4
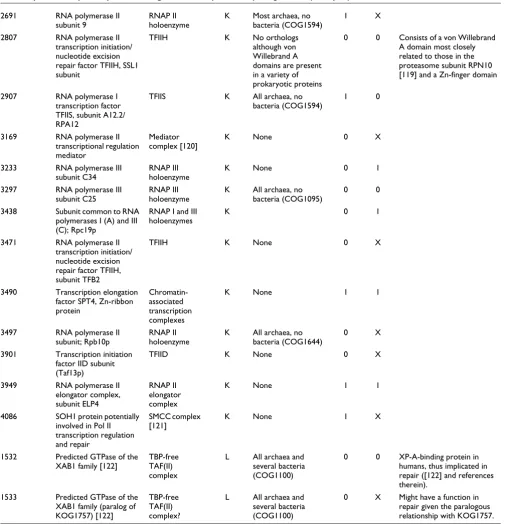
[image:9.612.57.547.111.743.2]2691 RNA polymerase II subunit 9
RNAP II holoenzyme
K Most archaea, no bacteria (COG1594)
1 X
2807 RNA polymerase II transcription initiation/ nucleotide excision repair factor TFIIH, SSL1 subunit
TFIIH K No orthologs although von Willebrand A domains are present in a variety of prokaryotic proteins
0 0 Consists of a von Willebrand A domain most closely related to those in the proteasome subunit RPN10 [119] and a Zn-finger domain
2907 RNA polymerase I transcription factor TFIIS, subunit A12.2/ RPA12
TFIIS K All archaea, no bacteria (COG1594)
1 0
3169 RNA polymerase II transcriptional regulation mediator
Mediator complex [120]
K None 0 X
3233 RNA polymerase III subunit C34
RNAP III holoenzyme
K None 0 1
3297 RNA polymerase III subunit C25
RNAP III holoenzyme
K All archaea, no bacteria (COG1095)
0 0
3438 Subunit common to RNA polymerases I (A) and III (C); Rpc19p
RNAP I and III holoenzymes
K 0 1
3471 RNA polymerase II transcription initiation/ nucleotide excision repair factor TFIIH, subunit TFB2
TFIIH K None 0 X
3490 Transcription elongation factor SPT4, Zn-ribbon protein
Chromatin-associated transcription complexes
K None 1 1
3497 RNA polymerase II subunit; Rpb10p
RNAP II holoenzyme
K All archaea, no bacteria (COG1644)
0 X
3901 Transcription initiation factor IID subunit (Taf13p)
TFIID K None 0 X
3949 RNA polymerase II elongator complex, subunit ELP4
RNAP II elongator complex
K None 1 1
4086 SOH1 protein potentially involved in Pol II transcription regulation and repair
SMCC complex [121]
K None 1 X
1532 Predicted GTPase of the XAB1 family [122]
TBP-free TAF(II) complex
L All archaea and several bacteria (COG1100)
0 0 XP-A-binding protein in humans, thus implicated in repair ([122] and references therein).
1533 Predicted GTPase of the XAB1 family (paralog of KOG1757) [122]
TBP-free TAF(II) complex?
L All archaea and several bacteria (COG1100)
[image:10.612.57.567.113.637.2]en
t
re
v
ie
w
s
re
ports
refer
e
e
d
re
sear
ch
de
p
o
si
te
d r
e
se
a
rch
interacti
o
ns
inf
o
rmation
1625 DNA polymerase α processivity subunit, inactivated phosphatase
DNA polymerase α holoenzyme
L Small subunit of archaeal DNA polymerase II (COG1311)
0 0 The small, regulatory subunit of DNA polymerase α also forms a pan-eukaryotic KOG3044, which is a paralog of KOG0861 (the only recent duplication in KOG3044 is seen in vertebrates). In contrast, another paralog, the small subunit of DNA polymerase
ε, is represented in animals, fungi and the early-branching protozoan Plasmodium, but not in plants or
Microsporidia. Thus, the history of this polymerase subunit apparently involved inactivation of the phosphatase (or nuclease) inherited from archaea, with subsequent duplications at early stages of eukaryotic evolution [123]
0479 DNA replication licensing factor MCM3
Pre-replication complex
L All archaea, no bacteria (COG1241)
0 X
0481 DNA replication licensing factor MCM5
Pre-replication complex
L All archaea, no bacteria (COG1241)
0 X
0482 DNA replication licensing factor MCM7
Pre-replication complex
L All archaea, no bacteria (COG1241)
0 0
0964 Structural maintenance of chromosome protein 3 (cohesin subunit SMC3)
Sister chromatid cohesion complex
L Many archaea and bacteria (COG1196)
0 X
0979 Structural maintenance of chromosome protein 5 (cohesin subunit SMC5)
Sister chromatid cohesion complex
L Many archaea and bacteria (COG1196)
0 X
1942 TBP-interacting protein TIP49 (DNA helicase)
chromatin remodeling complex
L Most of the archaea, no bacteria (COG1224)
0 0
1979 DNA mismatch repair ATPase, MLH1
Mismatch repair complex
L Most bacteria and some archaea (COG0323)
1 1
2267 DNA primase, large subunit
DNA polymerase
α:primase complex
L All archaea, no bacteria (COG2219)
0 0
2299 Ribonuclease HI Replisome L All archaea, most bacteria (COG0164)
1 X
2310 DNA repair exonuclease MRE11
MRN complex involved in double-strand break repair
L All archaea, most bacteria (COG0420)
1 1
2929 Origin recognition complex, subunit 2 (ORC2)
ORC L None 1 1
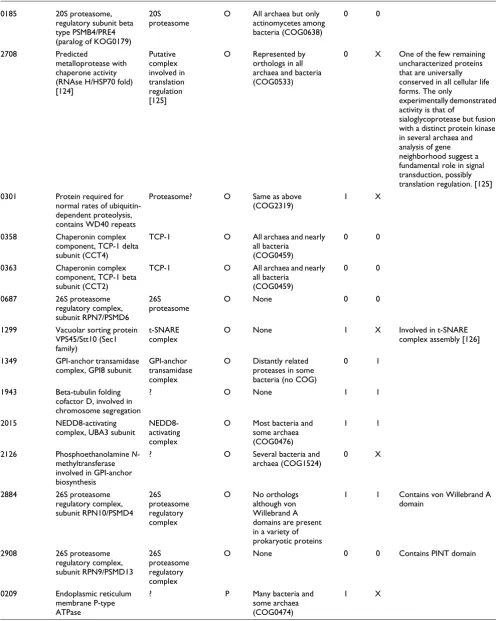
0179 20S proteasome, regulatory subunit beta type PSMB1/PRE7 (paralog of KOG0185)
20S proteasome
O All archaea but only actinomycetes among bacteria (COG0638)
[image:11.612.53.562.105.724.2]0185 20S proteasome, regulatory subunit beta type PSMB4/PRE4 (paralog of KOG0179)
20S proteasome
O All archaea but only actinomycetes among bacteria (COG0638)
0 0
2708 Predicted
metalloprotease with chaperone activity (RNAse H/HSP70 fold) [124]
Putative complex involved in translation regulation [125]
O Represented by orthologs in all archaea and bacteria (COG0533)
0 X One of the few remaining uncharacterized proteins that are universally conserved in all cellular life forms. The only
experimentally demonstrated activity is that of
sialoglycoprotease but fusion with a distinct protein kinase in several archaea and analysis of gene neighborhood suggest a fundamental role in signal transduction, possibly translation regulation. [125]
0301 Protein required for normal rates of ubiquitin-dependent proteolysis, contains WD40 repeats
Proteasome? O Same as above (COG2319)
1 X
0358 Chaperonin complex component, TCP-1 delta subunit (CCT4)
TCP-1 O All archaea and nearly all bacteria
(COG0459)
0 0
0363 Chaperonin complex component, TCP-1 beta subunit (CCT2)
TCP-1 O All archaea and nearly all bacteria
(COG0459)
0 0
0687 26S proteasome regulatory complex, subunit RPN7/PSMD6
26S proteasome
O None 0 0
1299 Vacuolar sorting protein VPS45/Stt10 (Sec1 family)
t-SNARE complex
O None 1 X Involved in t-SNARE complex assembly [126]
1349 GPI-anchor transamidase complex, GPI8 subunit
GPI-anchor transamidase complex
O Distantly related proteases in some bacteria (no COG)
0 1
1943 Beta-tubulin folding cofactor D, involved in chromosome segregation
? O None 1 1
2015 NEDD8-activating complex, UBA3 subunit
NEDD8-activating complex
O Most bacteria and some archaea (COG0476)
1 1
2126 Phosphoethanolamine N-methyltransferase involved in GPI-anchor biosynthesis
? O Several bacteria and archaea (COG1524)
0 X
2884 26S proteasome regulatory complex, subunit RPN10/PSMD4
26S proteasome regulatory complex
O No orthologs although von Willebrand A domains are present in a variety of prokaryotic proteins
1 1 Contains von Willebrand A domain
2908 26S proteasome regulatory complex, subunit RPN9/PSMD13
26S proteasome regulatory complex
O None 0 0 Contains PINT domain
0209 Endoplasmic reticulum membrane P-type ATPase
? P Many bacteria and some archaea (COG0474)
[image:12.612.57.553.115.736.2]en
t
re
v
ie
w
s
re
ports
refer
e
e
d
re
sear
ch
de
p
o
si
te
d r
e
se
a
rch
interacti
o
ns
inf
o
rmation
3379 Uncharacterized member of the histidine triad superfamily of nucleotide hydorlases
? R Most archaea and bacteria (COG0537)
1 X Only biochemical function predicted.
2635 Coatomer (COPI) complex delta subunit
COPI complex U None 0 0
2927 Membrane component of ER protein translocation apparatus (Sec62)
Sec complex U None 0 1
2978 Dolichol-phosphate mannosyltransferase
? U All archaea, most bacteria (COG0463)
0 X
3198 Signal recognition particle, subunit Srp19
Signal recognition particle
U All archaea, no bacteria (COG1400)
0 X
3315 Subunit of the targeting complex (TRAPP) involved in ER to Golgi trafficking
TRAPP U None 0 X
3369 Subunit of the targeting complex (TRAPP) involved in ER to Golgi trafficking
TRAPP U None 0 X
1992 Nuclear export receptor CSE1/CAS (importin beta)
? YU None 0 X
New functional predictions 2316 PP-loop family ATP
pyrophosphatase domain, which in fungi, plants and insects is fused to a duplicated translation inhibitor domain. The fusion, along with the phyletic pattern of the PP-ATPase domain, suggests an essential function in translation regulation
? A Orthologs of the PP-loop domain are present in all archaea (COG2102) but not in bacteria. Orthologs of the translation inhibitor domain are found in most bacteria and several archaea (COG0251)
1 X PP-loop ATPases have been previously implicated in base thiolation in various RNAs [127] and proteins in this K/ COG might have a similar function, which is likely to be conserved in eukaryotes and archaea. However, the fusion with translation inhibitor, which has been reported to have endoribonuclease activity [128] is a eukaryote-specific feature
2523 Predicted RNA-binding protein containing a PUA domain, probable role in RNA modification [129]
Putative novel RNA modification complex
A Orthologs present in all archaea
(COG2016) but not in bacteria
[image:13.612.62.555.109.663.2]0270, 0271, 1539
WD40-repeat proteins Processosome A WD40-repeat proteins are present in several bacterial lineages and are particularly abundant in cyanobacteria but are missing in most archaea; none of them appear to be obvious orthologs of this protein (COG2319)
all 0 X,1,X By analogy with other conserved WD40-repeat proteins, predicted to be subunits of rRNA processing/ ribosome assembly complexes
2321 Nucleolar protein, contains WD40 repeats
rRNA processosome?
A WD40-repeat proteins are present in several bacterial lineages and are particularly abundant in cyanobacteria but are missing in most archaea; none of them appear to be obvious orthologs of this protein (COG2319)
0 1 Probable subunit of an rRNA-processing complex
1763 Uncharacterized conserved protein containing a CCCH Zn-finger; possible role in RNA processing or splicing
? A None 1 1 CCCH fingers have been
shown to bind 3' untranslated regions in various mRNAs [130,131]
2837 Protein containing a U1-type, RNA-binding C2H2 Zn-finger. Probable role in RNA splicing/ processing
Spliceosome? A None 0 0 U1-type fingers are essential for the assembly of U1 RNP [132]
3073 Predicted RNA-binding protein containing PIN domain and involved in 18S rRNA processing
Pre-40S subunit A Most archaea, no in bacteria (COG1412)
0 1 Interacts with Nop14p and is required for 40S subunit biogenesis and 18S rRNA maturation (11694595). The presence of the PIN domain suggests RNA-binding and, possibly, RNAse activity
3154 Uncharacterized protein with potential function in translation or ribosomal biogenesis
Pre-40S subunit?
A? Most archaea, no bacteria (COG2042)
1 X The general functional prediction stems from the observation that the gene for this protein forms a predicted conserved operon with the gene for ribosomal protein L40E in several archaeal genomes 3214 Small protein containing
a Zn-ribbon, possibly RNA-binding; potential role in RNA processing or transcription regulation
? A? Conserved in
Crenarchaeota (COG4888)
1 1
3800 Predicted E3 ubiquitin ligase containing RING finger, subunit of transcription/repair factor TFIIH and CDK-activating kinase assembly factor
[image:14.612.55.555.110.722.2]en
t
re
v
ie
w
s
re
ports
refer
e
e
d
re
sear
ch
de
p
o
si
te
d r
e
se
a
rch
interacti
o
ns
inf
o
rmation
3176 Predicted α-helical protein, possibly involved in replication/repair; paralog of KOG3636
A novel complex with PCNA involved in replication?
L? Conserved in most (possibly all) archaea but not in bacteria (COG1711)
0 X A function in DNA replication/repair and/or transcription is suggested by the analysis of the genome context of archaeal orthologs which form an evolutionarily conserved association with the genes for replication sliding clamp (PCNA ortholog) (K.S.M. and E.V.K., unpublished work) 3303 Predicted α-helical
protein, possibly involved in replication/repair transcription; paralog of KOG3508
A novel complex with PCNA involved in replication?
L? Conserved in most (possibly all) archaea but not in bacteria (COG1711)
0 0 A function in DNA replication/repair and/or transcription is suggested by the analysis of the genome context of archaeal orthologs which form an evolutionarily conserved association with the genes for replication sliding clamp (PCNA ortholog) (K.S.M. and E.V.K., unpublished.work)
0396 Predicted E3 ubiquitin ligase
Ub ligase O None 1 1 The proteins in this KOG contain a modified RING domain, which might not be capable of metal-binding similarly to the U-box domain [133] that has been shown to function as E3 [134]
1443 Multitransmembrane protein, predicted drug/ metabolite transporter
? R Most archaea and bacteria (COG0697)
1 X
2647 Multitransmembrane protein, potential transporter
? R Most bacteria and some archaea (COG0628)
0 1
2488 Predicted N-acetyltransferase
? R Most archaea and bacteria (COG0454)
1 X Putative role in ribosomal maturation?
3347 Predicted nucleotide kinase; nuclear protein (Fap7p)
? R Conserved in all archaea but not in bacteria (COG1936)
0 1 Involved in oxidative stress reponse in yeast [135]
3974 Predicted sugar kinase Putative novel complex with KOG2585 proteins
R All archaea and most bacteria (COG0063)
1 1 Based on fusions seen in prokaryotes, predicted to interact functionally and, possibly, physically with uncharacterized proteins of KOG2585 (represented in all eukaryotes but includes paralogs in some species)
No functional prediction 2318 Uncharacterized
conserved protein
? S None 0 1
3237 Uncharacterized conserved protein containing coiled-coil domain
? S None 0 1 Coiled-coil domains are
often involved in complex assembly; this could be an uncharacterized component of the chromatin or the spliceosome
[image:15.612.54.550.108.705.2]Encephalitozoon, and as shown by comparison with the knockout phenotype data (Table 2 and see below), these pan-eukaryotic KOGs are of particular biological importance. For the great majority of these KOGs (113 of the 131), the function has been experimentally determined or confidently predicted to a varying degree of detail using computational methods (Table 2). However, around 20 KOGs from this set remained uncharacterized at the time of this analysis and, for all but two of these, substantial functional inferences could be drawn through a combination of sequence-profile analysis, structure prediction and genomic-context analysis of prokaryotic homologs (Table 2). Some of these predicted new functions are variations on well-known themes, such as two predicted PP-loop ATPases, which are probably involved in novel, essential RNA modifications (KOGs 2522 and 2316) or two predicted E3 components of ubiquitin ligases (KOGs 0396 and 3800). Other predicted functions appear to be com-pletely new, such as proteins in KOG3176 and 3303 which are likely to be essential components of eukaryotic replication and/or repair systems. Each of these uncharacterized but ubiquitous and largely essential eukaryotic genes is an attrac-tive target for experimental studies.
Examination of the experimentally characterized and pre-dicted functions of pan-eukaryotic, single-member KOGs leads to interesting conclusions. Nearly all the functionally characterized KOGs in this set consist of proteins that are subunits of known multiprotein complexes (Table 2). The most prominent of these are the complexes involved in rRNA processing and ribosome assembly, such as the recently dis-covered rRNA processosome and the pre-40S subunit, as well as the spliceosome, and various complexes involved in transcription (Table 2). Accordingly, this set of KOGs is mark-edly enriched for proteins involved in various forms of RNA processing, assembly of ribonucleoprotein (RNP) particles and transcription. In addition, KOGs in the single-member
TRAPP complex involved in protein trafficking [66]. Alto-gether, more than 80% of the yeast proteins in the pan-eukaryotic, single-member KOGs belong to known macromo-lecular complexes included in the MIPS database [67], as compared to around 64% for all yeast proteins in the KOGs, which is a moderate but statistically highly significant excess (data not shown). This preponderance of multiprotein com-plex formation among the single-member pan-eukaryotic KOGs is fully compatible with the balance hypothesis [64].
The most unexpected observation regarding the single-mem-ber, pan-eukaryotic KOGs, is probably that in 14 of these pro-teins, the only detectable domain was the WD40 repeat (Table 2). This is particularly notable because WD40-repeat proteins, which are extremely abundant in eukaryotes and are present in several prokaryotic lineages as well [68], are not generally known to form well-defined, one-to-one ortholo-gous relationships. The WD40 proteins in the pan-eukaryotic KOGs listed in Table 2 are exceptions, which is probably due to their unique and essential roles in the assembly of RNA-processing complexes. It has recently been demonstrated that, in S. cerevisiae, seven of these proteins are subunits of the 18S rRNA processosome, or at least are involved in ribos-omal assembly [69,70]. Taking these results together with the unusual phyletic pattern, it seems possible to predict with
[image:16.612.57.299.86.264.2]Distribution of the KOGs by the number of paralogs in each of the analyzed eukaryotic genomes
Figure 2
Distribution of the KOGs by the number of paralogs in each of the analyzed eukaryotic genomes. The species abbreviations are as in Figure 1.
Ecu Sce Spo Cel Dme Hsa
1 2 3 4 5 6 7 8 9 10 >10 0
500 1,000 1,500 2,000 2,500 3,000 3,500
Functional breakdown of the KOGs
Figure 3
Functional breakdown of the KOGs. Designations of functional categories: A, RNA processing and modification; B, chromatin structure and dynamics; C, energy production and conversion; D, cell-cycle control and mitosis; E, amino acid metabolism and transport; F, nucleotide metabolism and transport; G, carbohydrate metabolism and transport; H, coenzyme metabolism; I, lipid metabolism; J, translation; K, transcription; L, replication and repair; M, membrane and cell wall structure and biogenesis; O, post-translational modification, protein turnover, chaperone functions; P, inorganic ion transport and metabolism; Q, secondary metabolites biosynthesis, transport and catabolism; T, signal transduction; U, intracellular trafficking and secretion; Y, nuclear structure; Z,
cytoskeleton; R, general functional prediction only (typically, prediction of biochemical activity), S, function unknown. This breakdown is only for KOGs that included at least three species.
All
Six or seven species Metazoa
0 100 200 300 400 500 600 700 800
[image:16.612.315.557.429.581.2]en
t
re
v
ie
w
s
re
ports
refer
e
e
d
re
sear
ch
de
p
o
si
te
d r
e
se
a
rch
interacti
o
ns
inf
o
rmation
With some notable exceptions, such as the WD40 proteins, the KOGs in the single-member, pan-eukaryotic set show remarkable patterns of evolutionary conservation: they are either (nearly) ubiquitous in the three kingdoms of life, for example, RNA polymerase subunits, or are universally con-served in eukaryotes and archaea but missing in bacteria, such as most of the proteins implicated in RNA processing (Table 2). Thus, it appears that elaborate molecular machines central to the functioning of the eukaryotic cell have evolved, largely from ancestral archaeo-eukaryotic components, at the onset of eukaryotic evolution, and both loss and duplication of the respective genes have been strongly selected against throughout the rest of eukaryotic evolution.
Variation of evolutionary rates among KOGs
Genome-wide analysis of protein evolutionary rates shows a broad range of variation [71]. Here, we investigate the varia-tion of evoluvaria-tionary rates among the ubiquitous KOGs repre-sented in all seven analyzed genomes and the connection between the evolutionary rate and protein function in the KOG set. The characteristic evolutionary rate of each KOG, which included a member(s) from Arabidopsis, was deter-mined by measuring the mean evolutionary distance from Arabidopsis (the outgroup in the phylogenetic tree; see below) to the other species. Even among the KOGs that include all seven species and, accordingly, appear to repre-sent the conserved core of eukaryotic genes, the evolutionary rates differ by a factor of 20 between the fastest- and the slow-est-evolving KOGs. Excluding 5% of the KOGs from each tail of the distribution still leaves almost a fourfold difference in evolutionary rates (Figure 4a).
We then compared the distributions of evolutionary rates for different functional categories of KOGs (Tables 3,4 and Fig-ure 4b). Although all the distributions substantially overlapped, there was a statistically highly significant ence between the evolutionary rates for proteins with differ-ent functions (Tables 3,4 and Figure 4b). The slowest-evolving proteins are those involved in translation and RNA processing, the fastest-evolving ones are involved in cellular trafficking and transport, whereas components of replication and transcription systems have intermediate evolutionary rates (Tables 3,4 and Figure 4b).
A parsimonious scenario of gene loss and emergence in eukaryotic evolution and reconstruction of ancestral eukaryotic gene sets
Assuming a particular species tree topology, methods of evo-lutionary parsimony analysis can be used to construct a par-simonious scenario of evolution, that is, mapping of different types of evolutionary events onto the branches of the tree. With prokaryotes, the problem is confounded by the major
contributions from both lineage-specific gene loss and HGT to genome evolution, with the relative likelihoods of these events remaining uncertain [5,7]. The possibility of substan-tial HGT between major lineages of eukaryotes can apparently be safely disregarded, providing for an unambigu-ous most parsimoniunambigu-ous scenario that includes only gene loss and emergence of new genes as elementary events.
Some crucial aspects of the phylogenetic tree of the eukaryo-tic crown group remain a matter of contention. The consensus of many phylogenetic analyses appears to point to an animal-fungal clade and clustering of microsporidia with the fungi. However, a major uncertainty remains with respect to the topology of the animal tree: the majority of studies on protein phylogenies support a coelomate (chordate-arthropod) clade
[image:17.612.313.553.84.474.2]Variation of amino-acid substitution rates among KOGs
Figure 4
Variation of amino-acid substitution rates among KOGs. (a) Probability-density function for the distribution of evolutionary rates among the set of KOGs including all seven analyzed eukaryotic species. (b) Distribution functions for the evolutionary rates in different functional categories of KOGs. The designations of functional categories are as in Figure 3.
0 0.5 1 1.5 2 2.5 3
90% of KOGs 1.57/0.43 = 3.7
All KOGs 2.71/0.14 = 20.0
0 0.5 1 1.5 2 2.5 3
J L U S 0
0.2 0.4 0.6 0.8 1 1.2 1.4 1.6
[72-74], whereas rRNA phylogeny and some protein family trees point to the so-called ecdysozoan (arthropod-nema-tode) clade [75-78]. We treated the phyletic pattern of each KOG as a string of binary characters (1 for the presence of the given species and 0 for its absence in the given KOG) and constructed the parsimonious scenarios of gene loss and emergence during evolution of the eukaryotic crown group for both the coelomate and the ecdysozoan topologies of the phylogenetic tree. For the purpose of this reconstruction, the Dollo parsimony approach was adopted [79]. Under this approach, gene loss is considered irreversible; thus, a gene (a KOG member) can be lost independently in several evolution-ary lineages but cannot be regained. This assumption is justi-fied by the implausibility of HGT between eukaryotes (the Dollo approach is not valid for reconstruction of prokaryotic ancestors).
In the resulting parsimonious scenarios, each branch was associated with both gene loss and emergence of new genes, with the exception of the plant branch and the branch leading to the common ancestor of fungi and animals, to which gene losses could not be assigned with the current set of genomes (Figure 5a,b). There is little doubt that, once genomes of early-branching eukaryotes are included, gene loss associated with these branches will become apparent. The principal fea-tures of the reconstructed scenarios include massive gene loss in the fungal clade, with additional elimination of numerous genes in the microsporidian; emergence of a large set of new genes at the onset of the animal clade; and subsequent sub-stantial gene loss in each of the animal lineages, particularly in the nematodes and arthropods (Figure 5a,b). The esti-mated number of genes lost in S. cerevisiae after its diver-gence from the common ancestor with the other yeast species, S. pombe, closely agreed with a previous estimate produced by a different approach [57]. The switch from the coelomate topology of the animal sub-tree to the ecdysozoan topology resulted in relatively small changes in the distribution of gains and losses: the most notable difference was the greater number of genes lost in the nematode lineage and the smaller number of genes lost in the insect lineage under the ecdyso-zoan scenario compared to the coelomate scenario (Figure 5a,b).
The parsimony analysis described above involves explicit reconstruction of the gene sets of ancestral eukaryotic genomes. Under the Dollo parsimony model, which was used for this analysis, an ancestral gene (KOG) set is the union of the KOGs that are shared by the respective outgroup and each of the remaining species. Thus, the gene set for the common ancestor of the crown group includes all the KOGs in which Arabidopsis co-occurs with any of the other analyzed species. Similarly, the reconstructed gene set for the common ances-tor of fungi and animals consists of all KOGs in which at least one fungal species co-occurs with at least one animal species. These are conservative reconstructions of ancestral gene sets because, as already indicated, gene losses in the lineages branching off the deepest bifurcation could not be detected. Under this conservative approach, 3,413 genes (KOGs) were assigned to the last common ancestor of the crown group rates for different functional categories of KOGs*
Functional category
Number of KOGs
Mean rate, substitutions
per site
Standard deviation
J 227 0.98 0.37
H 62 0.98 0.30
A 167 1.01 0.36
C 140 1.01 0.43
O 307 1.01 0.40
F 50 1.05 0.34
E 130 1.07 0.38
L 139 1.11 0.38
B 56 1.13 0.33
Z 64 1.13 0.46
K 209 1.15 0.42
G 115 1.16 0.43
I 110 1.16 0.32
T 200 1.18 0.39
D 111 1.19 0.40
R 415 1.23 0.42
M 33 1.26 0.47
U 196 1.27 0.42
Q 30 1.27 0.37
P 69 1.28 0.45
N 2 1.30 0.78
S 348 1.40 0.41
All 3203 1.16 0.42
*Only the KOGs that included a member(s) from Arabidopsis were analyzed; the evolutionary rates are the average distances between the Arabidopsis representative in the given KOG and the proteins from other species (see Material and methods for details). The functional categories are designated as in Figure 5.
between selected functional categories of KOGs (t-test)
J L U S
J
-L 3 × 10-3
-U 1 × 10-12 3 × 10-4
[image:18.612.308.559.102.208.2] [image:18.612.56.298.130.459.2]-en
t
re
v
ie
w
s
re
ports
refer
e
e
d
re
sear
ch
de
p
o
si
te
d r
e
se
a
rch
interacti
o
ns
inf
o
rmation
(Figure 5a,b). More realistically, it appears likely that a cer-tain number of ancestral genes have been lost in all, or all but one, of the analyzed lineages during subsequent evolution, such that the gene set of the eukaryotic crown group ancestor might have been close in size to those of modern yeasts. In terms of the functional composition, the reconstructed core gene set of the crown-group ancestor resembled more the highly conserved KOGs than the animal-specific KOGs (Fig-ure 3) in being enriched in housekeeping functions such as translation, transcription and RNA processing (data not shown).
The functional profiles of the gene sets that were lost in differ-ent lineages showed substantial differences (Table 5). Thus, for example, in the lineage leading to the common ancestor of the animals, the greatest loss among genes assigned to functional categories was seen in amino acid and coenzyme metabolism; in contrast, in the fly and the nematode, more substantial degradation was observed among transcription factors and proteins with chaperone-like functions. Genes for proteins involved in RNA processing and translation are, in general, not heavily affected by loss except in the highly degraded parasite E. cuniculi. On many occasions, the switch
[image:19.612.57.554.85.495.2]Parsimonious scenarios of loss and emergence of genes (KOGs) in eukaryotic evolution
Figure 5
Parsimonious scenarios of loss and emergence of genes (KOGs) in eukaryotic evolution. (a) The coelomate topology of the phylogenetic tree of the eukaryotic crown group. (b) The ecdysozoan topology of the phylogenetic tree of the eukaryotic crown group. The numbers in boxes indicate the inferred number of KOGs in the respective ancestral forms. The numbers next to branches indicate the number of gene gains (emergence of KOGs) (numerator) and gene (KOG) losses (denominator) associated with the respective branches; a dash indicates that the number of losses for a given branch could not be determined. Proteins from each genome that did not belong to KOGs as well as LSEs were counted as gains on the terminal branches. The species abbreviations are as in Figure 1.
3,491
520
13,688
162
4,503
541
−
− 3,711
398
37
1,358
193
422 −
−
55
Dme Cel Hsa Sce Spo Ecu Ath 3,260
5,361
5,000 3,048
3,835
3,413 15
802
1,679
299 202 1,969
842 586
267
3,491
369
4,503
751
13,688
114
3,711
85
188
1,671
193
422
55
3,260 5,210
5,313 3,048
3,835
3,413 15
802
1,679
299 202 1,969
842 586
267
from the coelomate to the ecdysozoan topology replaces two independent, parallel losses in the insect and nematode clades with a single loss at the base of the ecdysozoan branch, although, on the whole, trees based on gene content support the coelomate topology [74]. In particular, the ecdysozoan topology, unlike the coelomate topology, implies early loss of several genes involved in translation, transcription and repair (Table 6). Notably, a large fraction of genes lost in each line-age has only a general functional prediction or no prediction at all (Table 5). This emphasizes the paucity of our current understanding of lineage-specific gene sets.
As noticed previously during the analysis of the genes lost in S. cerevisiae after its divergence from the common ancestor with S. pombe, functionally connected genes tend to be co-eliminated during evolution [57]. The present study general-izes this conclusion as many functionally coherent groups of
co-eliminated KOGs become apparent (Table 5). Importantly, different branches of the same complex systems tend to be eliminated in parallel in different lineages, for example, largely non-overlapping sets of genes for proteins of the ubiquitin-proteasome-signalosome systems are lost in the fungal-microsporidial lineage and in the nematodes (Table 6). It seems likely that elimination of these genes reflects independent trends for simplification of regulatory processes in these lineages.
An interesting trend seen in these data is the deterioration of the mitochondrial ribosome, which occurred in several eukaryotic lineages and appears to have been partly parallel (as it occurred independently in fungi-microsporidia and in animals) and partly consecutive: early loss in the ancestral animal line was followed by elimination of additional genes for ribosomal proteins in individual lineages (Table 6). C.
Functional category Lost genes (KOGs)
Hs* Dm* Coelomates/ Ecdysozoa
Ce* Animals Sc Sp Yeasts Ec Fungi-Ec
Total 162/114 520/369 37/188 541/751 193 299 202 55 1,969 802
RNA processing and modification 2/3 9/8 1/2 10/11 4 15 7 1 88 32
Translation 3/3 16/11 0/5 13/10 9 9 6 3 122 10
Transcription 5/2 16/12 0/4 29/33 2 16 9 4 83 40
Replication and repair 4/5 28/14 1/15 29/14 2 9 7 3 60 16
Chromatin structure and dynamics 1/1 8/6 0/2 8/6 0 5 3 1 29 11
Energy production and conversion 7/10 9/10 5/4 12/10 7 6 13 1 110 37 Cell cycle control and mitosis 3/3 11/6 0/5 15/11 3 12 3 1 61 16
Amino acid metabolism and transport 5/6 16/9 1/8 15/7 38 6 9 0 110 18
Nucleotide metabolism and transport 3/3 6/3 0/3 8/5 5 0 3 1 38 9 Carbohydrate metabolism and transport 3/3 13/10 1/4 18/14 8 10 16 3 70 41
Coenzyme metabolism 0/2 5/5 2/2 14/12 11 1 1 0 51 12
Lipid metabolism 1/5 27/19 4/12 18/6 4 9 19 2 74 33
Membrane and cell wall structure and biogenesis 5/4 10/10 2/2 9/11 7 5 3 0 37 15
Post-translational modification, protein turnover, chaperone functions
3/5 22/15 2/9 44/40 8 29 21 4 167 69
Inorganic ion transport and metabolism 2/4 8/8 2/2 8/7 9 2 6 4 50 14 Secondary metabolites biosynthesis, transport
and catabolism
1/2 6/5 1/2 5/3 2 4 1 0 23 5
Signal transduction 5/3 32/22 0/10 30/37 4 16 7 3 110 52
Intracellular trafficking and secretion 4/3 10/8 0/2 14/14 3 5 11 0 116 22
Nuclear structure 0/0 3/3 0/0 5/6 0 1 0 0 16 5
Cytoskeleton 0/0 2/2 0/0 6/8 0 9 0 3 44 6
General functional prediction only (typically, prediction of biochemical activity)
14/13 79/55 5/29 88/72 30 55 24 11 241 134
Function unknown 91/34 184/128 10/66 143/414 37 75 33 10 269 205
[image:20.612.58.557.119.485.2]