Acta Cryst.(2003). E59, o397±o398 DOI: 10.1107/S1600536803004446 Ning Shanet al. C9H8N+C4H3O4ÿ
o397
organic papers
Acta Crystallographica Section E
Structure Reports Online
ISSN 1600-5368
Quinolinium fumarate
Ning Shan,* Elaine Batchelor and William Jones
Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, England
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study
T= 140 K
Mean(C±C) = 0.002 AÊ
Rfactor = 0.034
wRfactor = 0.085
Data-to-parameter ratio = 14.3
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2003 International Union of Crystallography Printed in Great Britain ± all rights reserved
In the title complex, C9H8N+C4H3O4ÿ, fumarate anions are
linked by OÐH O hydrogen bonds, forming in®nite supramolecular chains along thec axis. Quinolinium cations are attached to the anionic chains via NÐH O and CÐ H O interactions. The crystal structure determination con®rms the protonation of the quinoline N atom and deprotonation of one of the carboxylic acid groups of fumaric acid.
Comment
Fumaric acid is an organic dicarboxylic acid which crystallizes in two polymorphic forms: one in the monoclinic space group P21/c(Brown, 1966) and the other in the triclinic space group
P1 (Bednowitz & Post, 1966). In both crystal structures, acid molecules are linked by carboxylic acidR2
2(8) hydrogen-bond
pairs, forming one-dimensional supramolecular tapes. Fumaric acid is of interest since it is known to form supramolecular assemblies with N-aromatic complexes (Batcheloret al., 2000). As part of our analysis of supramolecular architectures, the structure of the title complex, (I), was determined at 140 K.
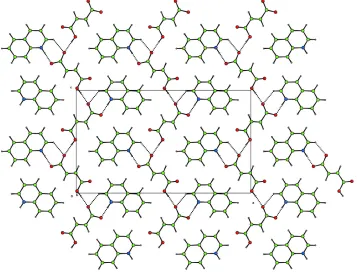
(I) consists of a 1:1 complex of fumarate anions and quinolinium cations. The asymmetric unit and atomic numbering scheme are shown in Fig. 1. Zigzag supramolecular acid chains are formed along the c axis via O1ÐH01 O3 hydrogen bonds (Table 2). In addition, quinolinium cations are linked to the acid chains (Fig. 2) by N1ÐH02 O4 hydrogen bonds and C9ÐH9 O3 interactions. Proton transfer is observed between the carboxylic acid group and the quinoline N atom (Table 1). Quinolinium cations form in®nite stacks along the b axis, the distance between adjacent mol-ecules within a stack beingca3.5 AÊ.
Experimental
Fumaric acid and quinoline were obtained from Aldrich without further puri®cation. 46 mg of the acid and 52 mg of the base were mixed and dissolved in a mixture of 9 ml of ethyl acetate and 10 drops of methanol. Crystals of (I) were obtained by slow evaporation at room temperature.
Crystal data
C9H8N+C4H3O4ÿ
Mr= 245.23
Orthorhombic,Pca21
a= 22.5838 (5) AÊ
b= 3.7273 (1) AÊ
c= 13.2912 (5) AÊ
V= 1118.81 (6) AÊ3
Z= 4
Dx= 1.456 Mg mÿ3
MoKradiation Cell parameters from 9880
re¯ections
= 1.8±27.5
= 0.11 mmÿ1
T= 140 (2) K Block, colourless 0.230.230.16 mm
Data collection
Nonius KappaCCD diffractometer Thin-slice!and'scans Absorption correction: multi-scan
(SORTAV; Blessing, 1995)
Tmin= 0.877,Tmax= 0.985
9868 measured re¯ections 2329 independent re¯ections
2118 re¯ections withI> 2(I)
Rint= 0.050 max= 27.5
h=ÿ23!29
k=ÿ4!3
l=ÿ17!14
Re®nement
Re®nement onF2
R[F2> 2(F2)] = 0.034
wR(F2) = 0.085
S= 1.08 2325 re¯ections 163 parameters
H-atom parameters constrained
w= 1/[2(F
o2) + (0.0494P)2
+ 0.0496P]
whereP= (Fo2+ 2Fc2)/3
(/)max= 0.001 max= 0.20 e AÊÿ3 min=ÿ0.23 e AÊÿ3
Table 1
Selected geometric parameters (AÊ).
O2ÐC10 1.207 (2)
O4ÐC13 1.282 (2) O3ÐC13O1ÐC10 1.233 (2)1.330 (2)
Table 2
Hydrogen-bonding geometry (AÊ,).
DÐH A DÐH H A D A DÐH A
N1ÐH02 O4i 0.88 1.67 2.553 (2) 179
C9ÐH9 O3i 0.95 2.79 3.348 (2) 119
O1ÐH01 O3ii 0.84 1.83 2.667 (2) 177
C3ÐH3 O2 0.95 2.35 3.284 (2) 168
Symmetry codes: (i)xÿ1
2;2ÿy;z; (ii) 2ÿx;2ÿy;zÿ12.
All H atoms bonded to C atoms were placed geometrically and re®ned using a riding model, with Uiso for each H atom taken as
1.2Ueqof the carrier atom. Atoms H01 and H02 were located from a
difference Fourier map and re®ned using a riding model. The abso-lute structure was not determined. Friedel opposites were merged prior to merging of data inPca21.
Data collection:COLLECT(Nonius, 1998); cell re®nement:HKL SCALEPACK(Otwinowski & Minor, 1997); data reduction: HKL DENZO (Otwinowski & Minor, 1997) and SCALEPACK; program(s) used to solve structure: SHELXS97 (Sheldrick, 1997); program(s) used to re®ne structure:SHELXL97 (Sheldrick, 1997); molecular graphics:XP(Sheldrick, 1993); software used to prepare material for publication:SHELXL97.
We are grateful for a DWEF Cambridge Scholarship and ORS Award (NS), as well as ®nancial assistance from the EPSRC for the purchase of the CCD diffractometer. We also thank Dr J. E. Davies for data collection.
References
Batchelor, E., Klinowski, J. & Jones, W. (2000).J. Mater. Chem.10, 839±848. Bednowitz, A. L. & Post, B. (1966).Acta Cryst.21, 566±571.
Blessing, R. H. (1995).Acta Cryst.A51, 33±38. Brown, C. J. (1966).Acta Cryst.21, 1±5. Flack, H. D. (1983).Acta Cryst.A39, 876±881.
Nonius (1998).COLLECT. Nonius BV, Delft, The Netherlands.
Otwinowski, Z. & Minor, W. (1997). Methods in Enzymology, Vol. 276,
Macromolecular Crystallography, Part A, edited by C. W. Carter Jr and R. M. Sweet, pp. 307±326. New York: Academic Press.
Sheldrick, G. M. (1993).XP. University of GoÈttingen, Germany.
Sheldrick, G. M. (1997). SHELXL97 and SHELXS97. University of GoÈttingen, Germany.
Watkin, D. J., Prout, C. K. & Pearce, L. J. (1996).CAMERON. Chemical Crystallography Laboratory, University of Oxford, England.
Figure 2
Projection on to (010), showing the zigzag supramolecular tapes formed by OÐH O hydrogen bonds and proton transfer between the carboxylic acid and the aromatic N atom (CAMERON; Watkinet al., 1996).
Figure 1
supporting information
sup-1
Acta Cryst. (2003). E59, o397–o398
supporting information
Acta Cryst. (2003). E59, o397–o398 [doi:10.1107/S1600536803004446]
Quinolinium fumarate
Ning Shan, Elaine Batchelor and William Jones
S1. Comment
Fumaric acid is an organic dicarboxylic acid which crystallizes in two polymorphic forms: one in the monoclinic space
group P21/c (Brown, 1966) and the other in the triclinic space group P1 (Bednowitz & Post, 1966). In both crystal
structures, acid molecules are linked by carboxylic acid R22(8) hydrogen-bond pairs, forming one-dimensional
supramolecular tapes. Fumaric acid is of interest since it is known to form supramolecular assemblies with N-aromatic
complexes (Batchelor et al., 2000). As part of our analysis of supramolecular architectures, the structure of the title
complex, (I), was determined at 140 K.
(I) consists of a 1:1 complex of fumarate anions and quinolinium cations. The asymmetric unit and atomic numbering
scheme are shown in Fig. 1. Zigzag supramolecular acid chains are formed along the c axis via O1—H01···O3 hydrogen
bonds (Table 2). In addition, quinolinium cations are linked to the acid chains (Fig. 2) by N1—H02···O4 hydrogen bonds
and C9—H9···O3 interactions. Proton transfer is observed between the carboxylic acid group and the quinoline N atom
(Table 1). Quinolinium cations form infinite stacks along the b axis with the distance between adjacent molecules within
a stack being ca 3.5 Å.
S2. Experimental
Fumaric acid and quinoline were obtained from Aldrich without further purification. 46 mg of the acid and 52 mg of the
base were mixed and dissolved in 9 ml of ethyl acetate and 10 drops of methanol mixture. Crystals of (I) were obtained
by slow evaporation at room temperature.
S3. Refinement
All H atoms bonded to C atoms were placed geometrically and refined using a riding model, with the Uiso values for each
H atom taken as 1.2 Ueq of the carrier atom. Atoms H01 and H02 were located from difference Fourier maps and refined
Figure 1
The molecular unit of (I), showing displacement ellipsoids at the 50% probability level (XP; Sheldrick, 1993).
Figure 2
Projection onto (010), showing the zigzag supramolecular tapes formed by O—H···O hydrogen bonds and proton transfer
between the carboxylic acid and the aromatic N atom (CAMERON; Watkin et al., 1996).
Quinolinium Fumarate
Crystal data
C9H8N+·C4H3O4− Mr = 245.23
Orthorhombic, Pca21 a = 22.5838 (5) Å
c = 13.2912 (5) Å V = 1118.81 (6) Å3 Z = 4
[image:4.610.128.485.285.564.2]supporting information
sup-3
Acta Cryst. (2003). E59, o397–o398 Dx = 1.456 Mg m−3
Dm = no Mg m−3
Dm measured by not measured Melting point: not measured K Mo Kα radiation, λ = 0.71073 Å Cell parameters from 9880 reflections
θ = 1.8–27.5° µ = 0.11 mm−1 T = 140 K Block, colourless 0.23 × 0.23 × 0.16 mm
Data collection
Nonius KappaCCD diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
thin–slice ω and φ scans
Absorption correction: multi-scan (SORTAV; Blessing, 1995) Tmin = 0.877, Tmax = 0.985
9868 measured reflections 2329 independent reflections 2118 reflections with I > 2σ(I) Rint = 0.050
θmax = 27.5°, θmin = 1.8° h = −23→29
k = −4→3 l = −17→14
Refinement
Refinement on F2 Least-squares matrix: full R[F2 > 2σ(F2)] = 0.034 wR(F2) = 0.085 S = 1.08 2325 reflections 163 parameters 1 restraint
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained w = 1/[σ2(F
o2) + (0.0494P)2 + 0.0496P] where P = (Fo2 + 2Fc2)/3
(Δ/σ)max = 0.001 Δρmax = 0.20 e Å−3 Δρmin = −0.23 e Å−3
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
N1 0.70065 (6) 0.3852 (3) 0.91328 (10) 0.0229 (3)
H02 0.6801 0.4547 0.8605 0.028*
O2 0.92188 (5) 0.7454 (3) 0.61425 (10) 0.0331 (3)
O4 1.14159 (4) 1.4168 (3) 0.75918 (9) 0.0311 (3)
O3 1.06865 (5) 1.2868 (3) 0.86560 (10) 0.0378 (3)
O1 0.99508 (5) 0.9657 (3) 0.51896 (9) 0.0319 (3)
H01 0.9738 0.8899 0.4717 0.038*
C7 0.76386 (7) 0.1615 (4) 1.08004 (12) 0.0259 (4)
H7 0.7852 0.0819 1.1375 0.031*
C2 0.78992 (7) 0.5428 (4) 0.82504 (12) 0.0252 (3)
H2 0.7674 0.6179 0.7684 0.030*
C9 0.67253 (6) 0.2611 (4) 0.99297 (14) 0.0266 (3)
H9 0.6305 0.2499 0.9923 0.032*
C13 1.08959 (7) 1.3009 (4) 0.78005 (12) 0.0242 (3)
C1 0.76118 (6) 0.4082 (4) 0.91110 (11) 0.0214 (3)
C3 0.85029 (7) 0.5651 (4) 0.82326 (14) 0.0302 (4)
H3 0.8697 0.6538 0.7649 0.036*
C5 0.85695 (6) 0.3265 (4) 0.99134 (14) 0.0279 (3)
H5 0.8802 0.2543 1.0474 0.034*
C8 0.70331 (7) 0.1452 (4) 1.07873 (12) 0.0282 (4)
H8 0.6823 0.0561 1.1354 0.034*
C12 1.05486 (7) 1.1844 (4) 0.68978 (13) 0.0240 (3)
H12 1.0709 1.2313 0.6250 0.029*
C10 0.96883 (6) 0.8973 (4) 0.60642 (12) 0.0247 (3)
C11 1.00317 (7) 1.0202 (4) 0.69517 (13) 0.0255 (3)
H11 0.9869 0.9780 0.7600 0.031*
C4 0.88398 (7) 0.4575 (4) 0.90732 (14) 0.0312 (4)
H4 0.9259 0.4764 0.9054 0.037*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
N1 0.0249 (7) 0.0232 (6) 0.0207 (7) 0.0025 (5) −0.0042 (5) 0.0000 (5)
O2 0.0252 (6) 0.0458 (7) 0.0283 (7) −0.0099 (5) 0.0005 (5) 0.0003 (5)
O4 0.0261 (6) 0.0434 (7) 0.0239 (6) −0.0082 (5) −0.0008 (5) −0.0017 (5)
O3 0.0381 (7) 0.0523 (8) 0.0230 (7) −0.0140 (5) 0.0026 (6) −0.0002 (5)
O1 0.0277 (6) 0.0455 (7) 0.0225 (6) −0.0095 (5) −0.0003 (5) −0.0034 (5)
C7 0.0347 (9) 0.0218 (7) 0.0211 (9) −0.0003 (6) −0.0040 (6) 0.0013 (6)
C6 0.0289 (8) 0.0176 (6) 0.0218 (8) 0.0013 (5) −0.0037 (7) −0.0017 (6)
C2 0.0323 (8) 0.0240 (7) 0.0193 (8) 0.0006 (6) −0.0020 (7) 0.0010 (6)
C9 0.0268 (7) 0.0266 (7) 0.0265 (8) −0.0005 (6) 0.0019 (8) −0.0012 (6)
C13 0.0273 (7) 0.0242 (7) 0.0212 (9) −0.0019 (6) −0.0012 (7) 0.0002 (6)
C1 0.0264 (8) 0.0167 (6) 0.0212 (8) 0.0008 (5) −0.0005 (6) −0.0033 (5)
C3 0.0335 (8) 0.0290 (8) 0.0281 (9) −0.0019 (6) 0.0058 (7) 0.0022 (7)
C5 0.0277 (8) 0.0250 (7) 0.0311 (9) −0.0008 (6) −0.0089 (7) 0.0005 (7)
C8 0.0364 (9) 0.0271 (8) 0.0211 (9) −0.0022 (6) 0.0035 (7) 0.0008 (6)
C12 0.0278 (8) 0.0238 (6) 0.0202 (8) 0.0007 (6) 0.0021 (6) 0.0007 (6)
C10 0.0232 (8) 0.0264 (7) 0.0245 (9) 0.0005 (6) −0.0023 (7) −0.0002 (6)
C11 0.0263 (8) 0.0303 (8) 0.0198 (8) −0.0017 (6) 0.0023 (6) 0.0003 (7)
C4 0.0254 (8) 0.0296 (8) 0.0385 (10) −0.0001 (6) −0.0001 (7) −0.0001 (7)
Geometric parameters (Å, º)
N1—C9 1.319 (2) C2—H2 0.9500
N1—C1 1.3702 (19) C9—C8 1.403 (2)
N1—H02 0.8800 C9—H9 0.9500
supporting information
sup-5
Acta Cryst. (2003). E59, o397–o398
O4—C13 1.282 (2) C3—C4 1.410 (3)
O3—C13 1.233 (2) C3—H3 0.9500
O1—C10 1.330 (2) C5—C4 1.363 (3)
O1—H01 0.8400 C5—H5 0.9500
C7—C8 1.369 (2) C8—H8 0.9500
C7—C6 1.414 (2) C12—C11 1.320 (2)
C7—H7 0.9500 C12—H12 0.9500
C6—C1 1.414 (2) C10—C11 1.484 (2)
C6—C5 1.414 (2) C11—H11 0.9500
C2—C3 1.366 (2) C4—H4 0.9500
C2—C1 1.408 (2)
C9—N1—C1 121.29 (14) C2—C3—C4 120.50 (15)
C9—N1—H02 119.4 C2—C3—H3 119.7
C1—N1—H02 119.4 C4—C3—H3 119.7
C10—O1—H01 109.5 C4—C5—C6 120.40 (16)
C8—C7—C6 119.72 (14) C4—C5—H5 119.8
C8—C7—H7 120.1 C6—C5—H5 119.8
C6—C7—H7 120.1 C7—C8—C9 119.44 (14)
C7—C6—C1 118.29 (13) C7—C8—H8 120.3
C7—C6—C5 123.20 (15) C9—C8—H8 120.3
C1—C6—C5 118.51 (15) C11—C12—C13 123.63 (15)
C3—C2—C1 119.72 (14) C11—C12—H12 118.2
C3—C2—H2 120.1 C13—C12—H12 118.2
C1—C2—H2 120.1 O2—C10—O1 123.86 (15)
N1—C9—C8 121.46 (13) O2—C10—C11 122.37 (15)
N1—C9—H9 119.3 O1—C10—C11 113.76 (12)
C8—C9—H9 119.3 C12—C11—C10 124.19 (16)
O3—C13—O4 124.44 (15) C12—C11—H11 117.9
O3—C13—C12 121.72 (14) C10—C11—H11 117.9
O4—C13—C12 113.84 (14) C5—C4—C3 120.62 (15)
N1—C1—C2 119.96 (14) C5—C4—H4 119.7
N1—C1—C6 119.80 (13) C3—C4—H4 119.7
C2—C1—C6 120.24 (13)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
N1—H02···O4i 0.88 1.67 2.553 (2) 179
C9—H9···O3i 0.95 2.79 3.348 (2) 119
O1—H01···O3ii 0.84 1.83 2.667 (2) 177
C3—H3···O2 0.95 2.35 3.284 (2) 168