Variation-Tolerant Capture and Multiplex Detection of Nucleic Acids:
Application to Detection of Microbes
Christina O¨ hrmalm,aRonnie Eriksson,a,bMagnus Jobs,a,cMagnus Simonson,bMaria Strømme,dKåre Bondeson,aBjörn Herrmann,e Åsa Melhus,eand Jonas Blomberga
Sections of Clinical Virologya
and Clinical Bacteriology,e
Department of Medical Sciences, Uppsala University Hospital, Uppsala, Sweden; Science Department, National Food Agency, Uppsala, Swedenb
; School of Health and Social Sciences, Högskolan Dalarna, Falun, Swedenc
; and Nanotechnology and Functional Materials, The Ångström Laboratory, Uppsala University, Uppsala, Swedend
In contrast to ordinary PCRs, which have a limited multiplex capacity and often return false-negative results due to target varia-tion or inhibivaria-tion, our new detecvaria-tion strategy, VOCMA (variation-tolerantcapturemultiplexassay), allows variation-tolerant, target-specific capture and detection of many nucleic acids in one test. Here we demonstrate the use of a single-tube, dual-step amplification strategy that overcomes the usual limitations of PCR multiplexing, allowing at least a 22-plex format with retained sensitivity. Variation tolerance was achieved using long primers and probes designed to withstand variation at known sites and a judicious mix of degeneration and universal bases. We tested VOCMA in situations where enrichment from a large sample vol-ume with high sensitivity and multiplexity is important (sepsis; streptococci, enterococci, and staphylococci, several enterobac-teria, candida, and the most important antibiotic resistance genes) and where variation tolerance and high multiplexity is impor-tant (gastroenteritis; astrovirus, adenovirus, rotavirus, norovirus genogroups I and II, and sapovirus, as well as enteroviruses, which are not associated with gastroenteritis). Detection sensitivities of 10 to 1,000 copies per reaction were achieved for many targets. VOCMA is a highly multiplex, variation-tolerant, general purpose nucleic acid detection concept. It is a specific and sen-sitive method for simultaneous detection of nucleic acids from viruses, bacteria, fungi, and protozoa, as well as host nucleic acid, in the same test. It can be run on an ordinary PCR and a Luminex machine and is suitable for both clinical diagnoses and micro-bial surveillance.
I
nfections are caused by diverse microbes. They often give similarsymptoms. Ideally, in a clinical situation, relevant pathogens, both common and uncommon, plus their antibiotic resistance and virulence, should be detected in one test. Variable nucleic acids (like in RNA viruses), large and expanding pathogen fami-lies, and diverse target molecules (antibiotic resistance genes, tox-ins) are important clinical challenges. They call for broadly tar-geted and multiplex DNA and RNA detection methods.
Another challenge is the need to encompass both large sample volumes (10 to 100 ml; for the diagnosis of septicemia or the
surveillance of food-borne pathogens [11,18]) and small sample
volumes (50 to 200l; for the diagnosis of many causes of
menin-goencephalitis in a small volume of cerebrospinal fluid). Sensitiv-ities down to a few target molecules per reaction and turnaround times of a few hours are often important. PCRs cannot normally be highly multiplex, due to mispriming. However, with conserved targets (16S rRNA and the second internal transcribed spacer [ITS2]), many different pathogens can be detected with one primer pair. Padlock probes are an alternative to PCR. They can be highly multiplex and can be combined with magnetic
nanopar-ticles (17) in a commercial portable read-out device (24),
al-though the short shanks make them especially vulnerable to pathogen variation. There is room for a new principle for the multiplex detection of nucleic acids.
Here we present a new multiplex diagnostic technique encom-passing mismatch tolerance, optional capture, and specific, as well
as generic, amplification steps. The variation-tolerant capture
multiplexassay (VOCMA) (Fig. 1; see also Table S1 in the
supple-mental material) combines several features in a novel way: (i) a combination of capture probe and primer (i.e., primer-probes), (ii) variation-tolerant primer-probes, (iii) a single-tube, dual-step
combination of target-specific preamplification at low primer concentrations and higher annealing temperature, followed by general amplification using generic primers at higher concentra-tions and lower annealing temperature, (iv) asymmetric amplifi-cation for the buildup of excess biotinylated strands, (v) variation-tolerant (VT) detection probe hybridization to target specific sequences, providing a large potential for multiplexing, as well as high sensitivity and specificity, and (vi) a simple and specific read-out. Variation tolerance is achieved by using long target-specific primers and probes containing degenerations and the universal base analog deoxyribose-inosine (dInosine) according to varia-tion analysis and principles elaborated in a previous publicavaria-tion
(the NucZip algorithm) (15).
Here we present the basic conditions for variation-tolerant capture, amplification, and detection, applied to two clinically im-portant situations, diagnosis of sepsis and gastroenteritis (gastro).
MATERIALS AND METHODS
Design of VT primer-probes and detection probes.The specific VT primer-probes and the specific VT detection probes (sequences and
mod-Received15 November 2011Returned for modification30 January 2012
Accepted11 July 2012
Published ahead of print18 July 2012
Address correspondence to Jonas Blomberg, [email protected].
Supplemental material for this article may be found athttp://jcm.asm.org/. Copyright © 2012, American Society for Microbiology. All Rights Reserved.
doi:10.1128/JCM.06382-11
The authors have paid a fee to allow immediate free access to this article.
on May 16, 2020 by guest
http://jcm.asm.org/
ifications in Tables S2 to S5 in the supplemental material) were designed using BLASTN (NCBI, NIH) (1), ClustalX (8,19), ConSort (J. Blomberg, described in the supplemental material), Mfold (25), VisualOmp (DNA Software, Ann Arbor, Michigan), and NucZip (15). Target sequences or whole genomes collected in BLASTN were further aligned to map nucle-otide conservation and variation with ConSort, which visualizes the fre-quency of variation and the variation of nucleotide composition in each base position, as well as the number of aligned sequences. The design was evaluatedin silicobefore thein vitroanalysis by adjusting and redesigning to avoid hairpin, homodimer, and heterodimer formation, while retain-ing target specificity. The predicted Gibb’s free energy (VisualOmp) and NucZip scores were used as tools to predict hybridization efficiency and to optimize variation tolerance. NucZip simulates nucleation, the initial contact between segments of a perfect match, followed by “zipping” the two strands upstream and downstream (15). The panel was thus built up stepwise, in a recursive loop involving reevaluation and redesign. Varia-tion tolerance was enhanced by using 50 to 70 nucleotide (nt) primer-probes and primer-probes, degenerations and dInosines at sites of target varia-tion, and locked nucleic acid (LNA) residues to increase affinity.
Synthetic targets.The synthetic targets (Biomers.net GmbH, Ulm, Germany) had a natural primer-probe and detection probe complemen-tary sequence derived from GenBank (see Tables S2 and S4 in the supple-mental material). The synthetic target for norovirus genogroup II was artificially designed to perfectly match the primer and probe sequences. Parts of the original sequences of the pathogens were deleted, because of constraints on synthetic oligonucleotide length (138 to 200 nt). Synthetic single-stranded DNA (ssDNA) targets were serially diluted 10-fold from 0.5⫻107to 0.5⫻100molecules/l either with 0.1⫻Denhardt=s solution
(Sigma-Aldrich Sweden AB, Stockholm, Sweden) in the sepsis VOCMA or with 20 ng/l yeast RNA in diethyl pyrocarbonate (DEPC) water (Ambion, Austin, TX) in the gastro VOCMA.
Nucleic acid extraction from clinical samples.The patient samples were provided and routinely handled, anonymously, according to Upp-sala University Hospital rules. The EasyMag extraction system (bio-Mérieux AB, Sweden) was used for fecal samples, reference bacteria, and bacteria and fungi from agar plates. The QIAamp DNA blood maxikit (Qiagen GmbH, Hilden, Germany) was used to extract DNA from patient samples (blood culture flasks) that had signaled positivity in the BacT/ ALERT blood culture system (bioMérieux). The DNA was extracted from 10 ml of blood and eluted in a total of 2 ml of elution (AE) buffer.
Capture hybridization.Synthetic ssDNA targets, 105copies, were
di-luted in different volumes of buffer ranging from 10l to 1,000l. A sample of 1l was drawn directly to VOCMA PCR, omitting the capture procedure. Capture was then performed on the remaining volumes by adding 105MyOne magnetic beads (coupled with the first specific VT
primer-probe, as described in the supplemental material) perl with shaking at 600 rpm at 50°C for 30 min in 1⫻PCR Gold buffer (AmpliTaq Gold, Roche, Branchburg, NJ). After hybridization, the samples were rinsed twice in PCR buffer by fixing the magnetic beads with magnets (MagnaBot 96 magnetic separation device; Promega, Madison, WI). After the final rinse, the beads, with the captured targets, were resuspended in the PCR mix. PCR was performed with the beads present in the reaction. TMAC (3 M tetramethylammonium chloride, 0.1% Sarkosyl, 50 mM Tris-HCl [pH 8.0], 4 mM EDTA [pH 8.0]; Sigma) was used as a PCR inhibitor to a final concentration of 2.7 M in samples containing 0.2⫻106
synthetic target molecules perl. Five microliters of the sample contain-ing TMAC went directly to VOCMA PCR while 5l went through the capture procedure before going to VOCMA PCR (as described above).
Amplification with the 7-plex gastro VOCMA.Two microliters of 0.5⫻105copies/l of synthetic ssDNA target, or 2l of extracted nucleic
acid was added directly to a 23-l one-step reverse transcriptase PCR (RT-PCR) master mix or to 5.0⫻105Dynabeads MyOne microspheres
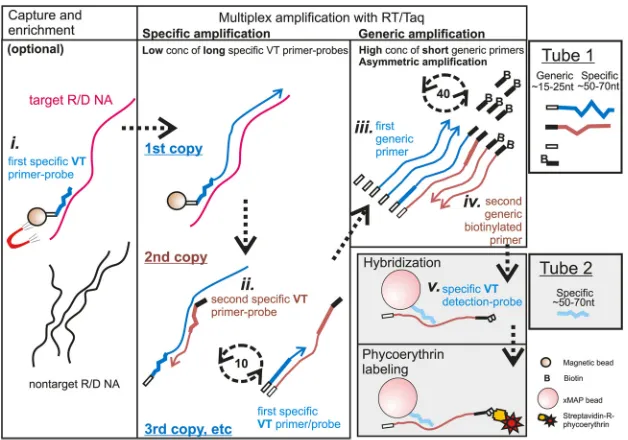
FIG 1Schematic summary of VOCMA principle. Optional capture is followed by amplification (in tube 1) and a liquid microarray detection step (in tube 2). The first specific VT primer-probe, bound to a magnetic bead (small brown circle), is used to capture the target. Beads are fixed by a magnet (red), and the sample is washed to remove unbound nucleic acid. A one-step PCR mixture containing a low concentration of the long first and second specific VT primer-probes and a high concentration of short generic primers is added. Subsequently, a high annealing temperature of 10 cycles allows the specific VT part of the magnetic bead-bound first specific VT primer-probe to elongate the first copy, which is then copied by the specific VT part of the second specific VT primer-probe. The following 40 to 50 cycles are run at a low annealing temperature, allowing asymmetric amplification by using a high concentration of short generic primers with an excess of the second biotinylated generic primer, generating biotinylated ssDNA. An aliquot is transferred to tube 2 where a Luminex bead-bound (large pink circle) specific VT detection probe hybridizes to the amplified target region. Streptavidin-R-phycoerythrin is added to the reaction to allow streptavidin (orange color) to bind to the biotin. The lasers from the Luminex 200 machine will identify the bead and measure the amount of the fluorescence reporter phycoerythrin (red star). The first specific VT primer-probe, the first generic primer, and the specific VT detection probe (all indicated in blue color) are antisense versus the first strand of the target sequence, whereas the second specific VT primer-probe and the second generic biotinylated primer are sense versus the target (both indicated in brown color).
on May 16, 2020 by guest
http://jcm.asm.org/
[image:2.585.136.450.66.286.2]coupled with the first specific VT primer-probe to undergo capture hy-bridization (see above). The one-step RT-PCR master mix contained a final concentration of 1⫻iScript buffer (Bio-Rad, Hercules, CA), 0.5l iScript reverse transcriptase, 300 nM generic first primer, 500 nM biotiny-lated generic second primer, and 50 nM each of the 14 first and second specific VT primer-probes to create a 7-plex gastro VOCMA mixture (see Table S3 in the supplemental material). Amplification was carried out on an MJ Research PTC-100 Peltier thermal cycler (sold by Scandinavian Diagnostic Services, Falkenberg, Sweden) as follows: 50°C for 20 min, 95°C for 5 min, followed by 10 cycles of 95°C for 15 s, ramping at 0.1°C/s from 75°C to 65°C, 65°C for 1 min, followed by 40 cycles of 95°C for 15 s, 52°C for 30 s, 60°C for 30 s, followed by 60°C for 5 min, 95°C for 1 min, and 4°C until the next step. A sample of 20 ng/l yeast tRNA (Sigma) was added as a no-template control (NTC) in the RT-PCR.
Amplification with the 22-plex sepsis VOCMA.Twol of 0.5⫻105
copies/l of synthetic ssDNA target or 2l of extracted nucleic acid from positive blood cultures, or other cultures or colonies of bacteria or fungi, was added to a 23-l master mix containing a final concentration of 2.5 mM MgCl2, 0.8 mM deoxynucleoside triphosphate (dNTP), 2.5l 10⫻
AmpliTaq Gold buffer, 0.2l (5 U/l) of AmpliTaq Gold enzyme (Am-pliTaq Gold, Roche, Branchburg, NJ), 300 nM generic first primer (16 nt long), 500 nM biotinylated generic second primer (16 nt long), and 5 nM each of the 42 first and second specific VT primer-probes (see Table S2 in the supplemental material). The fungiCandida albicansandC. dublinien-sisused the same VT primer-probes but different specific VT detection probes. Amplification was carried out as follows: 95°C for 9 min, followed by 10 cycles of 94°C for 30 s, 75°C for 1 s, ramping at 0.1°C/s to 62°C, 62°C for 1 min 30 s, 72°C for 30 s, followed by 55 cycles of 94°C for 20 s, 53°C for 30 s, 72°C for 30 s, followed by 72°C for 7 min, 95°C for 2 min, and 4°C until the next step. A sample of 0.1⫻Denhardt’s solution was added as an NTC in the PCR.
Hybridization of biotinylated target DNA to detection probe-linked xMAP beads.Five microliters of biotin-labeled VOCMA amplified target was mixed with hybridization buffer comprising 33l 4.5 M TMAC buf-fer, 12l 1⫻Tris-EDTA (TE) buffer (pH 8.0), and 0.05l (⬃2,500 microspheres) of each probe-coupled xMAP bead. The coupling proce-dure is detailed in the supplemental material. The gastro VOCMA had seven unique beads, each coupled with one of the 7 specific VT detection probes (46 to 68 nt long), and the sepsis VOCMA had 22 unique beads, each coupled with one of the 22 specific VT detection probes (50 nt long). The mixture was heated at 95°C for 2 min, followed by hybridization at 50°C for 30 min with shaking at 600 rpm on a Thermostar (BMG LabTech, Offenburg, Germany) microplate incubator. After centrifuga-tion (5 s, Galaxy minicentrifuge; VWR Internacentrifuga-tional), 40l was gently aspirated and a mixture of 38l 3 M TMAC-TE hybridization buffer with 2l (0.05 mg/ml) of streptavidin-R-phycoerythrin (Qiagen, Hilden, Ger-many) was added. The tubes were further incubated at 50°C for 15 min before analysis for internal bead and R-phycoerythrin reporter fluores-cence on the Luminex 200 flow meter (Luminex Corporation, Austin, TX). The quantity of the biotinylated target that hybridized to probe-linked beads was measured as median fluorescence intensity (MFI). The minimum number of beads analyzed per type of bead was set to 100.
Sequencing of VOCMA amplimers.The VOCMA PCR products of rotavirus and the norovirus-positive samples were purified using the QIAquick PCR purification kit (Qiagen GmbH, Hilden, Germany). The BigDye terminator v3.1 cycle sequencing kit (Applied Biosystems) was used for sequencing 2l of purified PCR product, 3.5l of 5⫻BigDye buffer, 0.32l of either forward or reverse primer (10 pmol/l), 1l of BigDye mix, and 18l of H2O. The sequencing PCR program was 95°C for 90 s, followed by 35 cycles of 96°C for 10 s, 50°C for 5 s, 60°C for 2 min, and 4°C indefinitely. The sequence readout was made in an Applied Bio-systems 3730 XL DNA analyzer machine. Sequencing primers were the generic first and second primers from the gastro VOCMA or sequencing primers for norovirus genogroup II (see Table S3 in the supplemental material).
RESULTS
To illustrate the concepts behind VOCMA (detailed in Table S1 in the supplemental material), two panels were developed: (i) a 22-plex sepsis VOCMA to demonstrate variation tolerance by cover-ing groups of resistance genes (e.g., CTX-M group I) or targetcover-ing
genes that are normally used for genotyping (e.g.,spagene
encod-ing protein A inStaphylococcus aureus), as well as comprising a
high degree of multiplexity, and (ii) a 7-plex gastro VOCMA to demonstrate that the method can detect RNA viruses, i.e., variable targets.
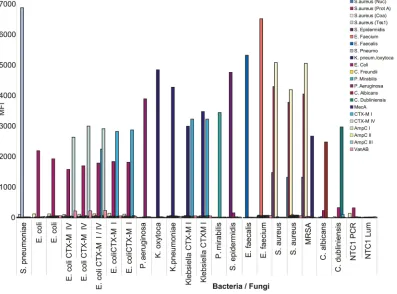
The multiplexity, specificity, and sensitivity of the sepsis VOCMA panel. A 22-plex sepsis VOCMA panel was created against bacteria and fungi known to be a common cause of sepsis (see Table S2 in the supplemental material). Reduction of the concentration to 10 nM or less of each specific VT primer-probes resulted in a reduction of primer dimers and increased specific signals (see Fig. S1A and B in the supplemental material). All de-tection probes showed strong specificity, with no or minimal primer dimer formation, using synthetic targets (see Fig. S2A and B). Two unexpected signals were occasionally seen, possibly due to contamination, but otherwise the specific target and probe com-bination always showed the highest signal. Nucleic acid from dif-ferent bacterial cultures from clinical samples and reference strains was extracted and analyzed. The 22-plex VOCMA has
three target genes for detectingS. aureus. Methicillin-resistantS.
aureus(MRSA) harbors the MecA gene, leading to simultaneous VOCMA amplification of four genes and therefore four median
fluorescent intensity (MFI) signals from the specific probes (Fig.
2). Further, differentEscherichia coliandKlebsiellastrains
harbor-ing antibiotic resistance genes belongharbor-ing to CTX-M groups I or IV
(14) were accurately detected. Conclusively, the 22-plex VOCMA
test showed high specificity (Fig. 2).
The analytic sensitivity was tested for all 22 synthetic ssDNA
targets in a 10-fold dilution series with 107
to 100molecules per
each reaction and resulted in 101to 103synthetic targets per
reac-tion (19 out of 22 targets) (see Table S6 in the supplemental ma-terial). It is noteworthy that the bead-bound probe displayed highly specific signals (see Fig. S3A and B in the supplemental material) even when the amplimers were very weakly detected in the ethidium bromide (EtBr)-stained agarose gel (see Fig. S3C in the supplemental material). During the expansion of the panel (from 5 sets to 10 sets and 22 sets of primer-probes and detection probes), there were no signs of a decrease in analytic sensitivity (data not shown). Higher multiplexities have not been tested with VOCMA, but it is likely that the limit has not yet been reached.
Blood culture-positive samples were tested and showed that
the MFI signal reached a plateau using 2l of nucleic acid extract,
while using 5l tended to decrease the signal more than increase
it (Fig. 3). All probes had a reproducible signal maximum, which differed among the specific probes. A small test of the clinical sensitivity, with a total of 11 different blood culture-positive sam-ples and one negative blood control, showed that the negative control became negative and seven of the positive samples were detected correctly (two were false negative). As mentioned previ-ously, two samples occasionally gave a false-positive signal for bothEnterococcus faeciumandE. coli, probably due to contamina-tion (data not shown).
Fidelity of detection of unequal copy numbers of simultane-ously present targets.The VOCMA system was tested for its
abil-O¨ hrmalm et al.
on May 16, 2020 by guest
http://jcm.asm.org/
ity to detect the presence of targets of different concentrations (see Fig. S4 in the supplemental material). Synthetic targets of the pro-tein A (Prot A) and coagulase (Coa) genes were coamplified with equimolar or 100-fold difference ratios. The equal ratio resulted in
approximately equal detection signals, representing the situation when several genes with the same copy number (from the same
chromosome or genome) are amplified. Interestingly, the 100⫻
difference in ratio still allowed detection of the target, with a lower
FIG 2Specificity of sepsis VOCMA against cultured bacteria and fungi. A 22-plex VOCMA experiment on cultured strains of bacteria and fungi, with or without antibiotic resistance, from reference strains or strains from patient samples. The panel of 22 probes is indicated by different colors. Thexaxis shows the cultures with different colors indicating different detection probes hybridizing. Theyaxis shows the MFI.
FIG 3Sepsis VOCMA with blood culture-positive samples. A 22-plex VOCMA experiment on blood culture-positive samples from patients with suspected sepsis. Theyaxis shows the MFI, and thexaxis shows the samples as duplicates with different amounts of purified nucleic acid added to the VOCMA PCR (2l and 5l of extract, respectively). The panel of 22 detection probes is indicated by the different colors in the figure.
on May 16, 2020 by guest
http://jcm.asm.org/
[image:4.585.96.493.69.363.2] [image:4.585.138.455.482.693.2]signal for the target present in the smaller amount, roughly in proportion to the target copy number in the reaction.
The multiplexity, specificity, and sensitivity of the gastro VOCMA panel.The 7-plex gastro VOCMA is designed to detect the most common pathogens causing viral gastroenteritis, includ-ing highly variable RNA viruses. Although the method detects most of the intended targets in clinical samples, it would benefit from further optimization and inclusion of an internal control. The MFI signals during optimization of the primer concentration of the first generic primer (200 to 500 nM) and the second generic biotinylated primer (300 to 600 nM) did not vary significantly, while higher concentrations than 50 nM each of the long specific VT primer-probes caused primer dimers and thereby lesser sensi-tivity (data not shown).
The specificity of the gastro VOCMA using synthetic targets was high, with no cross-hybridization (see Fig. S5 in the supple-mental material). Furthermore, the analytic sensitivity was tested
in a 10-fold dilution series of 105to 100copies of the synthetic
targets and resulted in positivity in dilutions with 100to 101
cop-ies/reaction for rotavirus and adenovirus, 102copies/reaction for
astrovirus and poliovirus, 102to 103copies/reaction for norovirus
genogroup II, and 104copies/reaction for norovirus genogroup I
and sapovirus (data not shown), which is comparable with the analytic sensitivity of the 22-plex sepsis VOCMA. The signal of
MFI reached a plateau atⱖ105copies/reaction (see Fig. S6 in the
supplemental material).
To better understand the clinical sensitivity, 64 successively collected samples from patients with gastroenteritis (February 2012) were tested (see Table S7 in the supplemental material). The RT-PCR for norovirus genogroup I and genogroup II revealed that 26 samples were positive for norovirus genogroup II. The VOCMA detected 12 of the 15 samples of which contained
⬎20,000 copies per ml, while the 11 samples with⬍11,000 copies
per ml were not detected, correlating with the analytic sensitivity for genogroup II (see Fig. S6 in the supplemental material). How-ever, astrovirus (MFI 539, confirmed by sequencing), rotavirus (MFI 667, confirmed by sequencing), adenovirus (MFI 385), and norovirus genogroup I (MFI 139) were detected in four RT-PCR norovirus genogroup II-negative samples, and a coinfection of genogroup I (MFI 264) and genogroup II (MFI 421) was detected in one sample.
Detection of rotavirus in clinical samples shows the advan-tages of multiplex testing and tolerance for variation.Clinical fecal samples from patients with gastroenteritis, previously tested negative with a specific norovirus genogroup I and genogroup II
quantitative RT-PCR (qRT-PCR) (4), were analyzed with the
gas-tro VOCMA (see Fig. S7 in the supplemental material). Of the 94 samples, 14 previously unrecognized rotavirus-positive samples (MFI signals 825 to 4477) were found and confirmed by sequenc-ing. A BLASTN search showed a high similarity to human rotavi-rus A. Two norovirotavi-rus-positive samples were also detected, but for unknown reasons, sequencing was unsuccessful. The 14-nt over-lap of the rotavirus detection probe and first specific VT primer-probe did not create problems, because they were oriented in the same sense. If any remaining free first specific VT primer-probe would bind the biotinylated target the detection probe would still have 53 nt of its total length of 67 nt to hybridize to the target.
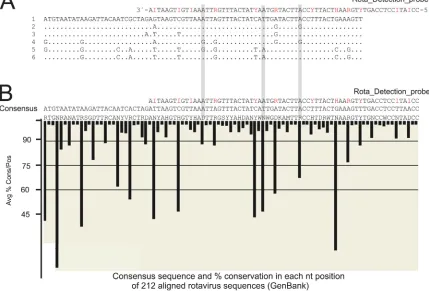
The sequences of the rotavirus-positive VOCMA amplicons resulted in six different sequences having 15-nt variant positions, of which 10 positions were within the target region of the
detec-tion probe (Fig. 4A). Importantly, the pattern of high variation
seen in the 212 aligned sequences from GenBank (Fig. 4B) and the
positions of the variations within the sequenced samples (Fig. 4A)
matched with the 5 dInosines and 7 degenerations in the detection probe, leaving at most only two uncovered mismatches. The vari-ation-tolerant design obviously sufficed for the detection of rota-viruses.
Detection of norovirus in clinical samples shows tolerance for variation.Different variants of a virus can circulate in a com-munity at the same time, and the virus is also changing over time. Twenty-one norovirus genogroup II samples, known to be
posi-tive in RT-PCR (4), were selected from a 2-year period (January
2009 to January 2011). The VOCMA could detect all but one of the norovirus PCR-positive samples (see Fig. S8 in the supplemental material). Sequencing showed that the probe covered the varia-tions in all the eight different variants of the 13 successfully se-quenced samples. The adenovirus coinfection detected in one of the samples was confirmed by adenovirus RT-PCR (threshold
cy-cle [CT], 34.0; 271 copies/ml). The tolerance for variation was also
demonstrated with the ability to detect different cultured entero-viruses (see Fig. S9 in the supplemental material).
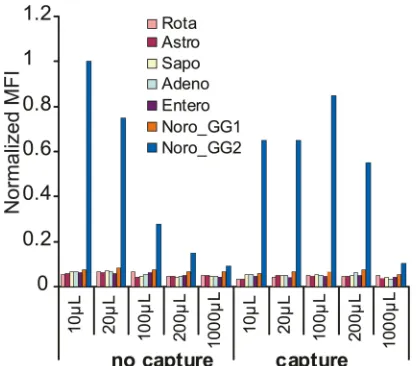
Capture.There are two aspects to the concept of “capture” in VOCMA. The first aspect is the enrichment of specific nucleic acid
targets from a large volume. InFig. 5, the trend of decreasing MFIs
with increasing sample volumes from samples not subjected to capture is clear (the “no capture” series), while the samples that were subjected to the capture procedure showed higher MFIs over the volume range. The signal with capture was half as strong as
that from the noncapture 10-l solution at a 20-fold higher
dilu-tion (200l). The second aspect is the rinsing of the nucleic acid of
interest from PCR inhibitors (demonstrated with TMAC as inhib-itor in Fig. S10 in the supplemental material) and from excess nontarget nucleic acid. A clean sample resulted in a clear MFI signal, while a TMAC-containing sample failed altogether. How-ever, subjecting the TMAC-containing sample to the capture pro-cedure before PCR rescued the sample from inhibition of the PCR.
DISCUSSION
VOCMA combines several features that are sought after in a diag-nostic tool: it is relatively sensitive, specific, tolerant to variations in the target, and multiplex, and it includes removal of inhibitors and enrichment of the target from a relatively large extract vol-ume. Moreover, it is relatively fast (PCR, 3 h; hybridization, 45 min; Luminex, 15 min, excluding hands-on time).
Multiplex testing is gradually becoming important in clinical
microbiology. Microarrays (21) and massively parallel (“deep”)
sequencing (e.g., see reference13) have great potential but still
have limited sensitivity and are time consuming. Multiplex PCR
(6,7,20) and multiplex padlock probes with rolling circle
ampli-fication (24) are close to routine diagnostic use. The Templex
multiplex PCR technique (3) has similarities with VOCMA but
handles specific and generic primers differently.
Probes in a suspension array have faster hybridization kinetics
(10) than probes on a surface. A prevalent form of suspension
array is based on probes bound to Luminex xMAP color-coded
beads (e.g., see references9,10, and12). Unlike real-time PCRs,
which are performed in closed tubes, a Luminex-based readout requires the opening of tubes after amplification, creating a con-tamination risk. Although we did not encounter significant am-plimer contamination during the development of VOCMA, this
O¨ hrmalm et al.
on May 16, 2020 by guest
http://jcm.asm.org/
step should be performed in a hood in a post-PCR environment, separated from the pre-PCR facility. An advantage of VOCMA
over the Luminex xTAG system (12) is that it uses a bead-bound
sequence-specific probe, which considerably diminishes the ten-dency for false-positive signals. VOCMA uses five hybridization events, of which three are target-specific, leading to a high overall
specificity (15). Moreover, unlike the xTAG system, VOCMA
am-plifies and labels in the same tube, without having to open it. It uses a change in the annealing temperature to activate the short biotinylated second generic primer.
Combining a highly multiplex PCR with Luminex technology allows for a multiplex system where the PCR design skill sets the limit for its performance. Standard PCR involves one target se-quence and two primer sese-quences, all in solution. If the
concen-tration of each primer is high (⬎200 nM), and/or there are many
different primers, the likelihood of unwanted interactions in-creases. In VOCMA, dividing amplification into two steps (target specific and generic) allows for the concentration of free target-specific VT primer-probes to be decreased to as low as 1 nM (see Fig. S1A and B in the supplemental material), simultaneous high multiplexity and specificity, and comparatively high sensitivity (see Table S1 in the supplemental material). The strategy of using a low concentration of the specific primers contributes to the spec-ificity, since the production of primer dimers from the long and numerous specific primers is suppressed (see Table S1). The tem-perature switch allows all primers to be added as one mix in the
tube, which minimizes the risk of contamination (see Table S1)
(2). As presented here, the concentration of each specific primer
was 5 nM in the 22-plex sepsis VOCMA experiment and 50 nM in the 7-plex gastro VOCMA experiment. To achieve an excess of a single-stranded biotinylated product, the biotinylated generic primer was in excess and the amplimers were denatured before
hybridization (see Table S1) (23).
The feature of using three different target-specific oligonucle-otides in VOCMA offers freedom of design. A VOCMA panel can include components with different degrees of variation tolerance and different combinations and numbers of specific primer-probes and detection primer-probes targeting highly conserved and more variable targets. This was partially demonstrated in our sepsis panel, which covered both fungal detection using the intervening transcribed sequences and bacterial genomic and antibiotic resis-tance gene detection components.
In a complex multiplex assay, it can be difficult to control all possible sources of false negativity. By using the same oligonucle-otide for capture and primary priming (the first specific VT
primer-probe) (Fig. 1), an additional diversity-constricting step is
avoided. Target nucleic acid will be greatly enriched versus non-target nucleic acid, and the first two PCR rounds will be bead bound, which will decrease mispriming (see Table S1 in the sup-plemental material). The MyOne beads were small enough to stay in suspension during the reactions but could still be collected by a magnetic separator.
FIG 4Variation tolerance of the rotavirus probe. Amplicons from the rotavirus-positive samples (see Fig. S8 in the supplemental material) were sequenced. (A) The resulting six sequence variants of 13 successfully sequenced clinical samples were aligned in ClustalX 1.83. Red letters in the rota detection probe sequence show the positions of nucleotide degeneration and dInosine. (B) A total of 212 sequences reported to GenBank were aligned utilizing the program ConSort (see the supplemental material). Black bars indicate the average percentage of conservation in each nucleotide position in the GenBank sequences. Gray bars indicate
the nucleotide positions that caused mismatching with the probe.
on May 16, 2020 by guest
http://jcm.asm.org/
[image:6.585.77.506.76.367.2]The demonstrated capture and enrichment process illustrated that, judging from MFI values (which are not linearly related to the amount of target nucleic acid), 50% of target nucleic acid was recovered from extract volumes that increased up to 20-fold. It is likely that capture from even larger volumes could be achieved by increasing the concentration of the magnetic beads carrying the first specific VT primer-probe. Capture should be especially useful for detection of bacteria in whole blood, of low abundance proto-zoa and virulence factor genes in feces, and of food-borne patho-gens, where sample volumes of over 10 ml may have to be used, giving large extract volumes. The nucleic acid concentration can be prohibitively high in such extracts. Others showed that a My-One magnetic bead-bound 70-nt long capture probe improves the limit of detection (LOD) 10-fold and 100-fold compared to using
a 50-nt or 25-nt long capture probe, respectively (16),
corroborat-ing our technique. We also show that long LNA-containcorroborat-ing primer-probes are especially suitable for capture.
Target variation, especially in RNA viruses, is a major problem in molecular diagnostics. The recently published NucZip
algo-rithm for long probe hybridization (15), together with the use of
the computer program ConSort for mapping the pattern and fre-quency of variation, was instrumental in the design of VT primer-probes and detection primer-probes. The greatest conservation is placed
at the 3=end for the primer-probes, and both the primer-probes
and the detection probes are typically 50 to 70 nt long to provide a high affinity to the target and tolerance to mismatches to the target sequence. The first hybridization event of the specific part of the primer-probe and the pathogen target will often include mis-matching nucleotides in several positions, but after the third and fourth PCR cycles, many primer-probes, including the generic part of the primer, will fit the amplimers perfectly. One of the basic rules for VOCMA is that a primer-probe and detection probe should have at least one stretch of nine perfectly matching nucle-otides or at least two stretches of five perfectly matching
nucleo-tides (15). Variable positions can be covered by a universal base
analog (UBA), e.g., dInosine, and, to a certain extent, the natural base guanosine. However, UBAs tend to decrease the affinity and cannot be used too generously (they should generally be kept be-low 30% of the length). The choice of target region is dependent on the frequency of variation and, more importantly, on the pat-tern of variation, since longer segments of perfect matches are preferred and dInosines are especially sensitive to neighboring
mismatches (15). Degenerated (redundant) nucleotide positions
can also increase variation tolerance and are included in recent
versions of NucZip (A. Danielsson, C. O¨ hrmalm, and J. Blomberg,
unpublished data). If the nucleotide positions are neighbored by invariant stretches of at least 3 nucleotides on both sides, there is hybridization cooperativity between degenerate probe positions
(15). Thus, the primer-probe and detection probe design
algo-rithm must balance the content of UBAs, guanosine, degenera-tions, and variable positions against each other to obtain both maximal mismatch tolerance and high affinity and specificity to the target.
Three molar TMAC hybridization buffer increases the melting
temperature (Tm) by increasing the binding strength of A·T base
pairs to approximately that of G·C base pairs (5,22). In our sepsis
VOCMA panel, all the probes were the same length (50 nt), which means that they will bind with comparable efficiency to a perfect matching target at the same hybridization temperature. The probes in the gastro VOCMA were variable in length (48 to 68 nt) because of the scarcity of conserved target stretches.
The generic first and second primers have artificial sequences which do not have homologous or heterologous complementarity to the other primers and probes. They are only utilized in solution, and their function is to finalize, and to take the brunt of, the
amplification. Amplification factors of 105 to 108 are typically
achieved with these primers.
At first sight, it appears that the generic primer pair could also amplify falsely. The very high amplification factors and their ge-neric nature could be suspected to cause nonspecificity. However, each target-specific primer-probe and each generic primer must act in succession in an interlocked way to start the second most productive phase of amplification, provided by the generic
prim-ers (Fig. 1). The specificity is further enhanced by the placement of
the label on the second generic primer. The sequence of the
ge-neric primers was testedin silicoto avoid hybridization from the
human genome and the primers and probes included in the panel. Unspecific amplification from the primer-probes or from the ge-neric biotinylated primer cannot yield unspecific reactions since target-specific detection probes are used in the detection step. However, it will reduce the efficiency of the PCR and thus reduce its sensitivity.
The limits of detection of the 22-plex sepsis VOCMA panel were 10 to 1,000 copies per reaction for 19 of the targets. The differentiation of artificially contaminating staphylococci in sepsis
cultures (coagulase-negative bacteria likeStaphylococcus
epider-midis) from more likely pathogens such asS. aureusrelies on the
detection of thenucgene. In this panel, two more genes, the
co-agulase and protein A genes, were included for the detection ofS.
aureus. The MecA gene is important for the diagnosis of MRSA. A
toxin gene,tss1, was also included to allow rapid identification of
highly pathogenic toxin-producingS. aureusstrains.
The antibiotic resistance genes from CTX-M groups I and IV and the AmpC genes were also included. To cover the variation in AmpC genes, the primers and probes were divided into three sets:
FIG 5Capture in increasing volumes. Copies (105) of synthetic norovirus
target were diluted in 10l to 1,000l. One microliter was drawn from each dilution and processed according to the VOCMA protocol without the capture procedure (no capture series). The remaining sample volumes were processed to include the capture procedure (capture series). The presented data are the means of two separate runs.
O¨ hrmalm et al.
on May 16, 2020 by guest
http://jcm.asm.org/
[image:7.585.60.268.66.249.2]AmpC I, AmpC II, and AmpC III. The CTX-M primers and probes were made to be specific for either CTX-M group I or group IV while still remaining tolerant to variations within the group. All components of the sepsis panel were proven to have enough sensitivity and specificity for rapid identification of sepsis bacteria after overnight culture of patient samples.
The limits of detection of the 7-plex viral gastro VOCMA panel were around 100 to 1,000 copies per reaction. Specificity was proven by detection of the synthetic targets and viruses in clinical samples. Although the norovirus-specific primer-probes and tection probe employed here cannot be considered a “final” de-sign, the system did find noroviruses in samples which had been negative in a conventional real-time PCR. The clearest demon-stration of the usefulness of multiplex testing of gastroenteritis was the frequent detection of rotaviruses in samples submitted for norovirus testing and the detection of astrovirus, rotavirus, ade-novirus, and norovirus genogroup I in the small clinical sensitivity study. Thus, the gastro VOCMA panel demonstrated the utility of both variation tolerance and multiplex testing.
ACKNOWLEDGMENTS
We thank Amal Elfaitouri, Clinical Virology, Uppsala University, for pro-viding cell medium supernatant from enterovirus-infected cells, from which the different enterovirus RNA extracts were prepared.
Financial support from the Young research award from the European Society for Clinical Microbiology and Infectious Diseases (ESCMID) is greatly appreciated. Support from Replico Medical AB, the regional re-search councils of the Uppsala, Gävle, Falun, Västerås, and O¨ rebro re-gions, the Knut and Alice Wallenberg Foundation, the Göran Gustafsson Foundation, Innovationsbron, and the Nordic Joint Committee for Agri-cultural Research is gratefully acknowledged.
REFERENCES
1.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ.1990. Basic local alignment search tool. J. Mol. Biol.215:403– 410.
2.Forsman A, et al.2003. Single-tube nested quantitative PCR: a rational and sensitive technique for detection of retroviral DNA. Application to RERV-H/HRV-5 and confirmation of its rabbit origin. J. Virol. Methods 111:1–11.
3.Han X, et al.2008. Simultaneously subtyping of all influenza A viruses using DNA microarrays. J. Virol. Methods152:117–121.
4.Hohne M, Schreier E.2004. Detection and characterization of norovirus outbreaks in Germany: application of a one-tube RT-PCR using a fluoro-genic real-time detection system. J. Med. Virol.72:312–319.
5.Jacobs KA, et al.1988. The thermal stability of oligonucleotide duplexes is sequence independent in tetraalkylammonium salt solutions: applica-tion to identifying recombinant DNA clones. Nucleic Acids Res.16:4637– 4650.
6.Jarvinen AK, et al.2009. Rapid identification of bacterial pathogens using a PCR- and microarray-based assay. BMC Microbiol.9:161.
7.Khamrin P. et al.A single-tube multiplex PCR for rapid detection in feces of 10 viruses causing diarrhea. J. Virol. Methods173:390 –393. 8.Larkin MA, et al.2007. Clustal W and Clustal X version 2.0.
Bioinfor-matics23:2947–2948.
9.Lee WM, et al.2007. High-throughput, sensitive, and accurate multiplex PCR-microsphere flow cytometry system for large-scale comprehensive detection of respiratory viruses. J. Clin. Microbiol.45:2626 –2634. 10. Liu J, et al.2011. Multiplex reverse transcription PCR Luminex assay for
detection and quantitation of viral agents of gastroenteritis. J. Clin. Virol. 50:308 –313.
11. Lucore LA, Cullison MA, Jaykus LA.2000. Immobilization with metal hydroxides as a means to concentrate food-borne bacteria for detection by cultural and molecular methods. Appl. Environ. Microbiol.66:1769 – 1776.
12. Mahony J, et al.2007. Development of a respiratory virus panel test for detection of twenty human respiratory viruses by use of multiplex PCR and a fluid microbead-based assay. J. Clin. Microbiol.45:2965–2970. 13. Moore RA, et al.2011. The sensitivity of massively parallel sequencing for
detecting candidate infectious agents associated with human tissue. PLoS One6:e19838. doi:10.1371/journal.pone.0019838.
14. Naas T, Oxacelay C, Nordmann P.2007. Identification of CTX-M-type extended-spectrum-beta-lactamase genes using real-time PCR and pyro-sequencing. Antimicrob. Agents Chemother.51:223–230.
15. Ohrmalm C, et al.2010. Hybridization properties of long nucleic acid probes for detection of variable target sequences, and development of a hybridization prediction algorithm. Nucleic Acids Res.38:e195. doi: 10.1093/nar/gkq777.
16. Parham NJ, et al.2007. Specific magnetic bead based capture of genomic DNA from clinical samples: application to the detection of group B strep-tococci in vaginal/anal swabs. Clin. Chem.53:1570 –1576.
17. Strömberg M, et al.2008. Sensitive molecular diagnostics using volume-amplified magnetic nanobeads. Nano Lett.8:816 – 821.
18. Swaminathan B, Feng P.1994. Rapid detection of food-borne pathogenic bacteria. Annu. Rev. Microbiol.48:401– 426.
19. Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. 1997. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25:4876 – 4882.
20. Tsalik EL, et al.2010. Multiplex PCR to diagnose bloodstream infections in patients admitted from the emergency department with sepsis. J. Clin. Microbiol.48:26 –33.
21. Wang D, et al.2003. Viral discovery and sequence recovery using DNA microarrays. PLoS Biol.1:e2. doi:10.1371/journal.pbio.0000002. 22. Wood WI, Gitschier J, Lasky LA, Lawn RM.1985. Base
composition-independent hybridization in tetramethylammonium chloride: a method for oligonucleotide screening of highly complex gene libraries. Proc. Natl. Acad. Sci. U. S. A.82:1585–1588.
23. Yu D, Wu S, Wang B, Chen Y, Li L.2011. Rapid detection of common viruses using multi-analyte suspension arrays. J. Virol. Methods177: 64 –70.
24. Zardán Gómez de la Torre T, et al.2011. Detection of rolling circle amplified DNA molecules using probe-tagged magnetic nanobeads in a portable AC susceptometer Biosens. Bioelectron.29:195–199.
25. Zuker M.2003. Mfold web server for nucleic acid folding and hybridiza-tion predichybridiza-tion. Nucleic Acids Res.31:3406 –3415.