Acta Cryst.(2003). E59, o141±o142 DOI: 10.1107/S1600536803000795 Leulmi Bendheifet al. 2C5H12NO2+2H2PO3ÿ
o141
organic papers
Acta Crystallographica Section E
Structure Reports
Online
ISSN 1600-5368
L
-Valinium hydrogenphosphite
Leulmi Bendheif,
Nourredine Benali-Cherif,* Lamia Benguedouar, Karim Bouchouit and Hocine Merazig
Laboratoire de Chimie MoleÂculaire, du ControÃle de l'Environnement et des Mesures Physico-Chimiques, Faculte des Sciences, DeÂpartement de Chimie, Universite Mentouri de Constantine, 25000 Constantine, Algeria
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study
T= 293 K
Mean(C±C) = 0.003 AÊ Disorder in solvent or counterion
Rfactor = 0.038
wRfactor = 0.100
Data-to-parameter ratio = 10.8
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2003 International Union of Crystallography Printed in Great Britain ± all rights reserved
The crystal structure of the title compound, C5H12NO2+
-H2PO3ÿ, can be described as a stacking of l-valinium and
hydrogenphosphite ions. The stability of such an arrangement results from a network of hydrogen bonds, which maintain the cohesion of the organic±inorganic layers in the crystal. The asymmetric unit contains two valinium residues and two hydrogenphosphite ions, one of which is disordered.
Comment
In recent years, organic±inorganic hybrid materials have attracted considerable attention as prefered materials in non-linear optics (NLO), such as second harmonic generation (SHG) and optical bistability, owing to their large optical non-linearities (Masse & Zyss, 1991; Zaccaroet al., 1998; Mossetet al., 1996). The very high SGH and NLO properties make these hybrid materials highly attractive for application to frequency doubling of the light produced by semiconductor lasers (Kondo et al., 1988). l-Valinium hydrogenphosphite, (I), results from our systematic investigation of organic±inorganic hybrid materials obtained by interaction between various phosphoric oxyacids and amino acids.
The structure can be described as alternating layers of organic cations (C5H12NO2+) and hydrogenphosphite anions
(H2PO3ÿ), both layers being parallel to theacplane. The main
feature of this stacking is the presence of strong hydrogen bonds, similar to those observed in other ionic compounds (Pecaut & Bagieu-Beucher, 1993; Averbuch-Pouchot, 1993). Within a layer, the distance between the H2PO3ÿ groups is
signi®cantly longer [PÐP = 4.904 (3) AÊ], because the (HPO3H)nchain is more stretched. These entities generate,
through strong hydrogen bonds, in®nite chains of (HPO3H)n
parallel to thebaxis. Hydrogenphosphite groups are hydrogen bonded to the organic cation in two ways, ®rst via the carboxylic acid group and secondviathe ammonium groups. We do not observe any hydrogen bonds either between organic cations or between inorganic anions. The valinium residues adopt a gauche II conformation and their mean backbone conformation angles 1(O2ÐC1ÐC2ÐN1) and 2(O1ÐC1ÐC2ÐN1) [ÿ11.7 (2)/168.8 (1) and ÿ12.1 (2)/
167.9 (1) for cations A and B respectively] (Table 1)] are similar to those observed in dl-valinium di-hydrogenphosphate (Ravikumaret al., 2002).
Experimental
Crystals of l-valinium hydrogenphosphite were prepared by slow evaporation, at room temperature, of an aqueous solution ofl-valine and phosphorous acid in a stoichiometric ratio of 1:1. After six months, crystals appeared as colourless prisms.
Crystal data
C5H12NO2+H2PO3ÿ Mr= 199.15 Monoclinic,P21=n a= 16.3590 (3) AÊ b= 6.2540 (2) AÊ c= 19.4560 (3) AÊ = 109.238 (1) V= 1879.37 (8) AÊ3 Z= 8
Dx= 1.408 Mg mÿ3 MoKradiation
Cell parameters from 12190 re¯ections
= 1.4±26.4 = 0.28 mmÿ1 T= 293 (2) K Prism, colourless 0.600.400.35 mm
Data collection
Nonius KappaCCD diffractometer 'scans
Absorption correction: none 12190 measured re¯ections 3629 independent re¯ections 3205 re¯ections withI> 2(I)
Rint= 0.065 max= 26.4 h=ÿ20!20 k=ÿ7!7 l=ÿ23!23
Re®nement
Re®nement onF2 R[F2> 2(F2)] = 0.038 wR(F2) = 0.100 S= 1.06 3629 re¯ections 335 parameters
H atoms treated by a mixture of independent and constrained re®nement
w= 1/[2(Fo2) + (0.0425P)2 + 0.5313P]
whereP= (Fo2+ 2Fc2)/3 (/)max= 0.034
max= 0.25 e AÊÿ3
min=ÿ0.24 e AÊÿ3
Table 1
Selected geometric parameters (AÊ,).
O2aÐC1a 1.2218 (19)
O1aÐC1a 1.2913 (18)
O4aÐP1a 1.5104 (12)
O4bÐP1b 1.4964 (12)
O3aÐP1a 1.4855 (11)
O3bÐP1b 1.4930 (12)
O5aÐP1a 1.5591 (12)
P1bÐO13b 1.484 (6)
P1bÐO5b 1.5369 (16)
O1bÐC1b 1.3121 (19)
O2bÐC1b 1.206 (2)
O2aÐC1aÐO1a 125.70 (14) O13bÐP1bÐO3b 114.3 (5) O13bÐP1bÐO4b 110.3 (5) O3bÐP1bÐO4b 115.76 (7) O3bÐP1bÐO5b 111.65 (10)
O4bÐP1bÐO5b 109.73 (10) O3aÐP1aÐO4a 116.11 (7) O3aÐP1aÐO5a 110.45 (7) O4aÐP1aÐO5a 109.37 (7) O2bÐC1bÐO1b 125.42 (14)
O2aÐC1aÐC2aÐN1a ÿ11.7 (2) O1aÐC1aÐC2aÐN1a 168.78 (13) N1aÐC2aÐC3aÐC4a 74.0 (2) N1aÐC2aÐC3aÐC5a ÿ159.58 (18)
N1bÐC2bÐC1bÐO2b ÿ12.1 (2) N1bÐC2bÐC1bÐO1b 167.89 (13) N1bÐC2bÐC3bÐC4b 80.56 (19) N1bÐC2bÐC3bÐC5b ÿ153.39 (17)
Table 2
Hydrogen-bonding geometry (AÊ,).
DÐH A DÐH H A D A DÐH A
O1aÐH1a O4a 0.83 (3) 1.69 (3) 2.506 (2) 167 (3) O1bÐH1b O4bi 0.74 (3) 1.82 (3) 2.538 (2) 165 (3) N1aÐH10a O3bii 0.87 (2) 1.95 (2) 2.783 (2) 159 (2) N1bÐH11b O3aiii 0.89 (2) 1.85 (2) 2.724 (2) 170 (2) N1aÐH11a O4biv 0.87 (2) 1.98 (2) 2.837 (2) 169 (2) N1bÐH10b O3av 0.88 (2) 2.11 (2) 2.845 (2) 141 (2) N1aÐH12a O3bvi 0.84 (2) 1.97 (2) 2.802 (2) 170 (2) N1bÐH12b O4a 0.91 (2) 1.98 (2) 2.852 (2) 159 (2)
O5aÐH14a O2a 0.82 1.82 2.626 (2) 167
O5bÐH14b O2biii 0.82 2.23 3.026 (2) 165 O13bÐH13b O1b 0.82 1.90 2.681 (11) 160
Symmetry codes: (i) x;yÿ1;z; (ii) 2ÿx;2ÿy;2ÿz; (iii) x;1y;z; (iv)
1
2x;52ÿy;zÿ12; (v) 2ÿx;1ÿy;2ÿz; (vi)12x;32ÿy;zÿ12.
One of the monohydrogenphosphite anions is disordered. The disorder can be described as a rotation of this anion around the axis which bisects the angle O4bÐP1bÐO3b. The re®ned model corre-sponds to a disordered distribution between OH and H, with occu-pation factors of 0.85 (1) and 0.15 (1). Some of the H atoms were found in difference Fourier maps and were re®ned with isotropic displacement parameters. The hydroxyl H atoms were constrained, with only torsional freedom. The H atoms of the disordered hydro-genphosphite group were constrained.
Data collection:KappaCCD Reference Manual(Nonius, 1998); cell re®nement: DENZO and SCALEPACK (Otwinowski & Minor, 1997); data reduction:DENZOandSCALEPACK; program(s) used to solve structure:SHELX97 (Sheldrick, 1998); program(s) used to re®ne structure:SHELX97; molecular graphics:ORTEP-3 (Farrugia, 1997); software used to prepare material for publication: WinGX (Farrugia, 1999).
This work is supported by Mentouri-Constantine Univer-sity, Algeria. We thank Dr M. Pierrot and Dr M. Giorgi (LBS-UMR 6517, Faculte des Sciences et Techniques de Saint JeÂroÃme, Avenue Escadrille Normandie Niemen, 13397 Marseille Cedex 20, France) for diffraction facilities.
References
Averbuch-Pouchot, M. T. (1993).Acta Cryst.C49, 815±818. Farrugia, L. J. (1997).J. Appl. Cryst.30, 565.
Farrugia, L. J. (1999).J. Appl. Cryst.32, 837±838.
Kondo, T., Ogasawara, N. & Ito, R. (1988).Acta Cryst.C44, 102±104. Masse, R. & Zyss, J. (1991).Mol. Eng.1, 141±152.
Mosset, A., Baruchel, P., Lecante, P., Trombe, J. C., Ahamdane, H. & Bensamka, F. (1996).J. Mater. Chem.26, 503±508.
Nonius (1998). KappaCCD Reference Manual. Nonius BV, Delft, The Netherlands.
Otwinowski, Z. & Minor, W. (1997). Methods in Enzymology, Vol. 276, Macromolecular Crystallography, Part A, edited by C. W. Carter Jr and R. M. Sweet, pp. 307±326. New York: Academic Press.
Pecaut, J. & Bagieu-Beucher, M. (1993).Acta Cryst.C49, 834±837.
Ravikumar, B. S., Sridhar, B. & Rajaram, R. K. (2002)Acta Cryst.E58, o879± o881.
Sheldrick, G. M. (1998). SHELX97, which includes SHELXS97 and SHELXL97. Release 97-2. University of GoÈttingen, Germany.
Zaccaro, J., Bagieu-Beucher, M., Espesso, J. & Ibanez, A. (1998).J. Cryst. Growth,186, 224±232.
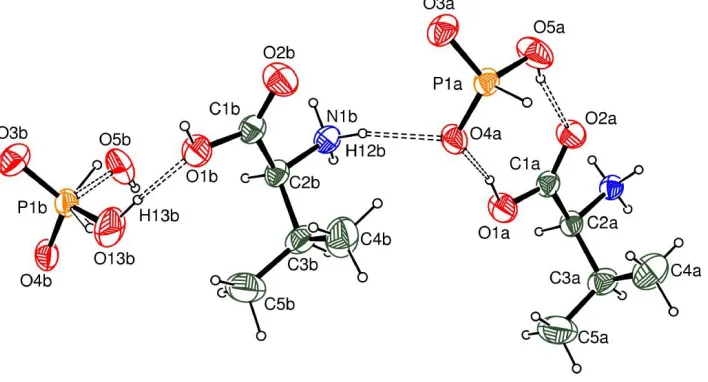
Figure 1
supporting information
sup-1
Acta Cryst. (2003). E59, o141–o142supporting information
Acta Cryst. (2003). E59, o141–o142 [doi:10.1107/S1600536803000795]
L
-Valinium hydrogenphosphite
Leulmi Bendheif, Nourredine Benali-Cherif, Lamia Benguedouar, Karim Bouchouit and Hocine
Merazig
S1. Comment
In recent years, organic–inorganic hybrid materials have attracted considerable attention as preferable materials in
linear optics (NLO), such as second harmonic generation (SHG) and optical bistability, owing to their large optical
non-linearities (Masse & Zyss, 1991; Zaccaro et al., 1998; Mosset et al., 1996). The very high SGH and NLO properties make
these hybrid materials highly attractive for application to frequency doubling of the light produced by semiconductor
lasers (Kondo et al., 1988). Bis(L-valinium) monohydrogenphosphite is a result of our systematic investigation on
organic–inorganic hybrid materials resulting from interaction between various phosphoric acids and amino acids. The
structure can be described as mixed layers built with organic cations (C5H12NO2+) and phosphite anions (H2PO3−), both
layers developing parallel to the [ac] plane. The main feature of this stacking is the presence of strong hydrogen bonds,
similar to those observed in other ionic compounds (Pecaut & Bagieu-Beucher, 1993; Averbuch-Pouchot, 1993). In a
layer, the distance between the H2PO3− groups is significantly longer [P—P = 4.904 (3) A°] because the (HPO3H)n chain is
more stretched. These entities form through strong hydrogen bonds infinite chains of (HPO3H)n parallel to the b axis.
Phosphite groups are hydrogen bonded to the organic cation in two ways, first via the carboxylic acid group [O5A—
H4A···O2A = 2.628 (2) Å] and second via the ammonium groups [N1A—H10A···O3B = 2.783 (2) Å, N1A—
H11A···O4B = 2.837 (2) Å and N1A—H12A···O3B = 2.802 (2) Å; N1B—H10B···O3A = 2.845 (2) Å, N1B—
H11B···O3A =2.724 (2) Å, and N1B—H12B···O4A = 2.852 (2) Å]. We did not observe any hydrogen bonds either
between organic entities or between inorganic anions. The valinium residues adopt a gauche II conformation and their
mean backbone conformation angles Ψ1(O2—C1—C2—N1) and Ψ2(O1—C1—C2—N1) [−11.7 (2)/168.8 (1) and
−12.1 (2)/167.9 (1)° for cations A and B respectively] (Table 1)] are similar to those observed in DL-valinium
di-hydrogenphosphate (Ravikumar et al., 2002).
S2. Experimental
Crystals of bis L-valinium monohydrogenphosphite were prepared by slow evaporation at room temperature of an
aqueous solution of L-valine and phosphorous acid in a stoichiometric ratio. After six months, crystals appeared as
colourless prisms.
S3. Refinement
One of the monohydrogenphosphite anions is disordered. The disorder can be described as a rotation of this anion around
the axis which bisects the O4B—P1B—O3B angle. The refined model corresponds to a disordered distribution between
OH and H, with occupation factors of 0.85 and 0.15. Some of the H atoms were found in difference Fourier maps and
were refined with isotropic displacement parameters. The hydroxyl H atoms were constrained using an AFIX 147
Figure 1
An ORTEP-3 (Farrugia, 1997) view with the atomic labelling scheme showing the asymmetric unit of the title compound.
Displacement ellipsoids are drawn at the 50% probability level.
(I)
Crystal data
2C5H12NO2+·2H2PO3−
Mr = 398.29
Monoclinic, P21/n
Hall symbol: -P 2yn a = 16.3590 (3) Å b = 6.2540 (2) Å c = 19.4560 (3) Å β = 109.238 (1)° V = 1879.37 (8) Å3
Z = 4
F(000) = 848 Dx = 1.408 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 12190 reflections θ = 1.4–26.4°
µ = 0.28 mm−1
T = 293 K Prism, colorless 0.60 × 0.40 × 0.35 mm
Data collection
Nonius KappaCCD diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
φ scans
12190 measured reflections 3629 independent reflections
3205 reflections with I > 2σ(I) Rint = 0.065
θmax = 26.4°, θmin = 1.4°
h = −20→20 k = −7→7 l = −23→23
Refinement
Refinement on F2
Least-squares matrix: full R[F2 > 2σ(F2)] = 0.038
wR(F2) = 0.100
S = 1.06 3629 reflections 335 parameters 6 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
supporting information
sup-3
Acta Cryst. (2003). E59, o141–o142w = 1/[σ2(F
o2) + (0.0425P)2 + 0.5313P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max = 0.034
Δρmax = 0.25 e Å−3
Δρmin = −0.24 e Å−3
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq Occ. (<1)
N1B 0.94569 (9) 0.7688 (2) 0.94100 (8) 0.0334 (3)
O2A 1.11810 (8) 0.56334 (19) 0.75490 (7) 0.0431 (3)
O1A 1.00386 (8) 0.7676 (2) 0.74927 (7) 0.0454 (3)
C1A 1.07410 (10) 0.7242 (2) 0.73555 (8) 0.0317 (3)
N1A 1.18909 (8) 0.8476 (2) 0.69167 (7) 0.0309 (3)
C2A 1.09959 (10) 0.8950 (2) 0.69071 (8) 0.0312 (3)
C3A 1.03663 (11) 0.9128 (3) 0.61199 (9) 0.0430 (4)
C4A 1.01512 (19) 0.6983 (5) 0.57475 (13) 0.0702 (7)
C5A 0.95530 (16) 1.0358 (5) 0.60860 (15) 0.0726 (7)
O4A 0.96181 (8) 0.50055 (19) 0.82765 (6) 0.0411 (3)
O4B 0.72263 (8) 1.39736 (19) 1.08334 (6) 0.0410 (3)
O3A 0.98837 (8) 0.16535 (19) 0.90586 (6) 0.0419 (3)
O3B 0.73116 (8) 1.06769 (19) 1.16091 (6) 0.0448 (3)
O5A 1.08408 (9) 0.2533 (2) 0.83260 (9) 0.0556 (4)
H14A 1.0949 0.3619 0.8136 0.083*
P1B 0.74461 (3) 1.16508 (7) 1.09540 (2) 0.03368 (14)
P1A 0.99145 (2) 0.27053 (6) 0.83830 (2) 0.03187 (14)
O1B 0.76483 (8) 0.6622 (2) 1.00204 (7) 0.0446 (3)
O2B 0.87394 (8) 0.4572 (2) 0.99502 (8) 0.0510 (3)
C2B 0.85797 (9) 0.8103 (2) 0.94514 (8) 0.0308 (3)
C1B 0.83356 (10) 0.6222 (3) 0.98358 (8) 0.0336 (3)
C4B 0.75966 (15) 0.6568 (5) 0.82464 (12) 0.0634 (6)
C3B 0.79278 (11) 0.8582 (3) 0.86876 (9) 0.0432 (4)
C5B 0.71834 (15) 0.9975 (4) 0.87349 (14) 0.0641 (6)
O5B 0.83698 (11) 1.1263 (3) 1.09437 (11) 0.0635 (5) 0.85
H14B 0.8521 1.2276 1.0746 0.095* 0.85
O13B 0.7050 (7) 1.0432 (19) 1.0270 (4) 0.051 (3) 0.15
H13B 0.7339 0.9358 1.0273 0.076* 0.15
H13C 0.8265 (14) 1.18 (2) 1.124 (6) 0.030* 0.15
H5C 0.6973 (16) 1.060 (5) 1.0410 (13) 0.030* 0.85
H2A 1.1035 (11) 1.020 (3) 0.7162 (10) 0.035 (4)*
H12A 1.1954 (12) 0.720 (4) 0.6803 (10) 0.041 (5)*
H12B 0.9469 (13) 0.656 (4) 0.9113 (12) 0.048 (5)*
H10A 1.2256 (12) 0.862 (3) 0.7356 (11) 0.038 (5)*
H11B 0.9650 (12) 0.890 (4) 0.9284 (11) 0.048 (5)*
H3A 1.0678 (14) 0.999 (3) 0.5870 (12) 0.058 (6)*
H11A 1.2041 (12) 0.934 (3) 0.6633 (11) 0.041 (5)*
H9B 0.6776 (17) 0.920 (5) 0.8938 (15) 0.084 (8)*
H10B 0.9826 (13) 0.741 (3) 0.9845 (12) 0.047 (5)*
H3B 0.8266 (13) 0.942 (4) 0.8457 (12) 0.057 (6)*
H7A 0.9212 (18) 0.946 (5) 0.6325 (16) 0.090 (9)*
H5A 1.0689 (19) 0.633 (5) 0.5752 (15) 0.087 (9)*
H6A 0.9782 (16) 0.718 (4) 0.5227 (15) 0.076 (7)*
H8A 0.9165 (19) 1.059 (5) 0.5602 (18) 0.098 (9)*
H7B 0.6794 (19) 1.049 (5) 0.8248 (18) 0.104 (10)*
H8B 0.7389 (17) 1.139 (5) 0.8987 (15) 0.083 (8)*
H4A 0.986 (2) 0.614 (6) 0.598 (2) 0.111 (12)*
H6B 0.8068 (17) 0.560 (4) 0.8229 (14) 0.077 (8)*
H1B 0.7575 (17) 0.571 (4) 1.0233 (15) 0.071 (8)*
H5B 0.7236 (17) 0.679 (5) 0.7726 (16) 0.081 (8)*
H4B 0.722 (2) 0.583 (5) 0.8469 (17) 0.097 (9)*
H1A 0.9971 (17) 0.670 (5) 0.7759 (15) 0.086 (9)*
H9A 0.9753 (19) 1.195 (6) 0.6354 (17) 0.102 (10)*
H13A 0.9486 (8) 0.163 (2) 0.7816 (7) 0.014 (3)*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
N1B 0.0359 (7) 0.0342 (7) 0.0326 (7) −0.0042 (5) 0.0145 (6) 0.0037 (5)
O2A 0.0483 (6) 0.0390 (7) 0.0524 (7) 0.0071 (5) 0.0306 (5) 0.0152 (5)
O1A 0.0489 (7) 0.0456 (7) 0.0541 (7) 0.0097 (5) 0.0339 (6) 0.0164 (6)
C1A 0.0383 (8) 0.0311 (8) 0.0293 (7) −0.0005 (6) 0.0162 (6) 0.0016 (5)
N1A 0.0375 (7) 0.0275 (7) 0.0305 (7) −0.0019 (5) 0.0150 (6) 0.0022 (5)
C2A 0.0380 (8) 0.0278 (8) 0.0328 (7) −0.0006 (6) 0.0184 (6) 0.0024 (6)
C3A 0.0393 (8) 0.0548 (11) 0.0367 (8) 0.0016 (7) 0.0150 (7) 0.0122 (7)
C4A 0.0668 (15) 0.0852 (18) 0.0446 (12) −0.0021 (13) −0.0005 (11) −0.0144 (11)
C5A 0.0557 (12) 0.092 (2) 0.0697 (15) 0.0274 (13) 0.0204 (11) 0.0304 (14)
O4A 0.0490 (6) 0.0375 (6) 0.0487 (6) 0.0093 (5) 0.0319 (5) 0.0098 (5)
O4B 0.0554 (7) 0.0346 (6) 0.0457 (6) 0.0059 (5) 0.0339 (5) 0.0053 (5)
O3A 0.0547 (7) 0.0383 (7) 0.0379 (6) −0.0057 (5) 0.0224 (5) 0.0072 (5)
O3B 0.0631 (7) 0.0380 (6) 0.0356 (6) −0.0061 (5) 0.0193 (5) 0.0026 (5)
O5A 0.0533 (8) 0.0433 (8) 0.0846 (10) 0.0144 (6) 0.0424 (7) 0.0235 (7)
P1B 0.0430 (2) 0.0306 (2) 0.0323 (2) 0.00354 (15) 0.01885 (17) −0.00120 (14) P1A 0.0378 (2) 0.0308 (2) 0.0311 (2) −0.00076 (15) 0.01700 (16) 0.00251 (14)
O1B 0.0504 (7) 0.0431 (7) 0.0516 (7) 0.0042 (5) 0.0324 (6) 0.0152 (5)
O2B 0.0539 (7) 0.0369 (7) 0.0719 (9) 0.0045 (6) 0.0340 (6) 0.0142 (6)
C2B 0.0355 (7) 0.0310 (8) 0.0290 (7) −0.0025 (6) 0.0147 (6) 0.0015 (6)
C1B 0.0380 (8) 0.0338 (8) 0.0310 (7) −0.0033 (6) 0.0142 (6) 0.0025 (6)
supporting information
sup-5
Acta Cryst. (2003). E59, o141–o142C3B 0.0388 (8) 0.0588 (11) 0.0327 (8) 0.0000 (8) 0.0129 (6) 0.0126 (7)
C5B 0.0583 (12) 0.0656 (15) 0.0650 (13) 0.0185 (11) 0.0156 (10) 0.0215 (11)
O5B 0.0532 (10) 0.0722 (13) 0.0755 (12) 0.0187 (9) 0.0351 (8) 0.0009 (9)
O13B 0.083 (7) 0.046 (5) 0.025 (4) 0.014 (4) 0.021 (4) −0.012 (3)
Geometric parameters (Å, º)
N1B—C2B 1.4863 (19) O3B—P1B 1.4930 (12)
O2A—C1A 1.2218 (19) O5A—P1A 1.5591 (12)
O1A—C1A 1.2913 (18) P1B—O13B 1.484 (6)
C1A—C2A 1.522 (2) P1B—O5B 1.5369 (16)
N1A—C2A 1.4879 (19) O1B—C1B 1.3121 (19)
C2A—C3A 1.542 (2) O2B—C1B 1.206 (2)
C3A—C4A 1.510 (3) C2B—C1B 1.517 (2)
C3A—C5A 1.519 (3) C2B—C3B 1.546 (2)
O4A—P1A 1.5104 (12) C4B—C3B 1.520 (3)
O4B—P1B 1.4964 (12) C3B—C5B 1.525 (3)
O3A—P1A 1.4855 (11)
O2A—C1A—O1A 125.70 (14) O4B—P1B—O5B 109.73 (10)
O2A—C1A—C2A 120.64 (13) O3A—P1A—O4A 116.11 (7)
O1A—C1A—C2A 113.66 (13) O3A—P1A—O5A 110.45 (7)
N1A—C2A—C1A 107.89 (12) O4A—P1A—O5A 109.37 (7)
N1A—C2A—C3A 111.05 (12) N1B—C2B—C1B 107.88 (13)
C1A—C2A—C3A 113.60 (13) N1B—C2B—C3B 110.75 (12)
C4A—C3A—C5A 111.5 (2) C1B—C2B—C3B 114.44 (13)
C4A—C3A—C2A 112.72 (16) O2B—C1B—O1B 125.42 (14)
C5A—C3A—C2A 111.60 (16) O2B—C1B—C2B 122.52 (13)
O13B—P1B—O3B 114.3 (5) O1B—C1B—C2B 112.07 (14)
O13B—P1B—O4B 110.3 (5) C4B—C3B—C5B 111.37 (18)
O3B—P1B—O4B 115.76 (7) C4B—C3B—C2B 112.77 (17)
O13B—P1B—O5B 92.8 (4) C5B—C3B—C2B 111.37 (16)
O3B—P1B—O5B 111.65 (10)
O2A—C1A—C2A—N1A −11.7 (2) N1B—C2B—C1B—O2B −12.1 (2)
O1A—C1A—C2A—N1A 168.78 (13) N1B—C2B—C1B—O1B 167.89 (13)
N1A—C2A—C3A—C4A 74.0 (2) N1B—C2B—C3B—C4B 80.56 (19)
N1A—C2A—C3A—C5A −159.58 (18) N1B—C2B—C3B—C5B −153.39 (17)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
O1A—H1A···O4A 0.83 (3) 1.69 (3) 2.506 (2) 167 (3)
O1B—H1B···O4Bi 0.74 (3) 1.82 (3) 2.538 (2) 165 (3)
N1A—H10A···O3Bii 0.87 (2) 1.95 (2) 2.783 (2) 159 (2)
N1B—H11B···O3Aiii 0.89 (2) 1.85 (2) 2.724 (2) 170 (2)
N1A—H11A···O4Biv 0.87 (2) 1.98 (2) 2.837 (2) 169 (2)
N1A—H12A···O3Bvi 0.84 (2) 1.97 (2) 2.802 (2) 170 (2)
N1B—H12B···O4A 0.91 (2) 1.98 (2) 2.852 (2) 159 (2)
O5A—H14A···O2A 0.82 1.82 2.626 (2) 167
O5B—H14B···O2Biii 0.82 2.23 3.026 (2) 165
O13B—H13B···O1B 0.82 1.90 2.681 (11) 160