Inflammation And
Atherosclerosis: The Role Of
Genetic Polymorphisms In
Cardiovascular
Pathophysiology
By
Dr David Joseph Brull
MB BS BSc MRCP (UK)
Thesis submitted for the degree of Doctor of Medicine to the Faculty of Medicine, University of London, September 2001
ProQuest Number: 10010078
All rights reserved
INFORMATION TO ALL USERS
The quality of this reproduction is dependent upon the quality of the copy submitted.
In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed,
a note will indicate the deletion.
uest.
ProQuest 10010078
Published by ProQuest LLC(2016). Copyright of the Dissertation is held by the Author.
All rights reserved.
This work is protected against unauthorized copying under Title 17, United States Code. Microform Edition © ProQuest LLC.
ProQuest LLC
789 East Eisenhower Parkway P.O. Box 1346
Acknowledgements
Table Of Contents
Abstract
Chapter 1
1.1 1.2 1.3 1.3.1 1.3.2 1.3.3 1.4 1.5 1.5.1 1.5.2 1.5.3 1.61.6.1
1.6.2 1.6.3 1.6.4 Page 1 1.7 1.6.5 1.7.1 1.7.2 1.7.3
Introduction
What Is Atherosclerosis? 2
What Are The Traditional Cardiovascular
Risk Factors? 4
The Systemic Inflammatory Response And
Atherosclerosis: Association Or Causality? 6
Fibrinogen And Coronary Artery Disease;
The Clinical Evidence 8
CRP, IL6 And Cardiovascular Disease.
Summary Of The Clinical Data 9
A Possible Role For Infectious Agents In The
Pathogenesis Of Coronary Disease 11
Interleukin-6 And The Acute Phase Response 12
Interleukin-6 The Pleiotropic Cytokine 13
Sources Of Interleukin-6 13
Biological Actions Of Interleukin-6 13
Molecular Regulation Of Interleukin-6 14
Acute Phase Proteins And Possible Atherogenic
Mechanisms 16
The Role Of Fibrinogen In Atherogenesis 16
The Role Of CRP In Atherogenesis 17
Interleukin-6, CRP And Endothelial Dysfunction 18
Interleukin-6, Obesity And Effects On Lipid
Metabolism 19
Psychological Stress And Interleukin-6 20
The Use Of Genetic Polymorphic Variants As Tools
To Investigate Disease Pathogenesis 22
What Are Genetic Polymorphisms And How
Do They Alter Gene Transcription And Translation? 23
Identification, Screening And In Vitro Testing Of
Genetic Polymorphisms 24
Page
1.7.4 Studies Investigating The Role O f p-Fibrinogen Gene
Polymorphisms And Cardiovascular Risk 27
1.7.5 CRP Gene Polymorphisms 28
1.7.6 “Stressing The Genotype” Analyses 29
1.7.7 Study Aims 31
Chapter 2
Materials And Methods
2.1.1 DNA Extraction From Blood
-The “Salting Out Method” 35
2.1.2 Cell And Nuclear Lysis 35
2.1.3 Deproteinisation 35
2.1.4 DNA Extraction 36
2.1.5 Precipitation And Washing 36
2.1.6 Handing DNA Samples That Fail To Spool 36
2.1.7 Dissolving The DNA 37
2.1.8 Generating Array Sheets 37
2.1.9 Creating Stock And Working Arrays 38
2.2 Polymerase Chain Reaction And Restriction
Enzyme Digestion 38
2.2.1 Sample Preparation For Polymerase Chain Reaction 39
2.2.2.1 IL6 -174 G>C Promoter Polymorphism 40
2.2.2.2 IL6 -572G>C And -597G>A Promoter Polymorphisms 40
2.2.3.1 P-Fibrinogen-455 G>A Promoter Polymorphism 41
2.2.3.2 P-Fibrinogen -854 G>A Promoter Polymorphism 41
2.2.4.1 CRP Prom oter-748A>G Polymorphism 41
2.2.4.2 CRP Exon 2 +1059G>C Polymorphism 42
2.2.4.3 CRP 3’ UTR +144T>C Polymorphism 42
2.3 Microtitre Array Diagonal Gel Electrophoresis 42
Page
Chapter 3
Coronary Artery Bypass Graft Surgery:
A Model Of Acute Inflammatory Response
3.1 Introduction 48
3.1.1 Subject Selection Criteria 49
3.1.2 Surgical Procedure 49
3.1.3 Sampling Protocol 50
3.1.4 DNA Extraction And Genotyping 50
3.1.5 Statistical Analysis 51
3.2 Results 51
3.2.1 Summary Of Genotype Data 53
3.2.2 Factors Influencing Withdrawal From Study 54
3.2.3 Factors Influencing Preoperative Inflammatory State:
Impact Of Smoking, Diabetic State And Drug Therapy 56
3.2.4 Post-CABG Acute Phase Response 57
3.2.5 IL6 Response Post-CABG By EL6 Genotype 59
3.2.5.1 IL6-174G>C Genotype 59
3.2.5.2 IL6 -572G>C Genotype 60
3.2.5.3 Combined IL6-174G>C and-572G>C Genotype 61
3.2.6 Fibrinogen Response Post-CABG By p-Fibrinogen
And IL6 Genotype 62
3.2.6.1 P-Fibrinogen-455G>A Genotype 63
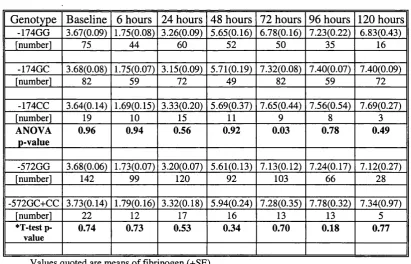
3.2.6.2 p-Fibrinogen -854G>A Genotype 65
3.2.6.3 IL6 -174G>C Genotype 66
3.2.6.4 IL6 -572G>C Genotype 66
3.2.6.5 Combined EL6 -174G>C And -572G>C Genotype 68
3.2.7 CRP Response Post-CABG By IL6 And CRP
Genotype 68
3.2.7.1 CRP -748G>A Genotype 69
32.1.2 CRP +1059G>C Genotype 69
3.2.7.3 CRP +1444C>T Genotype 70
3.2.7.4 IL6 Genotype And Post-CABG CRP Levels 71
Page
3.3.3 The Association Of EL6 And CRP Genotype
With Post-CABG CRP Response 76
3.3.4 The Association Of IL6 And p-Fibrinogen Genotype
With Post-CABG Fibrinogen Response 77
3.3.5 Disadvantages Of Using CAB G As A Model Of
Acute Inflammatory Response. 79
3.3.6 Statin Therapy And Post-CABG Outcome 82
3.4 Conclusions 83
Chapter 4
Strenuous Physical Exercise As A Model
Of The Acute Phase Response:
Impact Of Genetic Polymorphisms
4.1 Introduction 84
4.2 Overall Study Design And Methods 85
4.2.1 Blood Sampling Protocol And Serum Analysis 85
4.2.2 DNA Extraction And Genotyping 86
4.2.3 Statistical Analysis 86
4.3 Results 87
4.3.1 Genotype And Demographic Data 87
4.3.2 Effects of Sample Handling On 1L6 Levels 89
4.3.3 Acute Phase Response To Exercise 90
4.3.4 1L6 Response To Exercise By 1L6 Genotype 92
4.3.4.1 1L6-174G>C Genotype 92
4.3.4.2 1L6 -572G>C Genotype 92
4.3.5 Fibrinogen Response To Exercise By
Fibrinogen Genotype 95
4.3.5.1 Fibrinogen-455G>A Genotype 95
4.3.5.2 Fibrinogen -854G>A Genotype 96
4.3.6 Fibrinogen Response By 1L6 Genotype 98
4.3.6.1 1L6-174G>C Genotype 98
4.3.6.2 1L6 -572G>C Genotype 99
4.3.6.3 Combined 1L6 -174G>C and -572G>C Genotype 99
Page
4.3.8 CRP Response To Exercise By CRP Genotype 104
4.3.8.1 CRP+1444C>T Genotype 104
4.3.8.2 CRP -748G>A Genotype 105
4.3.8.3 CRP +1059G>C Genotype 105
4.4 Discussion 108
4.4.1 Advantages and Disadvantages Of Using Military
Recruits To Study The Inflammatory Response 109
4.4.2 Military Exercise Induces An Acute Phase Response 111
4.4.3 Exercise Models Of Inflammatory Response 111
4.4.4 Differences Between Past And Present Studies
Of Military Exercise 112
4.4.5 The Association Between IL6 Genotype And
Post-Exercise IL6 Response 113
4.4.6 The Effect Of P-Fibrinogen Genotype On
Fibrinogen Post-Exercise 115
4.4.7 The Effect Of IL6 Genotype On Fibrinogen
Post-Exercise 116
4.4.8 The Association Between IL6 And CRP Genotype
And Post-Exercise CRP Response 117
4.4.9 Pathophysiological Consequences Of Increased
Inflammatory Response 118
4.5 Conclusions 120
Chapter 5 What Is The Role Of Genotype In Cardiovascular
Pathophysiology?
5.1 Overview 122
5.2 Why Might IL6 Genotype Affect Gene Transcription? 122
5.3 Recent Studies Of The IL6 -174G>C Genotype 124
5.4 CABG As A Model Of Inflammatory Response 126
5.5 Exercise As A Model Of Inflammatory Response 128
5.6 The Association Between IL6 And CRP Genotype
Page
5.7 Why Do The Observations Of The CABG And
Exercise Models Generate Conflicting Findings? 130
5.8 Future Studies 132
5.9 Conclusions 133
Chapter 1
Figure 1
Figure 2
Figure 3
Figure 4
Appendix 1
Table 1
Table 2
Figures And Tables
Schematic Of The IL6 Signal Transduction Pathway
The IL6 Gene Promoter Region.
Schematic Representation Of The p-Fibrinogen
Gene Promoter
Schematic Representation of the CRP Gene
Summary Of Commonly Recognised Cardiovascular
Risk Factors
Previous Studies Of The P-Fibrinogen -455G>A
Polymorphism Page 15 26 28 29 33 34 Chapter 2
Figure 1
Figure 2
Appendix 2
Table 1
Table 2
Table 3
Table 4
Table 5
An Example Of A Standard Array “Grid”
IL6 -174G>C Genotype Resolved On MADGE
Stock Solutions For DNA Extraction
Oligonucleotide Primers Sequences
PCR Cycle conditions
Details Of Polymerase Chain Reaction And
Restriction Digests
Manufacture Of One 7.5% Non-Denaturing
Polyacrylamide Gel 37 44 45 46 46 47 47 Chapter 3
Table 1
Table 2
Table 3
Table 4
Baseline Patient Characteristics And Operative Details 52
Reasons For Patient Exclusion From Study. 52
IL6, P-Fibrinogen And CRP Genotype Distributions
And Rare Allele Frequencies 54
Comparison Of Subjects With Complicated And
Page
Table 5 Genotype Distribution Of Subjects With Complicated And
Uncomplicated Post-CABG Recovery 55
Table 6 Correlation Between Baseline Inflammatory And
Clinical Variables 56
Table 7 Effects Of Clinical Factors On Baseline Inflammatory
Markers 57
Table 8 Correlations Between Post-CABG Inflammatory Markers 58
Table 9 Mean IL6 Levels By IL6 Genotype 59
Table 10 Mean EL6 Levels By Combined IL6 -I74G>C
And -572G>C Genotype 62
Table 11 p-Fibrinogen Genotype And Post-CABG Fibrinogen
Response 63
Table 12. IL6 Genotype And Post-CABG Fibrinogen Response 66
Table 13 Combined IL6 -174G>C And -572G>C Genotype
And Post-CABG Fibrinogen Response 67
Table 14 CRP Genotype And CRP Levels Post-CABG 68
Table 15 1L6 Genotype And CRP Levels Post-CABG 71
Table 16 Combined 1L6 -174G>C and -572G>C Genotype 72
And Post-CABG CRP Response
Figure 1 Summary Of The Post-CABG Acute Phase Response 58
Figure 2 1L6 Response Post-CABG By 1L6-174G>C Genotype 60
Figure 3 1L6 Response Post-CABG By 1L6 -572G>C Genotype 61
Figure 4 Percentage Change In Fibrinogen Post-CABG
By -455G>A Genotype 64
Figure 5 Fibrinogen Response Post-CABG By p-Fibrinogen
-455G>A Genotype 64
Figure 6 Fibrinogen Response Post-CABG By P-Fibrinogen
-854G>A Genotype 65
Figure 7 1L6 -174G>C Genotype And Post-CABG Fibrinogen
Levels 67
Figure 8 CRP -748G>A Genotype And Post-CABG CRP Levels 69
Figure 9 CRP +1059G>C Genotype And Post-CABG CRP levels 70
Chapter 4 Page
Table 1 Genotype Distribution And Rare Allele Frequencies 88
Table 2 Baseline Demographics 89
Table 3 Effects Of Storage Conditions On IL6 Levels 89
Table 4 Correlation Between Baseline Inflammatory And
Clinical Variables 91
Table 5 Overall Acute Phase Response 91
Table 6 Correlations Between Post-FME Inflammatory Markers 91
Table 7 Mean 1L6 Levels By 1L6 Genotype 93
Table 8 Mean 1L6 Levels By Combined 1L6 -174G>C and
-572G>C Genotype 93
Table 9 Mean Fibrinogen Levels By P-Fibrinogen Genotype 96
Table 10 Fibrinogen Levels By Combined -455G>A And
-854G>A Genotype 97
Table 11 Mean Fibrinogen Levels By 1L6 Genotype 99
Table 12 Fibrinogen Levels By Combined -174G>C And
-572G>C Genotype 100
Table 13 Mean CRP Levels By 1L6 Genotype 102
Table 14 CRP Response By Combined 1L6 -174G>C And
-572G>C Genotype 103
Table 15 Mean CRP Levels By CRP Genotype 105
Figure 1 Summary Of The Post-Exercise Acute Phase Response 92
Figure 2 Post-Exercise 1L6 Response In Individuals Of
Different 1L6 -174G>C Genotype 94
Figure 3 Post-Exercise 1L6 Response In Individuals Of
Different 1L6 -572G>C Genotype 94
Figure 4 Post-Exercise 1L6 Response By Combined 1L6 -174G>C
And -572G>C Genotype 95
Figure 5 Post-Exercise Fibrinogen Response In Individuals of
Different p-Fibrinogen -455G>A Genotype 97
Figure 6 Post-Exercise Fibrinogen Response In Individuals of
Page
Figure 7 Post-Exercise Fibrinogen Response In Individuals of
Different IL6 -174G>C Genotype 100
Figure 8 Post-Exercise Fibrinogen Response In Individuals of
Different IL6 -572G>C Genotype 101
Figure 9 Post-Exercise Fibrinogen Response By Combined
IL6 -174G>C And -572G>C Genotype 101
Figure 10 Post-Exercise CRP Response In Individuals Of
Different 1L6 -174G>C Genotype 103
Figure 11 Post-Exercise CRP Response In Individuals Of
Different 1L6 -572G>C Genotype 104
Figure 12 Post-Exercise CRP Response In Individuals Of
Different CRP +1444C>T Genotype 106
Figure 13a Post-Exercise CRP Response In Individuals Of
Different CRP -748G>A Genotype 106
Figure 13b Post-Exercise CRP Response By -748G>A Genotype After
Removal of Data for Subjects Of Genotype +1444TT 107
Figure 14a Post-Exercise CRP Response In Individuals Of
Different CRP +1059G>C Genotype 107
Figure 14b Post-Exercise CRP Response By +1059G>C Genotype After
Individual Contributions To CABG Study
Study Design and LogisticsAlthough the initial idea to use coronary bypass surgery as a model of inflammatory
response was originally conceived by Professor Steve Humphries and Dr Hugh
Montgomery, I was entirely responsible for the final study design, sampling protocols
and arranged all the necessary logistics, as well as securing Ethics committee approval.
Patient Recruitment
I was responsible for patient recruitment during the study period and was helped by
Julie Sanders SRN during the final 6 months.
Plasma Sample and Clinical Data Collection
I was responsible for sample and clinical data collection during the study with
assistance from Julie Sanders (SRN) during the final 6 months.
Electronic Data Entry and Database Management
I collected and entered all the study data.
Serum Assays (IL6, CRP and fibrinogen)
Professor Gordon Lowe and Dr Ann Rumley (Department of Medicine, University of
Glasgow) kindly performed all the study serum analyses.
DNA Extraction and Genotyping
I performed all the DNA extractions and genotyping for the study.
Genotyping and data entry were checked by Dr Sukhbir Dhamrait
Statistical Analyses
I performed all the statistical analyses with advice from Professor Steve Humphries and
Individual Contributions to Army Study
Study Design and Logistics
Dr Hugh Montgomery conceived the original study design. However, I was wholly
responsible for its development, final study design, and implementation.
Subject Recruitment
I was responsible for recruiting all study subjects.
Sample Collection
I was responsible for the vast majority (>98%) of sample collections, helped at
weekends by Dr Sukhbir Dhamrait.
Clinical Data Collection, Electronic Data Entry And Database Management
I collected all the clinical data and was responsible for all data entry.
Serum Assays (IL6, CRP and fibrinogen)
Professor Gordon Lowe and Dr Ann Rumley (Department of Medicine, University of
Glasgow) kindly performed all the study serum analyses.
DNA Extraction and Genotyping
I performed all the DNA extractions and genotyping for the study.
Genotyping and data entry were checked by Dr Sukhbir Dhamrait
Statistical Analyses
I performed all the statistical analyses with advice from Professor Steve Humphries and
Abstract
IntroductionElevated markers of systemic inflammation are associated with cardiovascular risk - an
association that may be causally mediated by intereukin-6 (IL6), P-fibrinogen and C-
reactive protein (CRP). This hypothesis may be explored using polymorphic variants in
these genes as tools. The aim of this thesis was to address whether such polymorphisms
were functional in vivo.
Methods
Military exercise (MB) and coronary artery bypass graft surgery (CABG) were utilised
as models of inflammatory response to investigate whether the IL6 -572G>C and -
174G>C; p-fibrinogen -854G>A and -455G>A: and CRP -748G>A, +1059G>C and
+1444C>T polymorphisms were functional. DNA extracted from peripheral blood was
amplified by polymerase chain reaction and genotypes resolved by electrophoresis
following restriction enzyme digestion.
Results
One hundred and ninety three subjects undergoing elective CABG were studied. Serum
IL6, CRP and fibrinogen were measured preoperatively and during the first five post
operative days. Analyses suggested a significant effect of both IL6 polymorphisms on
peak post-CABG IL6 (highest for genotypes -174CC and -572GC/CC). Fibrinogen - 455AA genotype was associated with an earlier rise in fibrinogen but no effect on peak response, whilst peak CRP was significantly higher for subjects of CRP genotype +1444TT.
In the second study IL6, CRP and fibrinogen were measured before and after a 48-hour
military exercise in 250 male army recruits. Whilst there was no association between
Conclusions
These data support the in vivo functionality of the IL6 -174G>C and -572G>C; P-
fibrinogen -455G>A and CRP +1444C>T polymorphisms and suggest that they might
be applied in future candidate gene-association studies investigating cardiovascular
1
Introduction
1.1 What Is Atherosclerosis?
Atherosclerosis is a systemic disease of large and medium-sized arteries that can lead to
tissue ischaemia and infarction of subtended tissues. Over the last twenty-five years the
“response to injury hypothesis” (Ross and Glomset 1976; Ross 1986; Ross 1999)
describing repeated cycles of vascular endothelial injury followed by repair, has become
the prominent hypothesis for the underlying process driving the initiation and
progression of atherosclerosis.
Injury may occur secondary to a wide range of insults involving physical disruption of
the endothelium: such as oxidative stress, lipid ingress, arterial shear stress or smoking.
These factors induce a series of cellular changes that lead to a modified form of
inflammation in which monocytes and lymphocytes enter the vessel wall. Regardless of
the cause, endothelial dysfunction (ED) alters the normal homeostatic properties of the
endothelium, tipping the balance towards a prothrombotic, proatherogenic state.
The earliest event in atherosclerosis is the accumulation of low-density lipoprotein
(LDL) particles beneath the vascular endothelium where they undergo oxidation. These
modified LDL species can induce local vascular cells to produce chemokines that
promote monocyte recruitment and migration across the arterial walls. This is followed
by the attachment, adherence and spreading of peripheral blood monocytes, and T-
lymphocytes at sites throughout the arterial tree, particularly at branches and
bifurcations. Monocytes are chemotactically attracted into the subendothelial space
where they accumulate within the intimai layer. Once inside, they differentiate into
macrophages that become activated and secrete growth factors and cytokines. The
accumulation of monocytes and macrophages results in further LDL oxidation.
Oxidised LDL is recognised by scavenger receptors on macrophages and internalised to
form foam cells. In contrast to the uptake of native (unmodified) LDL, via the LDL
receptor, uptake of oxidised LDL by a macrophage scavenger receptor pathway is not
The formation of foam cells and their continued accumulation in the intima lead to the
first ubiquitous lesion of atherosclerosis, namely the fatty streak, that is common even
in infants and children (Napoli, D ’Armiento et al. 1997). Fatty streaks are areas of
intimai thickening, composed of macrophages distended by lipid droplets (foam cells),
as well as lymphocytes and vascular smooth muscle cells (VSMCs). The earliest
morphologically detectable cellular event in the inflammatory reaction is the adherence
of circulating blood monocytes to the intact endothelial surface. This inflammatory
response is then followed by a fibro-proliferative reaction, which in time leads to an
expanded intermediate or fibro-fatty lesion with the formation of a fibrous cap. This cap
consists of numerous VSMCs surrounded by collagen and elastic fibres covering the
more heterogeneous plaque core, which contains lipid debris.
The conventional concept that plaques grow inexorably over years by gradually
accumulating lipid, impeding blood flow until symptoms arise, has been brought into
question. Glagov (Glagov, Weisenberg et al. 1987) showed that coronary arteries could
accommodate large plaques without reducing lumen size by a process called
remodelling. During this process the vessel initially accommodates the expanding
atherosclerotic plaque, expanding outwards, before the bulk of atheroma ultimately
encroaches on the vessel lumen obstructing blood flow (Libby 1995). In this scenario
individuals might present with symptoms of stable progressive coronary ischaemia
(stable angina pectoris).
The current view is that atherosclerosis is a dynamic process of recurrent cycles of
plaque erosion and repair (Davies 1995; Davies 2000). Two major mechanisms lead to
vessel thrombosis- rupture of a plaque’s fibrous cap and superficial erosion of the
endothelium. Plaque disruption depends upon both active and passive phenomena.
Several morphological features characterise the unstable atheromatous plaque, including
a thin, eccentric fibrous cap and a large necrotic core of lipid and cellular debris. The
area of the plaque that is most vulnerable to disruption is the shoulder region, where the
fibrous cap is thinnest and where there is a high concentration of foam cells (Fuster,
Fayad et al. 1999). Several factors increase the risk of plaque rupture, including wall
stress, location, size and consistency of the atheromatous core and on blood flow
Many episodes of plaque rupture do not in fact lead to occlusive thrombus formation
and are clinically silent (Davies 1995). However thrombus is a rich source of
chemoattractants and mitogens for VSMCs, such as platelet derived growth factor
(PDGF) and thrombin. These factors recruit new VSMCs into the area to form a fresh
fibrous plaque. A new fibrous cap is thus generated rapidly but the bulk of atheroma
enlarges as a consequence. Thus in atherosclerosis there is a balance between the
reparative processes that lead to the development of a safe stable plaque and the
destructive inflammatory process that lead to its breakdown. Many patients experience
slow plaque expansion over many years, however, when the atherosclerotic plaque
reaches a critical size it may result in loss of vessel lumen, reducing tissue perfusion
leading to end-organ ischaemia: clinically manifest as cerebrovascular, peripheral
vascular, renovascular or coronary artery disease.
1.2 What Are The Traditional Cardiovascular Risk Factors?
Cardiovascular disease remains the major cause of death in the Western world and also
shows an increasing incidence in developing countries as they undergo industrialisation.
The concept of cardiovascular risk evolved from epidemiological studies of CAD
conducted in the 1940s and 1950s, such as the Framingham study (Kannel 2000). Using
a prospective epidemiological approach it has since been possible to demonstrate a
consistent association of characteristics observed in apparently healthy individuals with
the subsequent development of cardiovascular disease in these same people. Risk
factors were defined as measurable traits that would predict the future development of
disease. Whilst the observation of a statistically significant association between a trait
and disease does not provide proof of causality or establish a pathophysiological
relationship, such observations do contribute to our understanding of pathophysiology.
Over the last 50 years the list of major cardiovascular risk factors (Appendix 1,Table 1)
has continued to grow as our understanding of vascular biology improves. Despite the
identification of a variety of potent risk factors for the development of cardiovascular
disease, many of the underlying pathophysiological mechanisms remain occult.
Although patients with cardiovascular disease, such as coronary artery disease (CAD)
Well accepted risk factors for the development of CAD include increasing age, high
blood levels of total and low-density lipoprotein (LDL) cholesterol and triglycerides,
low levels of high-density lipoprotein cholesterol (HDL), elevated blood pressure,
cigarette use, diabetes mellitus, and evidence of left ventricular hypertrophy on
electrocardiography (Wilson 1994; Kannel and Wilson 1995; Kannel 2000).
Understanding of the importance of many traditional risk-factor measures has expanded
in scope over time. For instance, both systolic and diastolic blood pressure levels are
each associated with the occurrence of CAD, and a gradient of risk increases across the
entire range of blood pressure, even if it is below standard treatment thresholds
(Ramsay, Williams et al. 1999). Total (or LDL cholesterol) and HDL cholesterol are
highly associated with CAD incidence, as are levels of another lipoprotein particle
called lipoprotein-a (Lp(a)) (Kannel and Wilson 1995; Dahlen and Stenlund 1997; Wu
1999).
Diabetes mellitus has been consistently associated with CAD, with additional data to
demonstrate independent roles for obesity and regional adiposity, which are intimately
linked with the development of diabetes (Castelli 1996; Gensini, Comeglio et al. 1998).
High triglyceride associated with reduced HDL, indicating insulin resistance and small
dense LDL was shown to be associated with excess coronary disease. These risk factors
tend to cluster and are promoted by insulin resistance; itself induced by weight gain
(Reaven 1998; Reaven 1998).
In addition to plasma lipids, other factors such as elevated fibrinogen concentration and
leucocyte count have also been consistently associated with CAD in observational
studies. In particular, results from prevalence, case-control, and angiographic studies
identify elevated fibrinogen levels as a strong independent risk factor for the occurrence
of initial and recurrent cardiovascular events (Wilhelmsen, Svardsudd et al. 1984;
Meade, Mellows et al. 1986; Kannel, D ’Agostino et al. 1987; Yamell, Baker et al. 1991;
Ernst and Resch 1993; Woodward, Lowe et al. 1997; Woodward, Lowe et al. 1998).
Average fibrinogen levels are lower in women, contributing to their overall lower
cardiovascular risk than men, but levels are often raised in association with other risk
factors including hypertension, diabetes, cigarette smoking, obesity, elevated
haematocrit and dyslipidaemia (Balleisen, Bailey et al. 1985; Lee, Smith et al. 1990;
atherogenic cardiovascular risk factors further enhancing cardiovascular risk (Gensini,
Comeglio et al. 1998; Sakkinen, Wahl et al. 2000).
Prospective data indicate a relationship between fibrinogen and the subsequent
development of all the major atherosclerotic cardiovascular events including coronary
heart disease, stroke and peripheral artery disease. Fibrinogen exerts an independent
influence on the frequency of cardiovascular disease in general and coronary heart
disease in particular. For example, in both men and women, each standard deviation
increase in fibrinogen level (about 0.6 g/L) within the range of usual values is
associated with about a 20% age- and risk factor-adjusted increase in the incidence of an
initial cardiovascular event (Kannel 1997).
There is now a body of research to support the role of chronic low-grade infections in
the development of CAD. Certainly, chronic infection can increase coagulability by
inducing hyperfibrinogenaemia that may lead to atherogenesis if there is concurrent
endothelial damage or dysfunction (Vallance, Collier et al. 1997). Studies in humans
have suggested associations between a number of infections and CAD. These include
chronic gastritis (Mendall, Goggin et al. 1994; Patel, Mendall et al. 1995; Niemela,
Kartunen et al. 1996), periodontal disease (Loesche 1994; Beck, Garcia et al. 1996) and
low-grade respiratory infections (Saikku, Leinonen et al. 1992; Mendall, Carrington et
al. 1995; Patel, Mendall et al. 1995).
Finally it is important to note that cardiovascular risk tends to cluster in affected
families, where there are a number of identifiable risk factors. This supports the
importance of both a shared environment and shared genetic factors (Genest and Cohn
1995; Kannel and Wilson 1995; Kannel 2000; Sakkinen, Wahl et al. 2000).
1.3 The Systemic Inflammatory Response And Atherosclerosis: Association Or Causality?
Substantial evidence is now accumulating to suggest that inflammatory responses, be
they systemic, or local to the vessel wall, play a key mechanistic role in driving the
markers of inflammation, such as C-reactive protein (CRP) (Kuller, Tracy et al. 1996;
Ridker, Cushman et al. 1997; Koenig, Sund et al. 1998), fibrinogen (Meade, Mellows et
al. 1986; Kannel, Wolf et al. 1987; Yamell, Baker et al. 1991) and interleukin-6 (IL6)
(Ridker, Buring et al. 1998; Ridker, Hennekens et al. 2000; Ridker, Rifai et al. 2000),
with the development of coronary disease and with both prognosis and disease
progression. This is tme in healthy subjects and in those with established CAD:
including both stable CAD and unstable angina. Observational data thus suggest a role
for the inflammatory process in the development of progressive atherosclerosis, and in
the conversion to an acute coronary event. One might quite reasonably argue that the
inflammation is a response to rather than a cause of plaque generation or rupture.
Indeed, coronary lesions may themselves be pro-inflammatory. However, several lines
of evidence suggest that the inflammatory response is not merely a bystander, and
support a role for inflammatory processes in the pathogenesis of coronary artery disease
(Ross 1999).
It has long been recognised that the stable progressive atheromatous plaque seen in
chronic stable angina contains a variety of inflammatory cells including monocytes and
macrophages (van der Waal, Becker et al. 1996). Inflammation leads to the recruitment
of neutrophils, monocytes and activated macrophages in the cap of atherosclerotic
plaques. This has lead to the suggestion that they contribute towards plaque rupture by
secretion of matrix metalloproteinases (Alexander 1994; Galis, Sukhova et al. 1994).
Furthermore, the ruptured plaque associated with one of the acute coronary syndromes
(unstable angina or myocardial infarction) is also characterised by the presence of
activated inflammatory cells, including macrophages and lymphocytes at the site of the
rupture (Liuzzo, Biasucci et al. 1997). An “inflamed” vascular wall might increase the
levels of pro-inflammatory cytokines such as interleukin-1 (ILl), tumour necrosis
factor-a (TNF-a) and IL6. These in turn would modulate hepatic acute phase protein
synthesis.
The “inflammatory hypothesis” of CAD is further supported by the fact that a variety of
environmental factors associated with the development of coronary artery disease share
one feature: they all provoke an inflammatory response. Several of the factors known to
induce an acute inflammatory response, including severe exercise (Mittleman, Maclure
et al. 1993; Willich, Lewis et al. 1993) acute infection (Dobson, Alexander et al. 1988;
risk of acute myocardial infarction. In addition, psychological stress and emotional state
may have a profound influence on the development of CAD (Krantz, Kop et al. 1996;
Hemingway and Marmot 1999; Pashkow 1999; Reyes-Ortiz 1999; Krantz, Sheps et al.
2000), likely mediated through associated increases in white cell count, fibrinogen, and
circulating pro-inflammatory cytokines (Maes, Song et al. 1998; Nukina, Sudo et al.
1998). Interestingly, factors that reduce plasma inflammatory markers such as regular
physical exercise (Dufaux, Order et al. 1984; Ernst 1993; Rankinen, Rauramaa et al.
1993; Rankinen, Rauramaa et al. 1994; Mattusch, Dufaux et al. 2000) are associated
with reduced risk of coronary vascular disease (Berlin and Colditz 1990).
1.3.1 Fibrinogen And Cardiovascular Disease: The Clinical Evidence
In keeping with the putative role for inflammation in the pathogenesis of coronary
disease, elevated serum fibrinogen concentrations represent an independent risk factor
for ischaemic heart disease in healthy individuals (Wilhelmsen, Svardsudd et al. 1984;
Meade, Mellows et al. 1986; Kannel, D ’Agostino et al. 1987; Yamell, Baker et al. 1991;
Ernst and Resch 1993; Woodward, Lowe et al. 1997; Woodward, Lowe et al. 1998).
Fibrinogen is as powerful a predictor of future cardiovascular disease as other classical
risk factors such as hypercholesterolaemia, smoking and hypertension (Yamell, Baker et
al. 1991; Emst and Resch 1993), adding to their predictive value (Woodward, Lowe et
al. 1998).
In addition to the studies looking for an association between fibrinogen and CAD,
elevated fibrinogen levels also predict the development of peripheral vascular disease
(Lassila, Pltonen et al. 1993; Fowkes, Pell et al. 1994), cerebrovascular disease (Di
Minno and Mancini 1990) and aortic atherosclerosis (Tribouilloy, Peltier et al. 1998).
One fact worthy of note is that since there is a high degree of intra-individual variability
in fibrinogen levels (Pyke, Thompson et al. 1993), the real association between
fibrinogen and CAD might be stronger than reported so far.
There is a wealth of data examining the role of fibrinogen in patients with documented
CAD. The ECAT Angina pectoris Study Group found a significant positive correlation
between fibrinogen and the severity of angiographically documented CAD (Bolibar,
correlated with the transition from stable coronary disease to unstable angina (UA) and
myocardial infarction (Thompson, Kienast et al. 1995). There were similar results from
the TIMI-IIIB trial, where elevated fibrinogen at the time of hospital admission was
associated with an increased risk of in-hospital events in patients with UA (Becker,
Cannon et al. 1996). Similarly data from the FRISC study of patients with UA, showed
elevated fibrinogen levels at the time of admission were predictive of long-term risk of
death or myocardial infarction. The increased risk associated with elevated fibrinogen
was independent of, and additive to the prognostic influence of myocardial damage
(Toss, Lindahl et al. 1997; Lindahl, Toss et al. 2000). Thus, fibrinogen levels correlate
with disease progression, with the transition from stable coronary disease to unstable
angina and with myocardial infarction (Yamell, Baker et al. 1991; Thompson, Kienast
et al. 1995).
1.3.2 CRP, IL6 And Cardiovascular Disease: Summary O f The Clinical Data
A number of large prospective clinical studies have reported an association between
elevated CRP levels in healthy subjects and the future development of CAD. These
include the MRFIT (Multiple Risk Factor Intervention Trial) nested case control study
(Kuller, Tracy et al. 1996), where CRP was predictive of CAD mortality; the MONICA-
Augsburg Cohort Study (Koenig, Sund et al. 1998) and the Physicians Health Study
(Ridker, Cushman et al. 1997). These observations have been consistently observed in
other studies (Danesh, Whincup et al. 2000) and in the elderly (Tracy, Lemaitre et al.
1997). Most importantly, in all these studies the level of CRP that conferred an
increased risk of cardiovascular disease was within the “normal” reference range. This
suggests that even in the presence of a barely detectable low-grade chronic
inflammatory state, there is increased risk. More recently three studies have shown that
in addition to CRP, IL6 levels also predict the future development of cardiovascular
disease in health, both in men (Ridker, Rifai et al. 2000) and women (Ridker, Buring et
al. 1998; Ridker, Hennekens et al. 2000).
Besides the studies that have investigated the importance of inflammatory markers in
“healthy” individuals, several studies have explored the importance of inflammation in
patients with documented CAD. Not entirely unexpectedly, inflammatory markers are
elevated in subjects with documented CAD compared to angiographically normal
CRP predicts future cardiovascular events in both stable and unstable angina
(Haverkate, Thompson et al. 1997; Toss, Lindahl et al. 1997) even after percutaneous
coronary intervention (Gaspardone, Crea et al. 1998; Buffon, Liuzzo et al. 1999).
Patients with unstable angina in particular, show evidence of increased inflammatory
activity, documented by increased CRP and IL6 (Biasucci, Vitelli et al. 1996; Biasucci,
Liuzzo et al. 1999). The magnitude of this elevation exceeds that found in stable CAD
(Berk, Weintraub et al. 1990) and is predictive of poor outcome (Liuzzo, Biasucci et al.
1994; Toss, Lindahl et al. 1997). Elevated CRP and IL6 levels often persist for several
months following an acute coronary syndrome, and are associated with an increased
future cardiac event rate (Haverkate, Thompson et al. 1997; Biasucci, Liuzzo et al.
1999).
Combined, this evidence would suggest that the systemic inflammatory response might
be associated not only with the development of CAD, but also with the transition to
myocardial infarction or unstable angina. Consequently it appears that a gradient of
cardiovascular risk exists across the population as a whole, almost regardless of the
marker being measured: fibrinogen, CRP and IL6. The clinical associations of elevated
markers are similar, reflecting a universal underlying process, namely inflammation.
In addition to the data associating CRP levels with future risk of CAD, a number of
studies have show the same association to be true for both peripheral vascular disease
and stroke. Results from the Physician’s Health study showed that men in the highest
quartile of CRP had double the risk of stroke compared to those in the bottom quartile
(Ridker, Cushman et al. 1997). The Leiden 85-Plus stroke study also reported CRP to be
a strong predictor of fatal CVA (Gussekloo, Schaap et al. 2000), and in a subgroup of
subjects aged over 75 from the Helsinki Ageing Study, an increase in CRP predicted
total and cardiovascular mortality (Strandberg and Tilvis 2000). Finally, the Women’s
Health Study reported a five-fold increase in risk of CVA between subjects in the top
and bottom quartiles of CRP (Ridker, Buring et al. 1998). In contrast to the data
supporting a role for CRP in cerebrovascular disease, the association between CRP and
the development of peripheral vascular disease is less well understood. However data
again from the Physician’s Health study showed that baseline CRP was predictive of the
1.3.3 A Possible Role For Infectious Agents In The Pathogenesis O f Coronary
Disease.
Given that chronic low-grade infection may be associated with a continued
inflammatory response, it would seem plausible that such infection may be associated
with the development of coronary disease. There is now a body of research to support
the role of chronic low-grade infections in the development of CAD. Fabricant first
suggested that coronary disease might be due to an infectious agent after studying
arterial lesions provoked by Marek’s disease herpes virus in chickens (Fabricant,
Fabricant et al. 1978). Since then, numerous studies in humans have suggested
associations between chronic gastric (Mendall, Goggin et al. 1994; Patel, Mendall et al.
1995; Niemela, Kartunen et al. 1996), lung (Saikku, Leinonen et al. 1992; Mendall,
Carrington et al. 1995; Patel, Mendall et al. 1995), and gum (Loesche 1994; Beck,
Garcia et al. 1996) infections with both chronic inflammation and with the development
of CAD.
To date the most likely pathogenic candidates in human disease are Cytomegalovirus
(CMV), Helicobacter pylori (HP) and Chlamydia pneumoniae. However many of the
studies performed to date have been small, inadequately powered and lacking
appropriate control groups. Several recent publications have found no evidence of any
association between infection and CAD (Adler, Hur et al. 1998; Regnstrom, Jovinge et
al. 1998), including a recent meta-analysis looking at Chlamydia, which is perhaps the
strongest “candidate” infectious agent infection (Danesh, Whincup et al. 2000).
However an increasing body of data suggests that total cumulative pathogen burden is
associated with increased risk of atherogenesis (Zhu, Quyyumi et al. 2000; Zhu, Nieto
et al. 2001) Thus at present the role of infectious agents in the development of
1.4 Interleukin-6 And The Acute Phase Response
The acute phase response is an orchestrated response to a variety of insults that cause
tissue injury, involving a co-ordinated sequence of both local and systemic metabolic
changes. As such it is a common pathway in the body’s attempt at maintaining
homeostasis. The earliest description of the acute phase process came from the ancient
Greeks who noticed increased erythrocyte sedimentation in the blood of the severely ill,
however Abemethy and Avery first coined the term “acute phase response” in 1941
(Abemethy and Avery 1941). There are several characteristic features of the acute phase
response including fever, neutrophilia, altered lipid metabolism, increased
gluconeogenesis, protein catabolism, activation of the complement and coagulation
systems and induction of acute phase proteins (Kushner 1982; Baumann and Gauldie
1994).
Conditions that commonly lead to substantial changes in the plasma concentration of
acute phase proteins include infection, trauma, surgery, bums and malignancy. More
moderate changes occur after strenuous exercise, heat stroke and childbirth (Kushner
1982; Baumann and Gauldie 1994). Smaller changes occur after psychological stress
and in psychiatric illnesses (Maes, Bosmans et al. 1997; Maes, Delange et al. 1997).
The appearance or rapid increase in the concentration of a number of plasma proteins,
collectively known as “acute phase proteins" is one of the cardinal features of the acute
phase response. These proteins are mainly produced in the liver and serve a variety of
important functions. Acute phase proteins can broadly be classified into two main
groups. Firstly there are the group of positive acute phase proteins such as serum
amyloid A, C-reactive protein (CRP), complement C3 and a l acid glycoprotein,
fibrinogen, haptoglobin and al-antitrypsin, whose levels increase by at least 25%
during inflammatory disorders, and the so-called negative acute phase proteins such as
albumin, transferrin and HDL, whose levels decrease with the acute phase response
(Kushner 1982; Baumann and Gauldie 1994).
The production and regulation of the acute phase response is complex and regulated by
the interaction of a number of cytokines such as IL l, TN F-a and in particular IL6
Marinkovic, Jahreis et al. 1989; Ganapathi, Rzewnicki et al. 1991; Baumann and
Gauldie 1994; Szalai, van Ginkel et al. 2000).
1.5 Interleukin-6 The Pleiotropic Cytokine
L5.1 Sources O f Interleukin-6
Interleukin-6 is a glycoprotein molecule with a molecular mass of 21-28kD, which
varies depending on the degree of glycosylation and cell specific expression (Toasto,
Seamon et al. 1988; May, Shaw et al. 1991; Kishimoto, Akira et al. 1995). It is derived
from diverse tissues including monocytes and macrophages, lymphocytes, fibroblasts,
endothelial cells, hepatocytes, smooth muscle cells and is also found in atherosclerotic
plaques (Kishimoto 1989; Heinrich, Castell et al. 1990; Seino, Ikeda et al. 1994; Rus,
Vlaicu et al. 1996). Indeed high levels of IL6 mRNA have been detected co-localising
with macrophages at areas of plaque rupture (Schieffer, Schieffer et al. 2000).
Additionally adipose tissue has been shown to be a major source of IL6 in vivo,
producing up to 30% of the total circulating concentration of IL6 in healthy subjects
(Mohamed-All, Goodrick et al. 1997).
7.5.2 Biological Actions O f Interleukin-6
Interleukin-6 is perhaps the prototypic pleiotropic cytokine with a wide range of actions,
in addition to the regulation of the acute phase response, which is of most relevance to
atherogenesis.
IL6 induces platelet derived growth factor production from blood vessels and helps
promote smooth muscle proliferation. Additional actions include modulation of immune
reactions, with influence over both B-lymphocyte immunoglobulin production and T-
lymphocyte proliferation and differentiation; enhancement of several stages of
haematopoiesis (both megakaryocyte maturation and induction of macrophage
differentiation); modulation of bone resorption through stimulation of osteoclast
activity; stimulation of placental gonadotrophin secretion as well as a role in neuronal
differentiation (Andus, Geiger et al. 1987; Gauldie, Richards et al. 1987; Kishimoto
IL6 was initially known by a variety of names that were based on the multitude of
functions that were assigned to this cytokine. These include Interferon p2, IL-1-
inducible 26kD Protein, Hepatocyte Stimulating Factor, Cytotoxic T-cell Differentiation
Factor, B-cell Differentiation Factor (BCDF), monocyte-granulocyte inducer type 2,
monocyte-granulocyte-derived growth factor for human B-cells, cytolytic T-cell
differentiation factor and B cell Stimulating Factor 2. Once all the activities became
connected with one common protein, the actual name IL6 was proposed for this
molecule (Sehgal, Grienger et al. 1989).
1.5.3 Molecular Regulation O f Interleukin-6
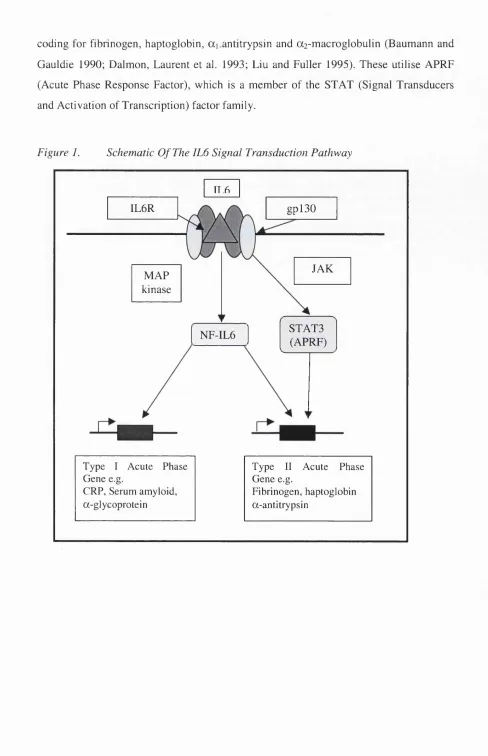
The signal transduction pathway of IL6 is complex but has been studied in depth
(summarised in Figure 1). The IL6 receptor consists of two transmembrane proteins: an
SOkD glycoprotein ligand-binding chain (IL6R) and a 130kD glycoprotein signal
transducer, gpl30 (Kishimoto, Akira et al. 1995; Heinrich, Behrmann et al. 1998).
Binding of 1L6 to its receptor gp80 triggers the association of gp80 with a signal
transducing subunit, gpl30, and the formation of a high affinity receptor.
The 1L6 receptor complex acts via either the Janus kinase pathway (JAK) or through
phosphorylation of nuclear factor 1L6 (NF-1L6) by mitogen activated protein kinase
(MAP-kinase) (Wegenka, Buschmann et al. 1993; Darnell, Kerr et al. 1994; Kishimoto
1994). Subsequent tyrosine phosphorylation of ST AT proteins (signal transducers and
activators of transcription), including STAT3, also known as APRF (Acute Phase
Response Factor), induces ST AT protein dimérisation and translocation into the
nucleus, where they bind response elements of other genes, through which 1L6 is known
to act (Wegenka, Buschmann et al. 1993).
lnterleukin-6 response elements (lL6REs) are present in the promoters of various acute
phase genes, present in two main types. Type 1 elements function in the promoters of
genes, including hemopexin, haptoglobin, C-reactive protein, and ai-acid glycoprotein
(Poli and Cortese 1989; Baumann and Gauldie 1990; Sehgal 1990; Won and Baumann
1990; Ray and Ray 1996). The transcriptional activators that bind to these elements are
members of a leucine zipper family of factors, the C/EBP (CCAAT-Enhancer binding
coding for fibrinogen, haptoglobin, ai.antitrypsin and az-macroglobulin (Baumann and
Gauldie 1990; Dalmon, Laurent et al. 1993; Liu and Fuller 1995). These utilise APRF
(Acute Phase Response Factor), which is a member of the STAT (Signal Transducers
and Activation of Transcription) factor family.
Figure 1. Schematic O f The IL6 Signal Transduction Pathway
IL6R
IT .6
gpl30
MAP kinase
JAK
NF-IL6 STATS
(APRF)
Type I Acute Phase Type 11 Acute Phase
Gene e.g. Gene e.g.
CRP, Serum amyloid. Fibrinogen, haptoglobin
1.6 Acute Phase Proteins And Possible Atherogenic Mechanisms
1.6.1 The Role o f Fibrinogen In Atherogenesis
Fibrinogen is a 340kD glycoprotein dimer composed of two identical subunits joined
together by disulphide bonds. Each subunit consists of three polypeptide chains Aa Bp
and Y which are joined at their amino acid ends (Ernst 1992). Each chain is coded for by
a separate gene that lies on a 50kb segment of DNA on the long arm of chromosome 4
(Marino, Fuller et al. 1986).
IL6 is extremely important in regulating the levels of fibrinogen, as there is an IL6
responsive element in the promoter region of all three fibrinogen genes (Dalmon,
Laurent et al. 1993; Hu, Harris et al. 1995; Mizuguchi, Hu et al. 1995; Zhang, Fuentes
et al. 1995). However as synthesis of the P-chain is thought to be the rate-limiting step
in fibrinogen synthesis (Roy, Mukhopadhyay et al. 1990; Zhang and Redman 1992), the
effects of IL6 on this sequence are likely to directly affect the rate of fibrinogen
synthesis.
The question arises as to how fibrinogen might promote atherogenesis? There are a
number of plausible biological mechanisms through which elevated plasma fibrinogen
levels might promote cardiovascular events (Resch, Ernst et al. 1992; Ernst and Resch
1993). Fibrinogen is a major determinant of plasma viscosity and red cell aggregation,
two factors that in turn predict cardiovascular events and stroke (Yamell, Baker et al.
1991; Lowe, Lee et al. 1997; Kesmarky, Toth et al. 1998; Koenig, Sund et al. 1998;
Woodward, Rumley et al. 1999). Data from the Caerphilly and Speedwell studies
showed that about 50% of the predictive value of plasma viscosity for ischaemic events
was attributable to fibrinogen (Sweetnam, Thomas et al. 1996). Increased viscosity may
increase platelet aggregation (Meade, Vickers et al. 1985) and adherence to the walls of
blood vessels (Errill 1969; Koenig 1999). This increases the likelihood of inappropriate
thrombosis and is associated with incident cardiovascular events (Gillum, Mussolino et
al. 1995).
Fibrinogen may also be involved in the earliest stages of plaque formation. Both
addition, fibrin degradation products have been shown to injure epithelial cells in
culture (Watanabe and Tanaka 1983). Furthermore they may also play a role in
recruiting smooth muscle cells from the media into the intima (Thompson and Smith
1989; Smith, Keen et al. 1990). Such focal smooth muscle cell proliferation is widely
perceived as one of the key events in the formation of stenosing atherosclerotic lesions
(Naito, Hayashi et al. 1989; Naito, Hayashi et al. 1990; Naito, Stirk et al. 2000).
Fibrinogen is an important clotting factor, necessary for the formation of thrombin, the
final step in the coagulation cascade whose elevated levels result in increased tendency
for thrombosis. However fibrinogen is not merely a coagulation factor, but also a
hepatically derived acute phase protein. Plasma levels rise acutely in response to many
stimuli including infection, injury or other trauma. (Green and Humphries 1989; de
Maat, Pietersma et al. 1996). Moreover, elevated fibrinogen levels have been associated
with a number of other traditional cardiovascular factors, including increasing age,
smoking, hypercholesterolaemia, diabetes, obesity, and the menopause (Balleisen,
Bailey et al. 1985; Lee, Smith et al. 1990; Folsom 1995). Increased plasma fibrinogen
may be an indicator of cardiovascular risk, because the level of fibrinogen reflects
underlying arterial wall inflammation. This is based on the theory of Ross that advance
atherosclerotic lesions result from an exuberant inflammatory-fibroproliferative
response to tissue injury (the response to injury hypothesis) (Ross and Glomset 1976).
L6.2 The Role O f CRP In Atherogenesis
Less is known about the actual mechanism through which CRP might promote
atherogenesis as compared to the data regarding fibrinogen. CRP, perhaps the
archetypal acute phase protein, is a member of the pentaxrin family of proteins and is a
highly conserved molecule characterised by a cyclic pentameric structure. In common
with all acute phase proteins, levels of CRP increase very rapidly in response to a range
of stimuli, often increasing by 1000-fold before decreasing with the resolution of the
acute phase stimulus. This enormous induction is the consequence of transcriptional up-
regulation of the CRP gene in the liver.
Whilst the exact function of CRP is not known, it is now recognised that CRP plays an
important role in host defence as part of the innate immune response (Szalai, Agrawal et
diminishing neutrophil chemotaxis (Kew, Hyers et al. 1990) and chemoattractant-
induced degranulation (Filep and Foldes-Filep 1989). Therefore it is possible that CRP
has a pivotal role during inflammation by modulating neutrophil recruitment. Moreover
CRP is deposited within acute myocardial infarcts (Kushner, Broder et al. 1978; de
Beer, Hind et al. 1982) and has been shown to play a role in inducing complement
mediated myocardial damage in an experimental model of myocardial infarction
(Griselli, Herbert et al. 1999).
Another important function of CRP is in selectively binding LDL (de Beer, Hind et al.
1982) especially oxidised and enzyme-modified LDL found within atheromatous
plaques (Torzewski, Bowyer et al. 1997; Torzewski, Torzewski et al. 1997; Torzewski,
Torzewski et al. 1998). This LDL-complexed CRP is able to activate complement
(Bhakdi, Torzewski et al. 1999) unleashing the opsonic and chemotactic functions of
the complement system. Finally, CRP has also been reported to stimulate tissue factor
production by peripheral blood monocytes, which in turn might have an important pro
coagulant effect (Cermak, Key et al. 1993; Nakagomi, Freedman et al. 2000).
1.6.3 lnterleukin-6, CRP And Endothelial Dysfunction
A further mechanism through which IL6 and hence also CRP, might influence the
development of atherosclerosis is through endothelial dysfunction (ED)(Anderson,
Gerhard et al. 1995; Vogel 1999). The vascular endothelium is a monolayer of cells
between the vessel lumen and the vascular smooth muscle cells. Far from being inert, it
is metabolically active and produces a variety of vasoactive mediators. Among these
mediators, endothelial derived nitric oxide is essential in the maintenance of vascular
homeostasis. Endothelium derived nitric oxide has a number of important anti^
atherogenic properties that include enhancement of vasodilatation, inhibition of platelet
aggregation, inhibition of leucocyte migration, inhibition of smooth muscle proliferation
and migration and inhibition of adhesion molecule activation and expression.
Endothelial dysfunction is one of the earliest manifestations of atherosclerosis. It
predates clinical evidence of disease by many years (Ross 1999), and potentially plays a
causative pathogenic role. There is now evidence directly linking inflammation with
levels indicative of a systemic inflammatory response are associated with a blunted
systemic endothelial vasodilator function. Moreover transient low-grade inflammation
is associated with prolonged ED (Hingorani, Cross et al. 2000), providing a potential
mechanism through which IL6 might drive the atherosclerotic process.
IL6 has been associated with several markers of ED and with the insulin resistance
syndrome (Hak, Stehouwer et al. 1999; Yudkin, Stehouwer et al. 1999; Festa,
D ’Agostino et al. 2000). There also appears to be a relationship between increasing CRP
levels with a number of metabolic disorders that have been associated with ED: namely
obesity, systolic hypertension and insulin resistance. Thus following endothelial injury,
IL6 release may stimulate chemokine and adhesion molecule expression and the ingress
of leucocytes (Romano, Sironi et al. 1997), stimulating fibroblast and smooth muscle
proliferation. This combination of cellular proliferation, LDL oxidation (itself a potent
stimulus to IL6 production (Liao, Matsumoto et al. 1999)) and leucocyte activation may
in turn lead to the generation of atherosclerosis.
1.6.4 lnterleukin-6. Obesity And Effects On Lipid Metabolism
Adipose tissue plays an active role in metabolism (Mohamed-Ali, Pinkney et al. 1998)
secreting significant amounts of IL6 into the systemic circulation (Mohamed-Ali,
Goodrick et al. 1997). Because the pro-inflammatory properties of IL6 include
induction of hepatic acute phase protein synthesis, the release of IL6 from adipose
tissue may induce a low-grade inflammatory response in obese individuals.
It has been hypothesised that IL6 is responsible for many of the lipid abnormalities seen
in the insulin-resistance syndrome (Pickup, Mattock et al. 1997). Indeed adipose tissue
secretes IL6, whose levels and those of CRP correlate with various measures of obesity
(Mendall, Patel et al. 1996; Tracy, Psaty et al. 1997) and with insulin resistance
(Yudkin, Stehouwer et al. 1999; Bastard, Jardel et al. 2000). This suggests two
important possibilities: namely that EL6 may be one of the means by which obesity
increases coronary risk (Folsom, Burke et al. 1991; Pi-Sunyer 1991; Rimm, Stampfer et
al. 1995; Folsom, Stevens et al. 1998) and that IL6 also has a role in the regulation of
body lipid metabolism, which is important in atherogenesis (Steinberg and Wiztum
1990). Indeed it is plausible that the increased cardiovascular risk associated with
1999; Cook, Mendall et al. 2000; Yudkin, Kumari et al. 2000; Visser, Bouter et al.
2001).
IL6 has been shown to reduce lipoprotein lipase activity in adipose tissue, leading to
reduced triglyceride uptake (Greenberg, Nordan et al. 1992). This may lead to
chronically raise circulating lipid levels that would contribute to atherogenesis.
Circumstantial evidence for this is provided by a study that found IL6 levels rose in
response to infection and resulted in altered serum lipid levels (Personen, Rapola et al.
1993). Although this study found lipid levels to be decreased rather than increased, the
decrease was more pronounced for cardioprotective high-density lipoprotein, rather than
for low-density lipoprotein and total cholesterol. This alteration in lipid ratio might
increase cardiovascular risk, but it seems likely to be independent of the action of IL6
on lipoprotein lipase. In man, IL6 is associated with elevated plasma free fatty acids
VLDL and triglycerides (Fernandez-Real, Broch et al. 2000). Experimental data also
suggest that IL6 induces a physiological “catabolic” state with increased resting energy
expenditure and elevated levels of free fatty acids (Stouthard, Romijn et al. 1995).
The effects of IL6 on lipids are complex. IL6 administration to healthy volunteers leads
to a reduction in circulating triglycerides, cholesterol and apolipoprotein B
(Papanicolaou, Tsigos et al. 1996). In addition 1L6 levels negatively correlate with
cholesterol in situations of high stress such as after major surgery (Akgun, Brtel et al.
1998) or myocardial infarction (Brugada, Wenger et al. 1996). Hence the overall effect
of 1L6 on lipid levels might depend on the absolute amount of cytokine present and the
differential effect on the bodies’ other endocrine organs. In the basal state, 1L6 may act
through inhibition of lipoprotein lipase, without influencing cholesterol levels.
Conversely at times of acute physiological stress the entire acute phase response is
stimulated together with the hypothalamic-pituitary axis and sympathetic nervous
system that may then lead to suppression of hepatic lipoprotein production.
L6.5 Psychological Stress And lnterleukin-6
Psychological stress is known to alter immune function with increases in white blood
count and fibrinogen similar to that of an acute inflammatory response. Stress can
al. 1997). IL6 in turn acts to stimulate the hypothalamic-pituitary axis increasing
hypothalamic secretion of corticotrophin secreting hormone and adrenal cortisol
secretion (Mastorakos, Chrousos et al. 1993). There is evidence of increased IL6
signalling in post-traumatic stress disorder that might suggest a role for cytokine
secretion in the modulation of anxiety reactions (Maes, Van Der Planken et al. 1999).
The circulating levels o f interleukin-IRa, IL6, interferon-y (IFN-y) and TNF-a are also
elevated in response to stress-induced anxiety (Maes, Song et al. 1998; Nukina, Sudo et
al. 1998; Goebel, Mills et al. 2000). The highest cytokine concentrations are detected in
those people with the highest perceived stress and anxiety levels (Maes, Song et al.
1998; Song, Kenis et al. 1999). Thus changes in pro-inflammatory cytokines such as
IL6 take part in the homeostatic response to psychological stress, in a similar way to
that following physical injury. In addition, brief episodes of mental stress have also
been shown to induce transient endothelial dysfunction providing a further mechanistic
link between stress and CAD (Ghiadoni, Donald et al. 2000).
Although an association between chronic psychosocial factors and CAD has long
recognized, it is only in recent years that this connection has being clarified (Krantz,
Kop et al. 1996; Hemingway and Marmot 1999; Pashkow 1999; Reyes-Ortiz 1999;
Krantz, Sheps et al. 2000). The first Whitehall Study showed an inverse social gradient
in mortality from CAD among British civil servants (Rose and Marmot 1981). There
were higher rates in men of lower employment grade, but only about a quarter of this
gradient could be attributed to classical coronary risk factors. Analysis of the 5-year
CAD incidence rates from the second Whitehall study showed that compared with men
in the highest grade (administrators), men in the lowest grade had an age-adjusted odds
ratio of developing new CAD of 1.50 (Marmot, Bosma et al. 1997). The largest
contribution to the socio-economic gradient in CAD frequency was from low control at
work. Much of the inverse social gradient in CAD incidence was thus attributed to
differences in psychosocial work environment. The imbalance between personal efforts
and rewards (“poor promotion prospects and a blocked career”) was associated with a
higher risk of CAD (Bosma, Peter et al. 1998). Furthermore, the adverse effect of this
“low-control” of the work situation continues to be manifest into retirement (Marmot
and Shipley 1996). A number of other psychological factors have also been shown to
affect the risk of an acute cardiac event, including anger, hostility (Williams 1987; Kop
1997; Gallacher, Yamell et al. 1999; Ketterer, Fitzgerald et al. 2000) and the pressor