A donor-specific epigenetic classifier for acute graft-versus-host disease severity in hematopoietic stem cell transplantation
Full text
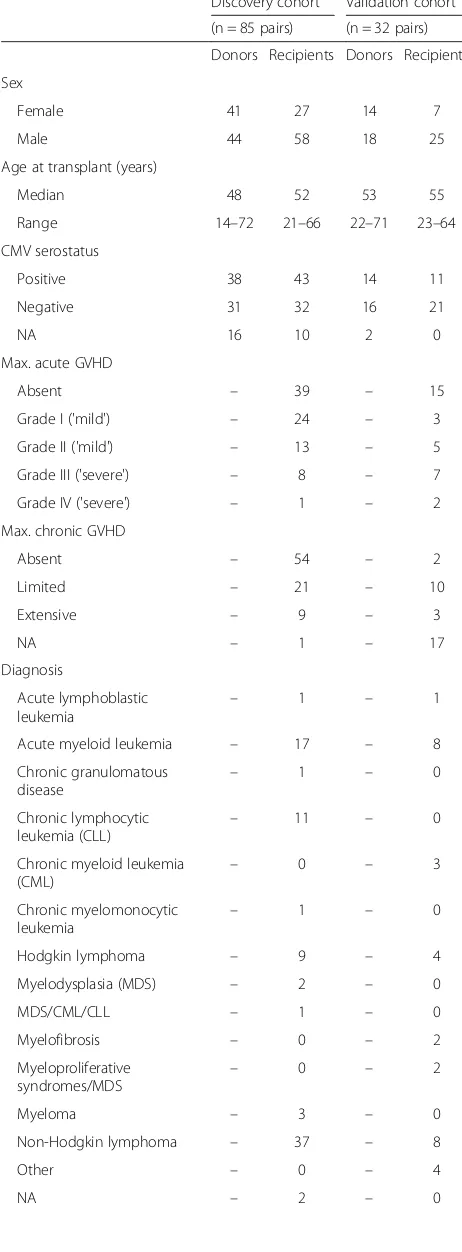
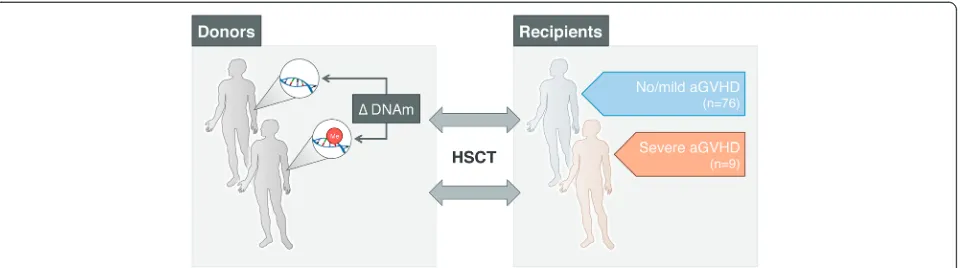
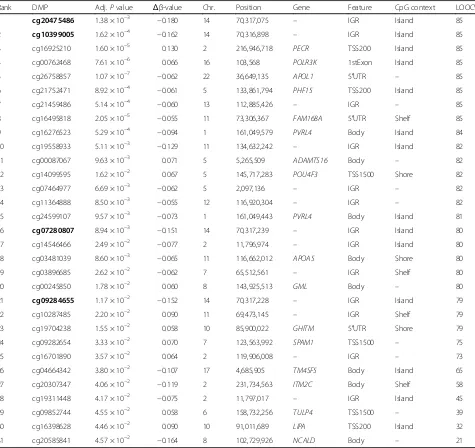
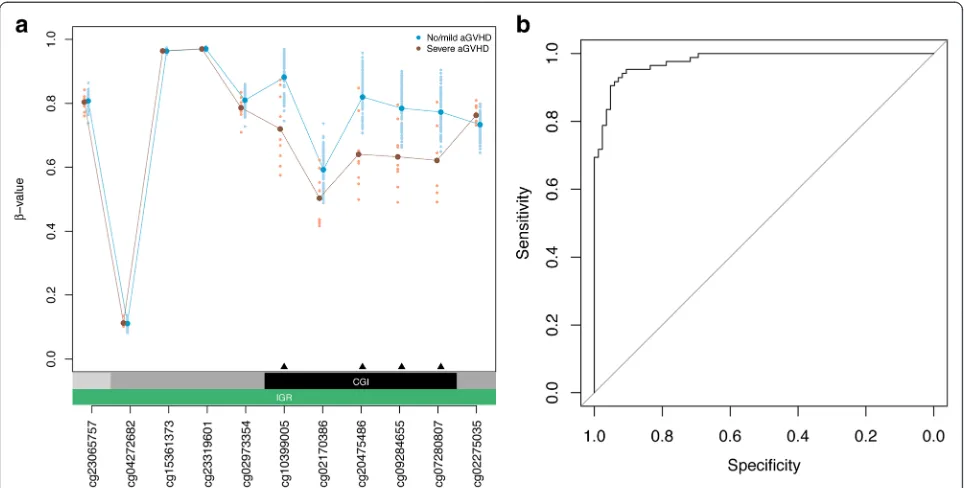
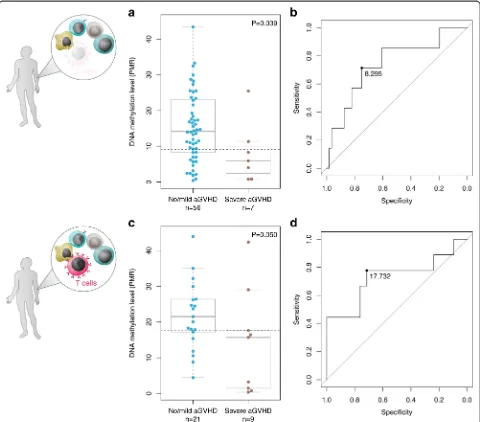
Figure
Related documents
If you select the Retain up-to-the-minute restore ability for older backups in other backup management groups option, transaction logs are deleted only from the specified
Dobson, Marriage is the Foundation of the Family , 18 Notre Dame J.L.. Ethics
Yet, in an increasingly urban landscape children often lack the opportunity to explore and learn about the natural environment right outside their classrooms (Louv, 2005; Pyle,
Finally a Simulink based model is proposed to reduce these harmonics from input current and hence to provide significant improvement in overall THD.The proposed methodology
Title of Study: Understanding College Men’s and Women’s Perceptions of Sexual Behavior Responsibility through the Lens of the Human Papillomavirus. Principal Investigator:
This paper looks at the costs associated with a large mine development, multiple coal transportation systems, a fleet of coal-fired power stations and the network
After the spatial science collaborator has performed the analysis, the resulting output can be shared back with the health organization, and can further be re-transformed using
