http://www.sciencepublishinggroup.com/j/ejb doi: 10.11648/j.ejb.20180601.12
ISSN: 2329-1745 (Print); ISSN: 2329-1737 (Online)
A Pipeline for Markers Selection Using Restriction Site
Associated DNA Sequencing (RADSeq)
Hanan Begali
Department of Life Science Informatics Master Program, Bonn-Aachen International Center for Information Technology B-IT at Bonn University, Bonn, Germany
Email address:
To cite this article:
Hanan Begali. A Pipeline for Markers Selection Using Restriction Site Associated DNA Sequencing (RADSeq). European Journal of Biophysics. Vol. 6, No. 1, 2018, pp. 7-16. doi: 10.11648/j.ejb.20180601.12
Received: October 19, 2017; Accepted: December 27, 2017; Published: January 20, 2018
Abstract:
Motivation: The discovery and assessment genetic variants for Next Generation Sequencing (NGS), includingRestriction site Associated DNA sequencing (RADSeq), is an important task in bioinformatics and comparative genetics. The genetic variants can be single-nucleotide polymorphisms (SNPs), insertions and deletions (Indels) when compared to a reference genome. Usually, the short reads are aligned to a reference genome at first using NGS alignment software, such as the Burrows- Wheeler Aligner (BWA). The alignment is usually stored into a BAM file, a binary format of standard SAM (Sequence Alignment/Map) protocol. Then analysis software, such as Genome analysis Toolkit (GATK) or SAMTools [30] [31], together with scripts written in R programming language, could provide an efficient solution for calling variants. We focused on RADSeq-based marker selection for Arabidopsis thaliana. RADSeq consists short reads that do not cover the whole reference genome. Finally, SNPs as output in Variant Call Format (VCF) have been visualized by Integrative Genomics Viewer (IGV) software. We found that the visualization of SNPs and Indels is helpful and provides us with valuable insights on marker selection. We found that applying Chi-Square test for all target genotypes, which are homozygous reference 0/0, heterozygous variants 0/1 and homozygous variants 1/1, to test Hardy-Weinberg Equilibrium (HWE) in order to reduce false positive rate significantly and we showed that our pipeline is efficient in RADSeq-based marker selection.
Keywords:
NGS-RADSeq, Arabidopsis thaliana (TAIR10), GATK, SAMTools, Chi-Square Test, HWE-P, Reliable SNPs1. Introduction
The critical state of identification genetics variants in the next generation sequencing (NGS), specifically, Restriction-site associated DNA sequencing (RADSeq) is well established in bioinformatics and comparative genomics [3] [4]. We are able to extract genetic markers information from data stemming using RADSeq, such as marker position and genotypes at the unique regions on the chromosome, by investigating single nucleotide polymorphisms (SNPs). SNPs can be defined as a difference in a single nucleotide of DNA at a particular location in the genome. Therefore, that necessitates the application of data processing in order to determine the reliable markers using RADSeq data, then evaluate them and obtain reliable SNPs [15] [17] [18].
Illumina [23], which is a recent technology, is used to sequence DNA and provides high- throughput sequencing (NGS) in order to study genomics. NGS is based on the
recent method commonly known as Restriction-site Associated DNA sequencing (RADSeq) [4]. RADSeq can be described as extensive parallel sequencing. RADSeq has been produced or generated by shearing DNA molecules into collections of numerous small fragments which are called a library. Moreover, the fragments are called reads which produce the contiguous strings. In order to discover SNP using RADSeq, mapping to an available reference
Arabidopsis thaliana (TAIR10) is performed [1] [14] [33].
2. Datasets and Data Analysis
2.1. Datasets
used [13]. Datasets, which are original material in FASTQ format [27 ] of RADSeq, have been derived from department lab for Illumina sequencing data. Datasets consist of 191 individuals from the next generation (F2). The F2 of these individuals has been produced by crossbreeding of two different strains: the mother strain, which is not yet published RADSeq 6216_2251, can be described as having a highly serrated leaf and the father strain, which is the common lab strain A. thaliana research: Col-0 the seed stock is N28167 [5] [26], has a simple leaf phenotype. Additionally, the father strain should be identical to the reference genome, however, since there were many generations in between, some SNPs may occur. Both strains are crossbred to identify target genotype DNA sequence polymorphisms in their offspring. Clustering of genetic markers (genetics variants) in their offspring is applied in order to compare the genetic variants of two populations.
The aim of this experimental design is to call variants in all individuals using RADSeq. The father strain, RADSeq Col-0: N28167 [5] [26], carries homozygous reference 0/0. Both alleles are the same as the reference alleles. The mother strain, RADSeq-2251, that carries homozygous variants 1/1, both alternate alleles are different from the reference alleles. Moreover, for every SNP at each position on the loci, the alleles frequencies must remain stable from generation to generation according to HWE [21].
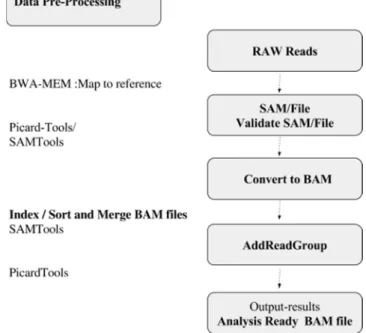
Figure 1. General workflow for this project.
2.2. Pre-Processing Data for Mapping Sequences
In order to prepare data using RAD Sequences, for downstream analysis, aligning sequences is performed. RAD sequences data is typically in a raw state. Data is present in the form of FASTQ files, which are used to store short reads data from high-throughput sequencing experiment before mapping and record each sequence with quality score for each nucleotide [27]. The preprocessing stage is the main step in preparing RAD sequences in order to continue usable variant discovery analysis. The goal of this step is to obtain an analysis-ready BAM file [30]. The Burrows Wheeler
Aligner, or BWA-MEM, which is the common software package, is used in order to identify low-coverage genomes in metagenome samples. Therefore, BWA-MEM is the preferred algorithm to be applied in this pipeline data analysis [25] for Illumina sequence reads [2].
The advantages of the BWA-MEM can be noted as the following: 1) it is more powerful, 2) it allows for local alignment to obtain the optimal solution for mapping problems, thereby decreasing false positives, and 3) it produces highly accurate results and relies on an algorithm that finds exact nucleotide matches [2] [10].
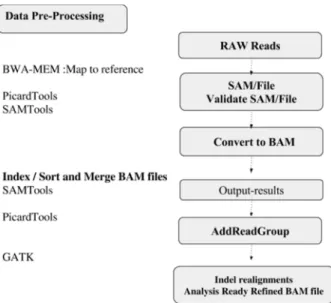
There are two procedures to generate a BAM file which is required in downstream analysis for calling variants, as shown in Figures 2 and Figure 3. The first step for mapping RAD sequences is to promptly place short reads of RADSeq along the reference genome, and then build an index for the fragments. Then, BWA-MEM is applied to find the most likely sequence location for each read. The next step is to convert the alignments to a more standard format. In order to quickly and conveniently extract reads from genome location, BAM index files (*.bam.bai) are created.
Figure 2. The workflow for generating BAM file.
Finally, SAM stands for Sequence Alignment / Map, which is the format, that is used to store large nucleotide sequence alignments in a human readable format. Although, both BAM and SAM formats are designed to contain the same information but SAM format is more human readable, and easier to process. In contrast, the BAM format provides binary versions of most of the same data, and is designed to compress reasonably well [22].
Figure 3. The workflow for generating realigned BAM file.
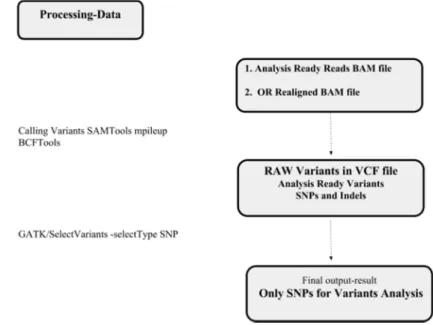
2.3. Processing Data for Calling Variants
Calling variants using RADSeq data can be defined as a computational method to establish an event of genetic variants resulting from NGS experiments. Furthermore, variant calling involves small-range variants such as SNPs, short insertions and deletions (indels). Moreover, variants calls are implemented in four procedures that are described in more detail in Figure 4 and Figure 5 [9] [23].
The drawback of variants discovery processing is that some of the variation have been observed due to mapping and sequencing artifacts, so that the biggest challenge is to balance the requirement for sensitivity versus specificity. That aims to minimize false negatives such as failing to
identify real variants and to minimize false positives such as failing to reject artifacts. This challenge is addressed by applying this process which aims to identify the sites where RAD-Seq data displays variation relative to the reference genome, then by calculating genotypes for each SNP at that site [15].
Several bioinformatics tools such as SAMTools and Genome analysis ToolKit (GATK) software are useful in order to incorporate different datasets for reliable variants calling. In theory, a SNP is identified when a nucleotide from an accession read differs from the reference genome at the same nucleotide position indicating an alteration in the most common DNA nucleotide sequence [15].
There are several approaches to call variants which are based on GATK software and SAM Tools. Furthermore, there are two workflow pipelines for each one as shown in Figure 4 and Figure 5 [3] [10].
First, the algorithm checks the active region containing the genetic variants for both haplotypes using the HaplotypeCaller with joint genotypes [32]. In other words, whenever the program detects a region that is showing signs of variation, the program generates an intermediate genomic gVCF for each individual. Then, joint genotyping are applied for all individuals in a manner that is considered to be highly efficient [3] [10].
The main goal of the Unified Genotyper Algorithm is to call SNPs and indels on a per-locus basis, which is based on a Bayesian genotype likelihood model, in order to determine simultaneously the most likely genotypes and allele frequency in RADSeq that are satisfied phred-scaled confidence value.
Figure 5. The workflow for calling variants by SAM Tools.
The main concept of SAMTools is to calculate the likelihood of mapping sequences, which contains the genotype, and relies on the summary information in the input BAMs. Then, these likelihoods are stored in the BCF format in which call variants have not yet been performed. Therefore, the SAMTools mpileup provides the summary of the coverage of mapped reads on a reference sequence at each single base pair [28]. The important application of BCF Tools is to perform the actual calling that is based on the prior likelihoods in the BCF format. Moreover, selecting variants is perfomed by using Select Variants -select_Type SNP/GATK software in order to extract only a subset of SNPs. The goal of that is to facilitate certain analyses such as filtering status [10].
By comparing between two outputs VCF files, which have been generated using GATK, we noticed that additional annotation, Haplotype-Score, of each SNP has been obtained by using Unified Genotyper Algorithm [19] [32]. Also because Haplotype Caller does a local reassembly, applying indel-realignment is not necessary. For other variant callers, such as Unified Genotyper, which do not perform a local reassembly, this process is recommended in performing indel-realignment [19]. By comparing VCF files which have generated by GATK and SAMTools, we noticed that VCF files which have been produced by SAMTools provide us with only one annotation, Mapping Quality score (MQ), for each SNP at loci.
2.4. Advanced Data Analysis for Evaluation Call-Sets SNPs
In order to generate highly accurate call sets, hard filtering is applied. The filtering is performed based on the annotation of each single variant in order to obtain a higher accuracy for each call. Also, filtering uses fixed thresholds on specific variant annotations, as the goal of this project is to obtain reliable SNPs [29].
After extracting the genetic variants SNPs from the call set by Select Variants–select Type SNP/GATK software, the
parameters for filtering SNPs are determined. This relies on extracting the essential information from a Variants Call Format (VCF) in a direct fashion by using Variants To Table/GATK software and gaining a comprehensive understanding of the information that is biologically significant. Then, filtering the SNP call-set is applied by Variant Filtration/GATK software under specific conditions (see table 1). The final product of this protocol is a VCF file containing high-quality variant calls that can be used in downstream analysis. Therefore, all SNPs which match any of these filtering conditions are considered as being of poor quality, are filtered out and are marked as FILTER in the output VCF file. In contrast, SNPs which do not match these fitering conditions are considered as good and marked PASS in the output VCF file [3] [10] [12].
R program language
In order to view the distribution of the annotations for each SNP at loci, kernel density plots is used. The distribution allows comparing the same annotations, such as Mapping Quality (MQ), in each file call-set, while the y-axis indicates proportion. Therefore, low scores of these parameters are examined and only reliable SNPs have to be kept.
Moreover, in order to obtain reliable SNPs with higher genotype quality, both Chi- Square test and P-HWE are computed by VCFTools [7]. However, selecting an appropriate level of certainty has been done initially. For example, a certainty, is referred as the P value or alpha value can be chosen; in this case, a certainty of 0.05 means that there is a 5% probability of there being a difference, when no such difference exists. For most scientific purposes, the level of certainty is arbitrarily set at 0.05, meaning there is only a 5% probability that the difference between observed and expected is due to chance alone [5] [14] [19] [20].
2.5. Visualization by Integrative Genomics Viewer (IGV)
major challenge with data visualization is dealing with large and diverse data which has been produced by sequencing. Nevertheless, the intuitive visualization of Integrative Genomics Viewer (IGV) relieves this problem.
IGV [29] is known as a desktop platform for visual interactive exploration of integrated genomic datasets such as all NGS alignments. It is focused on visualization of the best validation and confirmation analysis results. Also, IGV is considered as a supporting tool for visualization at all scales of the genome, particularly NGS data due to its main characteristic, breadth. Moreover, IGV is a high-performance visualization tool and written in the Java programming language, that runs on different platforms such as Linux, for supporting researchers in visualization of genomic data including NGS data, variant calls. The goal of IGV visualization is to determine SNPs at the position on the chromosome. Moreover, IGV are stable, easy to assay and it allows for distribution along the genome with higher density. Therefore, IGV provides extensive platforms for supporting viewing variants which are stored in the VCF format. This format allows for encoding all variant calls such as SNPs as well as the supporting genotype information for each individual RADSeq [29].
3. Results of Data Analysis
3.1. Pre-Processing Data for Mapping Sequences
Bioinformatics tool, SAMTools flagstat, is used for statistical analysis such as which is used in order to determine the number of reads, the number of mapped reads, the number of base pairs and the length of fragments. Moreover, SAMTools depth calculation is used to determine coverage per locus as well as the Lander/Waterman equation [8] [11]. The range length of all RADSeq is between 30 and 150 base pairs (bp). However, the number of mapped reads can vary from each other as not all of the reads have been mapped. We have observed that some of the mapped reads are less than the total
coverage of reads. The highest number of reads and mapped reads are detected in the same sample of next generation sequences (F2). Furthermore, the total number of reads is indicated, including mapped and unmapped reads. The maximum number of reads that is found in RADSeq datasets is 1,127,969 in a sample RADSeq 334. In contrast, the minimum number of reads is 1,713 in sample RADSeq 285. Moreover, 28.63% is the low map quality of the dataset that is detected in sample RADSeq 198 which then is excluded before calling variants. The reason for the lower map quality score is contamination during extraction of the RADSeq in the laboratory. In contrast, a higher map quality score reflects the presence of a sufficient amount of data, such as in the case of a sample RADSeq 301 (F2) where a score of 99.36% is determined. A higher coverage in shotgun sequencing is desired because it can overcome errors in base calling. Furthermore, it indicates a sufficient dataset can be obtained such as in a sample RADSeq 334 which is observed with the highest score of 46.48% that means each base in the reference genome has been sequenced between 46 and 47 times on average.In contrast, if the fragment has low depth of coverage, it is removed out before downstream analysis. The reason for filtering is to obtain a higher accuracy genotype which requires an increase in the average depth of coverage across all loci. For example, the sample RADSeq 276 is detected with an average coverage of only 1.515% as a minimum score which means that the average of each base in the reference genome has been sequenced only between one and two times in this sample.
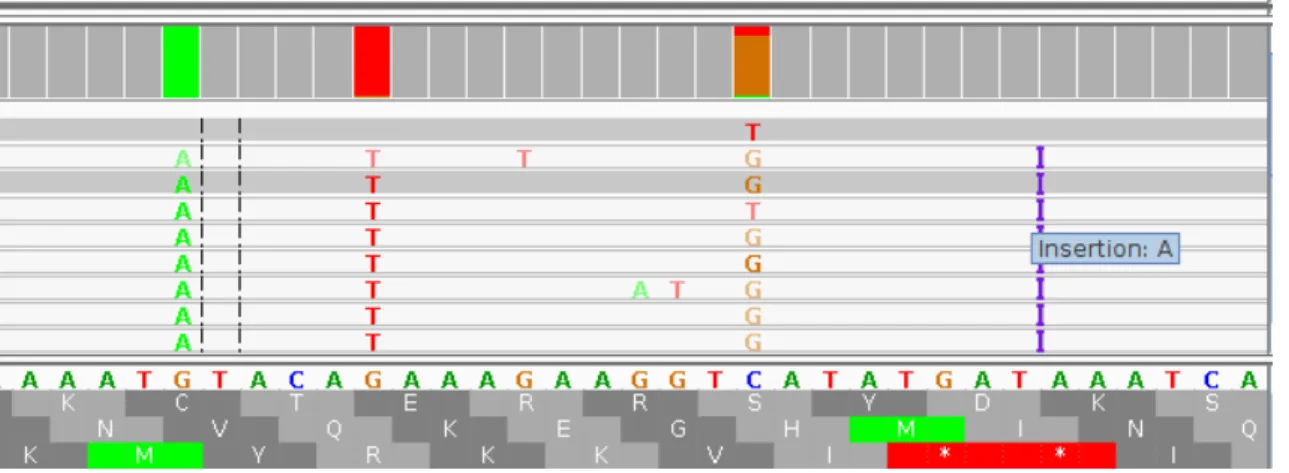
An example of IGV visualization of BAM file is shown in Figure 6 that insights into divergence regions for the alignments sequences are depicted. The visualization shows that for both mismatched bases, the A and G nucleotides have higher allele frequencies, while the T nucleotide has low allele frequency, such as in RADSeq 259, RADSeq 307 and RADSeq 338.
Figure 6. Screenshot from IGV for mismatched and an insertion of a base A at Chr 5: 13,520,387-13,520,442.
3.2. Processing Data for Calling Variants
Both Genome Analysis Toolkit software and SAMTools are given the VCF, which is the main goal for variants
able to detect mismatched nucleotides, some of these mismatches are introduced as errors.
The reasons of these errors can be noted as the following: 1) preparing the sample libraries at the wet lab, 2) machine errors when sequences are generated from the libraries, and 3) software and mapping artifacts when the reads are aligned. Therefore, quality filtering of variant calls is an essential step that is performed due to the fact that not all variants that we called are necessarily of good quality. Therefore, understanding the annotations comprehensively is useful for filtering data.
3.3. Advanced Data Analysis for Evaluation Call-Sets SNPs
By determining the parameters for the hard-filtering step, a higher quality of normalization of the depth of sample reads can be obtained by Qual by Depth (QD). Additionally, the null hypothesis indicates that the number of heterozygotes under HWE has been studied. Moreover, Moreover, when the mapping qualities, or the root mean square (RMS), are around 60, the site is considered to be good which indicates that the RMS has a higher accuracy. In practice, when evaluating the variant quality, we only filter out low negative values for all of the annotations of MQ Rank Sum, Read Pos-Rank Sum Test and Base Quality Rank Sum. The reason for filtering these low negative values is to filter out variants for which the quality of the data supporting the alternate allele is comparatively low. Moreover, there are some missing values in some tests such as Rank Sum Test due to that this test has only been applied on heterozygous alleles, with a mix of reads bearing the reference and the alternate alleles [23]. Additionally, hard filtering allows to update the new recalibrated VCF file which stores only variants with higher accuracy.
3.3.1. Hard Filtering Results
All SNPs`annotaions that are used in order to filter out bad SNPs as shown in Table 1.
Table 1. 1st caller-Haplotyper with joint genotypes, 2nd-caller Unified Genotyper, 3rd SAMTools, 4th refinement SAMTools.
Annotations 1st 2nd 3rd 4th
QD < 2.0 < 2.0 - -
Excess Het > 8.0 > 8.0 - -
MQ > 62 | | <57 > 60 | | <58 <57.4 <57.4
MQ Rank Sum < -0.5 < -6.0 - -
Read Pos Rank Sum < -1.0 < -16.0 - -
Base Quality Rank Sum < -2.0 < -4.0 - -
FS > 15.0 > 6.0 - -
SOR < 2.0 | | >2.5 <1.45 | | >1.75 - -
Haplotyper Score - > 1.8 - -
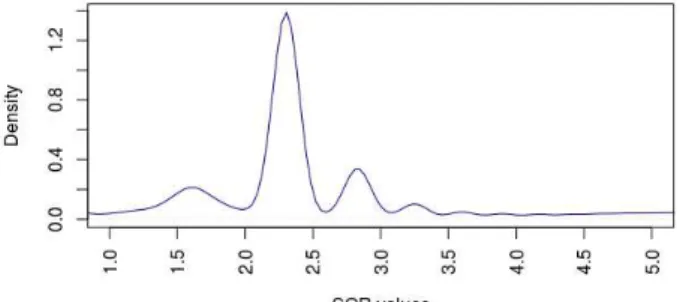
Two examples for data distribution in Figure 7 and Figure 8 which show values for SOR test and QD test for all SNPs which have been called by Haplotype Caller with Joint Genotypes.
Figure 7. SOR values distribution for unfiltered variants.
Figure 8. QD values distribution for unfiltered variants.
The generic filtering recommendation for QD is to filter out variants with QD below 2. This is because homozygous variants RADSeq contribute twice as many reads supporting the variant than do heterozygous variants. Moreover, The hard filtering recommendations tell us to fail variants with an SOR value greater than 2.5 or less than 2.
The goal of statistical analysis is to calculate how many genotypes of each SNP present at each loci. In order to be more specific, the times of genotype at each chromosome are computed. The most prevalent genotype repeats indicates the genotype majority in a population for the RADSeq, while the minority of individuals is represented by the least prevalent genotype repeats at this loci. The main aspect to determine genotype frequencies in a population is to test how they are in the next generation sequences. Therefore, Hardy-Weinberg equilibrium (HWE) principle is studied comprehensively because it informs us about the probability of genotype frequencies in that population [15]. Finally, both the Chi-Square and P-value according to HWE are calculated by VCFTools in order to indicate that the frequency of alleles in a population remains stable from generation to generation. Moreover, the statistical test, Chi-Square test, is considered as the goodness of fit to determine whether the SNPs at each position have a significant difference between the number of actual (observed) genotypes and expected genotypes or not [33].
3.3.2. Results of Critical Values for Chi-Square Test
Figure 9. Density distribution for critical value of Chi-Square test for each SNPs.
3.3.3. Results of P-Values
The four plots shown in all Figure 10, Figure 11, Figure 12 and Figure 13 show the distribution of all P-values calculated for each SNP at loci. Reliable SNPs have P-value greater than 0.05. In contrast, all SNPs that have P-values < 0.05 are rejected (false positive). We noticed that only by Haplotype Caller with joint genotypes algorithm have more reliable SNPs, which have been detected.
Figure 10. P-values by first procedure.
Figure 11. P-values by second procedure.
Figure 12. P-values by third procedure.
Figure 13. P-values by fourth procedure.
3.4. Visualization for Reliable SNPs
Figure 14 shows a first example for visualizing genomic data of RADSeq. This variant has a higher genotype quality score more than 20 that indicates higher accuracy at this position, which has been called by Unified Genotyper algorithm.
Figure 14. Screenshot from IGV for visualization shows the homozygous reference (gray color) with only one heterozygous variant (dark blue), RADSeq 254.
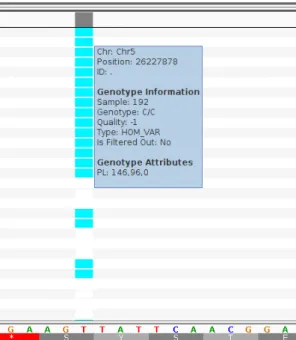
Figure 15. Screenshot from IGV for homozygous variants such as in RADSeq 192 at this position.
The third example, Figure 16, of RADSeq 197 shows homozygous reference (gray color) and homozygous variants (cyan color) with heterozygous variants (dark blue) which has a higher genotype quality score more than 60 and indicates higher accuracy of (more than 99.9999%) that has been called by Haplotype Caller algorithm.
Figure 16. Screenshot from IGV for visualization data in RADSeq 197.
Results of Chr C:
The fourth example is shown in Figure 17 of SNPs in genetic material on the chromosome of chloroplast (Chr C). These should all be 1/1 homozygous variants as mother`s alleles such as the genetic variants at this position has been
called by Unified Genotyper procedure of Chr C.
Figure 17. Screenshot from IGV for homozygous variants for SNPs in genetic material for Chr C.
4. Conclusions & Suggestions
The goal of this new genomic study was to identify high density of markers across genomes using NGS techniques such as RAD-Seq, particularly in organisms for which few genomic resources presently exist. RAD-seq data was used to identify SNPs and distinguish homozygous and heterozygous variants in a population of Arabidopsis thaliana obtained by crossbreeding two strains. To accomplish this, a customized pipeline was used that included four different variant callers which are based on the Genome Analysis Toolkit (GATK) [30] and SAMTools/BCFTools [11] [15] [30]. Both GATK and SAMTools produce genotype information in groups of individuals. These tools are meant to operate on top of a genome, for example by detecting nucleotide variants through matches to the reference sequence.
Other previous pipeline is available to provide several common output formats to integrate Stacks-generated genotype data of RAD-Seq with downstream analysis packages. In contrast, SAMTools/BCFTools and GATK can call SNPs in multiple samples of RAD-Seq and then can generate allele frequencies, but all populations are managed by hand as merging all BAM files in order to obtain one VCF file that consists of multiple samples, as compared to the integrated way that this occurs in Stacks. Furthermore, Stacks
was developed to have at its core a catalogue that works as an internal reference for each project regardless according to the presence of a genome [6].
analyses but with some difficulty for the following reasons. First, some SNPs have been detected due to errors during variant calling. We could divide the SNP calling errors into three classes: 1) preparing the sample libraries at the wet lab, 2) machine errors when sequences are generated from the libraries, 3) software and mapping artefacts when the reads are aligned. Therefore, quality filtering of variant calls is an essential step that has been applied in order to minimize false positive. Therefore, in order to obtain high specificity, a number of filters were applied to eliminate false positives. Second, parameters/thresholds required for the filters needed to be set comprehensively in dependence of the data obtained. Third, the final step consisted of the visualization of the variants using Integrative Genomics Viewer (IGV) is required. In contrast, a Stacks analysis starts with raw sequencing reads and then progress through all analysis steps to generate allele and genotype calls, a number of core population genetics statistics and formatted output files [6].
Finally, the results in this study were very consistent and indicated new pipeline for variants calling using NGS data, especially RAD-Seq is developed. Thus, the pipeline presents a valuable tool for exploring homozygous and heterozygous variants. We concluded that results from our study will provide practical and comprehensive guidance to more accurate and consistent variant identification. On this basis further research might be conducted as in the following: 1) A comprehensive study of those genotypes for variants that have been detected and refined. As most SNPs are found within protein coding regions, to characterize the function impact of each SNP would be interesting. For example, if the coded amino acid does not change the protein, it is called a synonymous SNP (as the codon is a 'synonym' for the amino acid), while if the coded amino acid causes a change in the protein, it is called a non-synonymous SNP, 2) the identification of genetic markers that have typically been involved in marker discovery of SNPs using RAD-Seq could be used in the future to associate with the phenotypes. That is to study variants which are associated with a phenotype could be comprehensively studied in the future.
Acknowledgements
We are grateful to Max Planck Institue for Plant Breeding Reascher, Xiangchao Gan, PhD and Professor Dr Miltos Tsiantis. Wellness for their offering this job with new topic Master project: bioinformatics and comparative genomics Nr: Summer-2017.
References
[1] Allen, R. S., Nakasugi, K., Doran, R. L., Millar, A. A., & Waterhouse, P. M. (2013). Facile mutant identification via a single parental backcross method and application of whole genome sequencing based mapping pipelines. Frontiers in plant science, 4.
[2] Almgren, P., BENDAHL, P., Bengtsson, H., Hössjer, O., & Perfekt, R. (2003). Statistics in genetics. Lecture notes, Lund.
[3] Andrews, K. R., Good, J. M., Miller, M. R., Luikart, G., & Hohenlohe, P. A. (2016). Harnessing the power of radseq for ecological and evolutionary genomics. Nature Reviews Genetics, 17 (2), 81–92.
[4] Baird, N. A., Etter, P. D., Atwood, T. S., Currey, M. C., Shiver, A. L., Lewis, Z. A.,. Johnson, E. A. (2008). Rapid snp discovery and genetic mapping using sequenced rad markers. PloS one, 3 (10), e3376.
[5] Bergelson, J., Kreitman, M., & Nordborg, M. (n. d.). Columbia col-0: n28167 or cs28167 [Computer software manual]. Retrieved from
https://www.arabidopsis.org/abrc/catalog/natural a ccession 5 html.
[6] Catchen, J., Hohenlohe, P. A., Amores, S. B. A., & Cresko, W. A. (2014 Nov 25). Stacks: An analysis tool set for population genomics. NIH Public Access, PMC.
[7] Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A.,... others (2011). The variant call format and vcftools. Bioinformatics, 27 (15), 2156–2158.
[8] Davey, J., & Blaxter, M. L. (2011). Radseq: next-generation population genetics. Briefingsin Functional Genomics, 9, 108.
[9] De Pristo, M. A., Banks, E., Poplin, R., Garimella, K. V., Maguire, J. R., Hartl, C.,... others (2011). A framework for variation discovery and genotyping using next-generation dna sequencing data. Nature genetics, 43 (5), 491–498.
[10] De Summa, S., Malerba, G., Pinto, R., Mori, A., Mijatovic, V., & Tommasi, S. (2017). Gatk hard filtering: tunable parameters to improve variant calling for next generation sequencing targeted gene panel data. BMC bioinformatics, 18 (5), 119.
[11] Emigh, T. H. (1980). A comparison of tests for hardy-weinberg equilibrium. Biometrics, 627–642. Group, S. F. S. W., et al. (2014). Sequence alignment/map format specification. Tech. rep. Version 1. 2015. url: http://samtools. github. io/hts-specs/SAMv1. pdf (visited on 01/04/2015).
[12] Herzeel, C., Costanza, P., Ashby, T., & Wuyts, R. (2013). Performance analysis of bwa alignment (Tech. Rep.). Technical Report Exascience Life Lab. 59.
[13] Ishii, K., Kazama, Y., Hirano, T., Hamada, M., Ono, Y., Yamada, M., & Abe, T. (2016). Amap: A pipeline for whole-genome mutation detection in arabidopsis thaliana. Genes & genetic systems, 91 (4), 229–233.
[14] Kosugi, S., Natsume, S., Yoshida, K., MacLean, D., Cano, L., Kamoun, S., & Terauchi, R. (2013). Coval: Improving alignment quality and variant calling accuracy for next-generation sequencing data. PLoS one.
[15] Li, H. (2010). Mathematical notes on samtools algorithms. October.
[16] Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with bwa-mem. arXiv preprint arXiv.
[17] Li, H. (2014). Toward better understanding of artifacts in variant calling from high-coverage samples. Bioinformatics, 30 (20), 2843–2851.
[19] Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N.,... Durbin, R. (2009). The sequence alignment/map format and samtools. Bioinformatics, 25 (16), 2078–2079.
[20] Marrano, A., Birolo, G., Prazzoli, M. L., Lorenzi, S., Valle, G., & Grando, M. S. (2017). Snp-discovery by rad-sequencing in a germplasm collection of wild and cultivated grapevines (v. vinifera l.). PloS one, 12 (1), e0170655.
[21] McCormick, R. F., Truong, S. K., & Mullet, J. E. (2015). Rig: recalibration and interrelation of genomic sequence data with the gatk. G3: Genes, Genomes, Genetics, 5 (4), 655–665.
[22] McKenna, A., et al. (2016). The genome analysis toolkit: A mapreduce framework for analyzing next-generation dna sequencing data. genomet+ research. published in advance jul. 19, 2010.
[23] Molnar, M., & Ilie, L. (2015). Correcting illumina data. Briefings in bioinformatics, 16, 588–599.
[24] Nielsen, R., Paul, J. S., Albrechtsen, A., & Song, Y. S. (2011). Genotype and snp calling from next-generation sequencing data. Nature Reviews Genetics, 12 (6), 443–451.
[25] Ossowski, S., Schneeberger, K., Clark, R. M., Lanz, C., Warthmann, N., & Weigel, D. (2008). Sequencing of natural strains of arabidopsis thaliana with short reads. Genome research, 18 (12), 2024–2033.
[26] Peter J. A. Cock, Christopher J Fields, Naohisa Goto, Michael Lheuer, Peter M Rice. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina
FASTQ variants. Nucleic Acids Research, Vol (38), No (6). 16 December 2009.
[27] Runs, E. S. (n. d.). Estimating sequencing coverage.
[28] Thorvaldsdóttir, H., Robinson, J. T., & Mesirov, J. P. (2013). Integrative genomics viewer (igv): high-performance genomics data visualization and exploration. Briefings in bioinformatics.
[29] Van der Auwera, G. A., Carneiro, M. O., Hartl, C., Poplin, R., del Angel, G., Levy Moonshine, A.,... others (2013). From fastq data to high-confidence variant 60 calls: the genome analysis toolkit best practices pipeline. Current protocols in bioinformatics, 11–10.
[30] Wang, J., Scofield, D., Street, N. R., & Ingvarsson, P. K. (2015). Variant calling using ngs data in european aspen (populus tremula). In Advances in the understanding of biological sciences using next generation sequencing (ngs) approaches (pp. 43–61). Springer.
[31] Warden, C. D., Adamson, A. W., Neuhausen, S. L., & Wu, X. (2014). Detailed comparison of two popular variant calling packages for exome and targeted exon studies. Peer J, 2, e600.
[32] Weigel, D., & Mott, R. (2009). The 1001 genomes project for arabidopsis thaliana. Genome biology, 10 (5), 107.