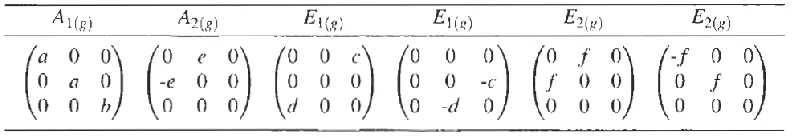International Journal of Emerging Technology and Advanced Engineering
Website: www.ijetae.com (ISSN 2250-2459,ISO 9001:2008 Certified Journal, Volume 5, Issue 9, September 2015)
315
Simulating Raman Spectrum of C
60
Cl
6
through Ab initio
Method
Rashid Nizam1,Narender Singh2, Richa Saxena3, Shabina Parveen4 1,2,3Department of Physics, IFTM University, Moradabad, India 4
Department of Botany Pt.L. M. S. Govt. Autonomous P. G. College, Rishikesh. India
Abstract-- The Raman spectrum of C60Cl6 has been calculated using ab initio method. It has been observed that the simulated spectrum of C60Cl6 is matching with the
available experimental data in literature. It is interested to note that by simple analysis of the normal coordinates of C60Cl6 , the four principal regions of its vibrational spectrum
100–130 cm-1, 140–400 cm-1, 400-900 cm-1 and 900–1600 cm-1 are assigned. In the first 100–130 cm-1 range, the chlorine-atom motion predominantly in the infrared spectrum of C60Cl6. In the second range 140–400 cm-1carbon with chlorine
atoms equally participate in the vibrations of C60Cl6.The
intensities of this range are unusual vibrations are low, and also are observed in the experimental spectra as weak features. In the third range 400-900 cm-1 the vibrations of C60Cl6 are restricted on the carbon skeleton of the molecule
with insignificant chlorine- atom motion. In the fourth range 900–1600 cm-1 the vibrations of the C
60Cl6 carbon skeleton
have considerable tangential character stretching vibrations in the infrared spectrum.
Keywords- Raman spectrum, Ab initio method, Infrared spectrum
I. INTRODUCTION
Halogenated fullerenes are of considerable interest because they are prospective originators for the sample preparation of fullerenes derivatives. Chloro-fullerenes were poorly investigated in comparison with other
halogenated fullerenes. Single crystal X-ray
crystallography has been synthesized and structurally characterized C60 chlorinated derivatives such as C60Cl24 (Th), C60Cl28 (C1), C60Cl30 (C2) and C60Cl30 (D3d) [1-3].The production of C60Cl6 was reported and this chloride was utilized for the preparation of organic derivatives of C60. [4–8] In the past decade, considerable progress has been made in the understanding of fullerene derivatives.
IR vibrational is a unique sensitivity spectroscopy to molecular structure, so it has an application can assist substantially in structure elucidation, particularly when other commonly used direct methods of structural analysis have failed to provide unambiguous answers. We report that comprehensive IR spectroscopic studies of C60Cl6 with the help of ab initio method.
II. COMPUTATIONAL DETAILS
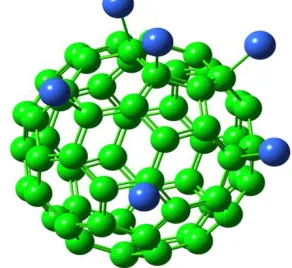
[image:1.612.370.516.372.506.2]The simplest structure of C60Cl6 molecule model has sixty carbon nuclei reside on a sphere and six chloride atoms attached with six carbon atoms of as shown in figure -1. The C60Cl6 structure has C1 symmetry and the bond length of each carbon-carbon is 1.35 Å and the bond length of carbon-chloride atom is 2.08 Å. The two different C-C bond lengths in C indicate that the л electrons are evenly delocalized over all bonds. The green colour and blue colour represent the carbon and chloride atoms respectively.
Figure 1 shows C60Cl6 structure configurations
III. CALCULATION
The frequencies are obtained in a frequency analysis based on the time independent Schrodinger equation for the stochastic explanation of the nuclei within the Born-Oppenheimer approximation. This harmonic approximation
that replaces the potential by
, where are Cartesian
nuclear coordinates and is the Cartesian Hessian matrix. Then the nuclear Schrodinger equation in the basis
of mass-weighted normal coordinates within this approximation is given by
( )
el
E
R
†
( )
c c cel
E
R
R
F
R
R
jc c
F
International Journal of Emerging Technology and Advanced Engineering
Website: www.ijetae.com (ISSN 2250-2459,ISO 9001:2008 Certified Journal, Volume 5, Issue 9, September 2015)
316
(1)
where is the diagonalized Hessian with elements
(2) According to Placzek’s classical theory of polarizability the Raman intensity is defined as the ratio of the radiation
power in a conical beam of solid angle [17]. Within classical electrodynamics the intensity of an oscillating dipole is given by [16]
(3)
Here is the angle between the directions of
propagation of the incident beam with the observation, is the wave number of the incident radiation, and
is the amplitude of the induced dipole moment which oscillates with the same frequency
as the incident radiation. In general the intensity of the radiation source is large, so quantum effects on the radiation field are negligible [18]. Still, for the molecular properties a quantum mechanical treatment is required. This approach is known as a “partial quantum mechanical treatment” of scattering phenomena.
It is compulsory to compute the time-independent
amplitudes of the oscillating dipole moment, which are
obtained as matrix elements of the induced dipole moment
[17]
(4)
(5)
is the polarizability of the molecule and is the amplitude of the incident electromagnetic field. The thesis has taken only the linear response of the molecule-radiation interaction; it means that the effects concerning first and higher hyper-polarizabilities are not considered. In
addition, it is supposed that the electric field vector is constant within the complete molecule. Now the rotational
contributions to the nuclear wave function defined in equation
(6)
This give total nuclear wave function where is a product of the product of the (3n-6) [(3N-5) for linear molecules, respectively] vibrational single mode wave function which will characterize by the rotational quantum numbers J and MJ. This is compulsory because as the matrix element in eq. (5) depends on the orientation of molecule with the radiation beam. But the translational contribution is not regard as it does not affect the matrix element. Considering the product for example
(7)
with defined in eq. (6). Any configuration of
structure is only valid if and ,
where are the frequencies for rotational,
vibrational, and electronic transitions of the respectively molecule, and it amounts to the neglect of coupling matrix elements between vibrational and rotational motion. One more requirement for this treatment is that the electronic ground state of the system is non degenerate.
So is a tensor that has to be concerned with matrix elements of the tensor components of the respective configuration structure.
† †
2
2
q q q q q tot tot
tot
R
F
R
E
q
F
2
(
q)
el q
ij q q
i j
E
R
F
R
R
d
d
4
2 2
0 0, 0
0
sin
2
ind
c
J
I
ssr
0
0,ind 0
0
0
c
0,
.
0ind tot tot
fi f i
0 tot totf i
Cm
00
tot
.
tot rot trans
; ,
.
,
tot
J
J M
J M
0 rot vib
0
elec,
,
rot vib elec
International Journal of Emerging Technology and Advanced Engineering
Website: www.ijetae.com (ISSN 2250-2459,ISO 9001:2008 Certified Journal, Volume 5, Issue 9, September 2015)
317
(8)
Where the integration over the electronic coordinates has
already been calculated out, in such a way that is the electronic expectation value for a given set of nuclear coordinates. The constituents of the polarizability tensor depend upon the selection of the space-fixed coordinate system. They are related to the components in the
respective molecule fixed axes by
(9)
It is interesting that the components of the polarizability in the molecular coordinate system do not change under molecular rotation, just compared to the ones in the space-fixed system. It makes that the integrations separately over vibrational and rotational coordinates and is given by
(10)
Here the matrix elements
only depend upon the vibrational quantum numbers
with the electronic state of the molecule, since in
the case of the transition dipole moment. As is not
known as a precise function of either, but estimation
of the matrix elements require integration over nuclear coordinates, so a Taylor series expansion is necessary [25]. The calculation of the matrix elements for the components of the polarizability tensor can be performed in similar way as for the components of the dipole moment, as shown. In equations (13) to (15) we
made use of the properties of the Hermite polynomials (
and are electronic matrix elements). Taylor series expansion of electronic matrix elements
(11)
This evaluation includes the total matrix elements with nuclear wave functions [neglecting quadratic and higher order terms from equations (11)]
(12)
The evaluation of matrix elements and
is done as follows
(13)
(14)
(15)
Summary of the above equations for fundamental
transitions
(16) 2
,
,
, pq,
,pq fi f f J f f z i i
Cm
J
M
J
J
V
,
, ,
p q
x y z
pq
', '
p q
' ', , , , ' ', cos ' cos ' ,
p q fi
pq fi f J f i J i
p q
J M xx yy J M
' '
' ', p q
p q fi f i
i,
f
' ' p q
qR
, pq fi
0 2 0 q q k k q k kR
O R
R
0 0 qfi f i q f k i
k k
R
R
f i
qf
R
k i
3 6
, , 1
N
f i p f p i
p
, , , 3 61 p f vp i N v p
3 6
, , , ,
1,
N
q q
f k i k f k k i p f p i
p p k
R
R
1 2 , , , 1 2 , , , , ,1
;
1
1
;
1
0;
k i vk k f k i
q
k f k k i k i vk k f k i
b
R
b
otherwise
,
1
,k i k i
1 2,
0
1
fi vk k i q
k
b
R
' ' ' 'cos
' cos
'
pq p q
p q
pp
International Journal of Emerging Technology and Advanced Engineering
Website: www.ijetae.com (ISSN 2250-2459,ISO 9001:2008 Certified Journal, Volume 5, Issue 9, September 2015)
318
(17)
It should be noted that is the product of the individual harmonic oscillator wave functions for all normal modes of the molecule through harmonic approximation in the molecule. Since also the Taylor series
for and are cut after the quadratic terms (electrical harmonicity), the procedure presented here is called the double harmonic approximation.
Equation (15) it gives that non vanishing part for the scattering intensity that occurs for all transitions with
. The case corresponds to
Rayleigh scattering, which means the frequency of the incident radiation remains unchanged.
For Raman scattering, one has to confine to the Stokes
case where , the wave number of the
oscillating induced dipole is no longer. The wave number
of the incident beam projecting on the structure
configuration has to be replaced by where is
the wave number of the vibrational transition for normal mode p.
Let us presume a phonon with a diagonal Raman tensor
with three elements .In addition, the
scattering configuration in the laboratory frame is (ZZ). To find the Raman intensity one integrate as well as average over all possible orientations of the crystal. By Euler's angles [20, 21].
(18)
(19)
Integrating and rearranging give up
(20)
With
(21)
The result in Eq. (19) embraces for all parallel polarizations of the incoming with outgoing linearly polarized light as well as for every Raman tensor excluding
that . For perpendicular linear polarization, e.g., the
integration yields .
Now it turn out to be noticeable that the symmetry can also the partially presumed from experiments on unoriented materials.
The intensity on unoriented substances pursues directly from the transformation of a tensor under rotation. A second-rank tensor can be divided with respect to the rotation group into a scalar (tensor of rank zero), an antisymmetric matrix (rank one), and a symmetric traceless matrix (rank two). These irreducible components have well defined quantum numbers and transformation properties under rotation. The matrix element for a fixed orientation is obtained from the Wigner-Eckart theorem and the integration over all crystal orientations is determined by the tensor invariants. Different researchers use slightly different invariants in Raman scattering. By Neslor and spiror [19] we define the isotropic invariant
(22) The antisymmetric anisotropy
2
2
2
4
8
vk
k k
b
c
tot
0, 1
k
k
0
, ,
1
k f k i
0
in
in p
p1 2 3
a
a
a
2 ,2 ,2
2 0
1
.R .e
sin
8
ZZ i s
I
e
d
d
d
2 1
,2 ,2
2 2 0
3
sin sin
sin sin
1
cos sin
cos sin
sin
8
cos
cos
ZZ
a
I
a
d
d
d
a
2 2
45
4 '
45
s zz
I
1 2 3
1
3
a
a
a
2
2
2 21 2 2 3 3 1
1 '
2
s a a a a a a
2
'
s
I
xz2
3 ' 45
s xz
I
1
International Journal of Emerging Technology and Advanced Engineering
Website: www.ijetae.com (ISSN 2250-2459,ISO 9001:2008 Certified Journal, Volume 5, Issue 9, September 2015)
319
(23)
And the symmetric anisotropy
(24)
[image:5.612.109.507.305.374.2]Where are the elements of the Raman matrix as given in Table-1 for carbon nanotubes.
Table - 1
shows Raman tensors of the carbon structure
A linear combination of the tensor invariants can be expressed in the scattering intensity on an unoriented sample in any scattering configuration. For linear parallel ( ) with perpendicular ( ) polarization of the incoming and scattered light the intensities are given by (apart from a constant factor
(25)
Which is the generalized result of Equation (19). The
quotient is known as the depolarization ratio . Under Placzek’s polarizability approximation, it is known that the depolarization ratio of a totally symmetric vibrational mode is less than 0.75, and that of the other modes equals 0.75. A Raman band whose depolarization ratio is less than 0.75 is called a depolarization band, and a band with a 0.75 depolarization ratio is called a U-depolarization band.
The Raman intensities for any polarization on arbitrarily oriented systems can be implicit through the matrix element for a particular Raman tensor and taking the averaged over Euler's angles. For generality, the result of fullerene Raman tensor of into any two rank tensor.
This transformation of tensor are can be prescribed by irreducible spherical tensors, that is a break up into the
rotation group. The irreducible spherical tensors have quick j and m quantum numbers under turning round that can be simply transform according to
(26)
Where is the matrix expression of the rotation group (rotation matrices). The rank k tensor can reduce into irreducible tensor of ranks 0, 1, 2, 3…k with help of the Clebsch-Gordan coefficients. A normalized Raman tensor
set are converted into irreducible tensors is given by
2
2
22 3
4
as xy yx xz zx yz zy
2
2
22
1
2
s xx yy yy zz zz xx
2 2 2
3
4
xy
yx
xz
zx
yz
zy
,
, ,
ij
i j
x y z
ll
2 2
45
4
ll s
I
I
3
s2
5
as2ll
I
I
j m
T
j j j
, ,
m p pm
p
T
T
D
j
D
0 1 2
R T
T
T
0 0
1
(
)
3
xx yy zzT
1
0
1
2
xy yxT
2
11
2
zx xz zy yzInternational Journal of Emerging Technology and Advanced Engineering
Website: www.ijetae.com (ISSN 2250-2459,ISO 9001:2008 Certified Journal, Volume 5, Issue 9, September 2015)
320
(27)
In a fixed scattering configuration, the intensity matrix
element is computed through the Wigner-Eekart
theorem. An average of irreducible spherical tensors over the randomly oriented graphene oxide molecules is specified by.
(28)
(29)
Where are the
Clebsch-Gordan coefficients with the selection rule M = mi – m. This gives
(30)
The rotation matrices are orthonormal
(31)
So, each of the contribution of different irreducible tensors rank J can solve break up. Thus the orthonormality of the turning round matrices diminishes to
(32)
The sum over p is self-determining of the angular momentum quantum numbers m which formulate to be calculated only once for every J under consideration. As the mixed elements in the squared sum do not necessarily cancel so care should be taken while adding over p by using orthogonality of the rotation matrices in Eq. (19)
For calculation has to sum J = 0, 1,
2 with the irreducible tensors in Equation (18).
Depolarization and P- polarization Raman spectrum
In Raman spectroscopy, depolarization Raman spectrum consists of depolarization ratio. This depolarization ratio is the intensity ratio between the perpendicular component and the parallel component of the Raman scattered light [22, 23]. The electric field of the incident light stimulates the Raman scattered light. Hence the direction of the vibration of the electric field, or polarization direction, of the scattered light might be estimated to be the same direction as that of the incident light. But in real world some part of the Raman scattered light has a polarization direction and some part remain perpendicular to that of the incident light. This component is known as the perpendicular component. The part component of the Raman scattered light whose polarization direction is parallel to that of the incident light is known as the parallel component, so the Raman scattered light consists of the parallel component and the perpendicular component. The ratio between these two components is expressed as the depolarization ratio.
(33)
The value of the depolarization ratio of a Raman band depends upon the symmetry of the molecule and the normal vibrational mode. As already discuss above the Placzek’s polarizability approximation, it is known that the depolarization ratio if of a totally symmetric vibrational mode is less than 0.75, and that of the other modes equals 0.75. Hence the Raman spectrum with greater than 0.75 depolarization ratio is said to be p - depolarization.
IV. RESULT &DISCUSSIONS
C60Cl6 molecule has C1 symmetry and it takes 396 steps to converge the simulated Raman spectrum of C60Cl6 molecule.
1
1
1
2
zx xz zy yzT
2
2
1
2
xx yy xy yxT
i
2
0
1
2
6
zz xx yyT
LS
I
2
2
Re
Je
LS l s l s
J
I
e
e T
d
2
,
J LS i i M S S
J M
I J m T J m d
J J
i S
m m J
i S
M
2
s i
J J
LS i S i S S i p p m m
p
I
J J
m m J m
m
T
D
d
1 2
1 1 2 2 1 2 1 2 1 2
*
1
2
1
j j
k k j j k k
D
D
d
j
21
22
1
J J
LS i S i S S i p
p
I
J J
m m J m
m
T
J
J LS
I
2 J p p
T
ll
I
I
International Journal of Emerging Technology and Advanced Engineering
Website: www.ijetae.com (ISSN 2250-2459,ISO 9001:2008 Certified Journal, Volume 5, Issue 9, September 2015)
321
The most significant change in the Raman spectrum induced by addition of chloride is in fullerene, and the redistribution with new the Raman spectrum intensity among the normal modes of fullerene is as given in the table. Fullerene derivative (C60Cl6) molecule has 192 degrees of freedom which is slightly greater than C60 normal degrees of freedom because of addition of six chlorides in the fullerene to form fullerene derivative. The rotational constants in x, y, and z axis in the fullerenes derivative (C60Cl6) is 0.06, 0.05 and 0.05 (GHz) respectively. The C60Cl6 has 354 symmetry adapted basis functions, 1062 primitive gaussians, 354 cartesian basis functions, 321 alpha electrons and 321 beta electrons with the nuclear repulsion energy 12972.07 Hartrees. The fullerenes derivative (C60Cl6)has done SCF E (RHF) = -4970.33 A.U. after 21 cycles with convergence density matrix= 0.4894D-08.
The main results from the calculation of the Raman spectrum of the C60Cl6 is as follows
1. Each chloride atom of C60Cl6 perturbed in a small fullerene cage. This makes small frequency shifts demonstration in the fullerene derivative spectrum. 2. The reduction of symmetrical shape in the fullerene
derivative split the degenerate vibrations of C60Cl6 quite large. Thissplitting increases vibration which produced by the intermolecular interactions in the crystal. Hence the Raman intensity among the various atoms of C60Cl6 distributes non-uniformly in the Raman Spectrum and some cases the intensity is concentrated to a single component.
3. The ab initio method is used to simulate many the Raman intensities of C60Cl6 which are not observed in experimentally [15]. But most of them are observed available experimental spectrum data at room temperature is matching with simulated Raman data as explain in table II.
4. The four main groups of simulated Raman peaks are seen in Raman spectrum. This Raman spectrum gives coordinates of C60Cl6 facilitated to assign four principal regions of its vibrational spectrum namely the first range from 100 to 130 cm-1, the second range from 140 to 400 cm-1, the third range from 400 to 900 cm-1 and the fourth range 900 to 1600 cm-1. In the first 100–130 cm-1 range, the chlorine-atoms motion are observed in the Raman spectrum of C60Cl6. In the second range 140–400 cm -1carbon and the chlorine atoms equally take part in the vibrations of C60Cl6. Unusually, the intensities of this range of Raman spectrum vibrations are medium to strong, and also are observed in the experimental spectrum in weak features.
In the third range 400-900 cm-1 the vibrations of C60Cl6 are restricted on the carbon skeleton of the molecule without a significant chlorine atoms motion. But the internal coordinates associated with chlorine atoms (C– Cl stretches and bends) do contribute to the vibrations of the C60Cl6 molecule. In general, this range may be characterized as particularly rich in the Raman spectrum with a large number of strong and medium intensity bands but half of intensities are relative lower. Radial displacements of the carbon atoms are more important as result in the changes of C–Cl distances and C-C-Cl angles. In the fourth range 900–1600 cm-1 the vibrations of the C60Cl6 carbon skeleton have considerable tangential character stretching vibrations in the Raman spectrum. The frequencies could not be precisely determined from the experimental spectra in this range due to their low intensities and overlap of the bands. Though the intensities of the C–C bands were quite low, yet the C=C stretches exhibited significant Raman activity with two principal bands at 1462.63 and 1559.32 cm-1 corresponding to the Raman spectrum.
5.It is worth wise that, the intensity of the Raman-active fundamentals of C60 decreases appreciably in the fullerene derivative (C60Cl6). It is found that most of the bands gain assigned as inactive fullerene fundamentals. So it can be taken as evidence that they are indeed fundamentals. In some regions of the simulated spectrum, the overall intensity decreases to an extent that the bands become barely detectable to compare with the available experimental spectrum data.
6.The minimum and maximum simulated infrared of C60Cl6 peak is 122.64 and 1559.32 respectively while experimental available infrared minimum and maximum data are 114.714 and 1572.71 respectively.
V. CONCLUSION
The C60Cl6 structures are very sensitive to electron correlation treatment with basis set that are employed. So the proper electron correlation level and basis set play an important role in calculating accurate structure. It is interesting to note that the C60Cl6 structure of matching the available experimental data very much even it is showing other important peaks which are not seen in the experimental data.
REFERENCES
[1] N. B. Shustova, A. A. Popov, L. N. Sidorov, A. P. Turnbull, E. nitz and S. I. Troyanov, Chem. Commun., 2005, 1411.
International Journal of Emerging Technology and Advanced Engineering
Website: www.ijetae.com (ISSN 2250-2459,ISO 9001:2008 Certified Journal, Volume 5, Issue 9, September 2015)
322
[3] P. A. Troshin, R. N. Lyubovskaya, I. N. Ioffe, N. B. Shustova, E.Kemnitz and S. I. Troyanov, Angew. Chem., Int. Ed. Engl., 2005, 44, 234.
[4] P. R. Birkett, A. G. Avent, A. D. Darwish, H. W. Kroto, R. Taylor and D. R. M. Walton, J. Chem. Soc., Chem. Commun., 1993, 1230. [5] K. I. Priyadarshi, H. Mohan, P. R. Bikett and J. P. Mittal, J. Phys.
Chem.,1996, 100, 501.
[6] A. G. Avent, P. R. Birkett, J. D. Crane, A. D. Darwish, G. J. Langley, H. W. Kroto, R. Taylor and D. R. M. Walton, J. Chem. Soc., Chem.Commun., 1994, 1463.
[7] A. K. Absul-Sada, A. G. Avent, P. R. Birkett, H. W. Kroto, R. Taylor and D. R. M. Walton, J. Chem. Soc., Perkin Trans. 1, 1998, 393.
[8] P. R. Birkett, A. G. Avent, A. D. Darwish, I. Hann, H. W. Kroto, G. J. Langley, J. O’Lounglin, R. Taylor and D. R. M. Walton, J. Chem. Soc., Perkin Trans. 2, 1997, 1121.
[9] N. R. Serebryanaya, V. D. Blank, V. A. Ivdenko, L. A. Chernozatonskii, 2001, "Pressure-induced superhard phase of C60" Solid State Commun. 118, 183.
[10] I. G. Batirev, K. H. Lee, W. R. Lee, H. M. Lee, J. A. Leiro, Chem. Phys. Lett. 1999, 262, 247.
[11] W. Heitler, The Quantum Theory of Radiation; Dover: New York, 1994, p. 136.
[12] D. P. Craig, T. Thirunamachandran, Molecular Quantum Electrodynamics; Dover: New York, 1998, pp. 17, 84, 89.
[13] D. A. Long, Raman Spectroscopy; McGraw-Hill: New York, 1977, pp. 31, 81–82.
[14] W. G. Bickley, Math Gaz 1941, 25, 19.
[15] I. V. Kuvychko, A. V. Streletskii, A. A. Popov, S. G. Kotsiris,T. Drewello, S. H. Strauss, O. V. Boltalina Chem. Eur. J. 2005, 11, 5426 – 5436
[16] D. A. Long, Raman Spectroscopy; McGraw-Hill: New York, 1977, pp. 31, 81–82.
[17] GZ. Placzek, Physik 1931, 70, 84.
[18] L. A. Pugh, K. N. Rao, In Molecular Spectroscopy: Modern Research, Vol. II; Academic Press: New York, 1976, p. 165. [19] J. Nestor and T. G. Spiro, J. Raman Spectroscopy 1,539 (1973). [20] V. Heine, Group Theory in Quantum Mechanics: An Introduction to
its Present Usage (Pergamon, Oxford, 1977).
[21] D. M. Brink and G. R. Satchler, Angular Momentum (Oxford, New York, 1993), 3 edn.
[22] I. G. Batirev,K. H. Lee, J. A. Leiro, Atomic structure and chemical bonding of boro and azafullerene dumb-bells, Chem. Phys. Lett. 2000, 695-699.
[23] D. W. Snoke, M. Cardona, S. Sanguinetti, G. Benedek, Solid State Commun. Phy. Rev. B 87, 121 (1993).
[24] X. Xu, Z. Shang, Li Ruifang , Z. Cai, X. Zhao, J. Phys. Chem. 2002, 106, 9284.
[25] D. A. Long, Raman Spectroscopy; McGraw-Hill: New York, 1977, pp. 31, 81–82.
Table - II
Experi. Data
Simul. Raman Data
Depol. Ratio
Polari. Ratio
Experi. Data
Simul. Raman Data
Depol. Ratio
Polari. Ratio
116 122.64 0.72 0.84 838.69 0.75 0.85
123.6 0.7 0.82 847.9 0.75 0.85
125.2 0.75 0.85 853.94 848.49 0.74 0.85
131.85 0.72 0.84 860.53 0.52 0.68
166 162.98 0.74 0.85 861.72 0.35 0.52
214 867.514 863.66 0.75 0.85
237 245.38 0.75 0.85 871.42 0.75 0.85
247.59 0.68 0.81 878.06 0.29 0.45
270.07 0.75 0.85 880.91 0.44 0.61
279 275.33 0.57 0.73 881.38 0.75 0.85
297.88 0.64 0.78 882 882.76 0.56 0.71
321 894.61 894.01 0.75 0.85
350 345.07 0.03 0.07 896.5 0.61 0.76
358.19 0.03 0.06 906.56 0.29 0.46
384 391.85 0.04 0.08 909.52 0.2 0.34
404.17 0 0.01 910.39 0.75 0.85
435.004 427.32 0.02 0.04 922.91 0.59 0.74
International Journal of Emerging Technology and Advanced Engineering
Website: www.ijetae.com (ISSN 2250-2459,ISO 9001:2008 Certified Journal, Volume 5, Issue 9, September 2015)
323
455.357 455.65 0.27 0.42 925.55 0.59 0.74
496 482.88 0.66 0.79 1062 1041.91 0.02 0.04
501.22 0.03 0.07 1050.43 0.75 0.85
504.51 0.75 0.85 1052.22 0.17 0.29
507.09 0.31 0.48 1065.71 0.02 0.05
515 516.86 0.75 0.85 1103.27 0.03 0.06
541 570.95 0.75 0.85 1108.94 0.02 0.04
581 579.91 0.1 0.18 1111 1109.09 0.75 0.85
597.8 0.06 0.12 1129.93 0.75 0.85
612 611.67 0.22 0.36 1136.91 0.75 0.85
628 627.4 0.01 0.03 1171 1136.98 0.75 0.85
633.86 0.75 0.85 1186.39 1203.01 0.75 0.85
638.591 634.22 0.13 0.23 1204.43 0.6 0.75
672.03 0.21 0.35 1229.42 0.43 0.61
694.94 0.34 0.51 1240 1239.87 0.75 0.85
711.405 718.2 0.09 0.16 1242.37 1260.26 0.11 0.2
755 755.1 0.75 0.85 1255.99 1267.29 0.75 0.85
757.47 0.73 0.84 1448 1451.24 0.75 0.85
786.21 785.42 0.67 0.8 1467 1462.63 0.65 0.79
799 1464.88 1473.24 0.72 0.84
816 1478.04 0.75 0.85
823 832.6 0.32 0.49 1488.7 1489.52 0.32 0.49
828.67 835.46 0.69 0.81 1568 1559.32 0.54 0.7
Corresponding author: