A stu d y of tech n iq u es fo r th e assessm en t and co n tro l of perform ance in p re p a ra tiv e scale liquid chrom atographic se p ara tio n s
A th e sis subm itted fo r th e d eg ree of
Doctor of Philosophy by
Jo sh u a Matthew Philip King B.Eng (Hons.)
D epartm ent of Chemical and Biochemical E ngineering U n iv ersity CoUege London
ProQuest Number: 10045907
All rights reserved
INFORMATION TO ALL USERS
The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed,
a note will indicate the deletion.
uest.
ProQuest 10045907
Published by ProQuest LLC(2016). Copyright of the Dissertation is held by the Author. All rights reserved.
This work is protected against unauthorized copying under Title 17, United States Code. Microform Edition © ProQuest LLC.
ProQuest LLC
789 East Eisenhower Parkway P.O. Box 1346
At th e end of th e room were a couple of long tables sm othered in, a t th e last count, six Macintosh com puters. In th e middle was th e Mac II on which a red w ire-fram e model of his sofa was lazily revolving with a blue w ire-fram e model of his staircase, complete with b an ister rail, rad iato r and fuse-box details, and of course th e awkward tu rn halfway up.
The sofa would s ta r t out spinning in one direction, hit an o b stru ctio n , tw ist itself in an o th er plane, hit an o th er o b stru ctio n , revolve round a th ird axis u n til it was stopped again, th en cycle th ro u g h th e moves again in a differen t o rd er. You d id n ’t have to watch th e sequence for v e ry long before you saw it rep e at itself.
The sofa was clearly stu ck .
A b s tr a c t
In th is stu d y tech n iq u es have been developed to control the perform ance of p rep a rativ e scale liquid chrom atographic separations. Analysis of chrom atograms has been carried out to determ ine how th e individual components comprising th e chromatogram overlap with one another. A mathematical model, consisting of one tem plate function to d escrib e th e elution profile of each individual component, has been developed to d escrib e th e chromatogram and has been successfully applied to chrom atographic data generated by a cation exchange of hen egg white. Param eters for th e functions have been obtained by use of a d irected search optimisation algorithm and re s u lts compared with those obtained by scanning SDS polyacrylamide gels of column fractio n s. The effect of th e size of th e difference between th e actual and predicted chrom atograms on th e accuracy of th e description of th e sep aratio n gained has been in v estig ated in term s of th e su b seq u en t effect on th e quality of possible control decisions made.
Fuzzy logic tech n iq u es have been applied to in te rp re t information obtained from chromatogram analysis in o rd er to identify th e position of th e p roduct within th e overall chromatogram. Sources of failure related to e ith e r chrom atographic phenomena or to mathematical fe a tu re s of th e fuzzy method have been in v estig ated .
Table of Contents Page
1 In tro d u ctio n 19
1.1 In tro d u ctio n to liquid chrom atography 20
1.1.1 Ion exchange chrom atography 20
1.1.1.1 Adsorption eq u ilib ria 21
1.1.2 Affinity chrom atography 22
1.1.3 Gel perm eation chrom atography 23
1.1.4 H.P.L.C - High perform ance liquid
chrom atography 23
1.2 Chrom atographic o u tp u t 24
1.2.1 Causes and effects of inefficiencies 26
1.2.1.1 Extra column e ffects 26
1.2.1.2 In tra column band broadening 27
1.2.1.2.1 Eddy diffusion 28
1.2.1.2.2 Mobile phase mass tr a n s fe r 28 1.2.1.2.3 S tag n an t mobile phase mass tra n s fe r 28 1.2.1.2.4 S tatio n ary phase mass tr a n s fe r 28
1.2.1.2.5 Longitudinal diffusion 28
1.2.2 B and-broadening and scale-u p 29
1.3 Chromatogram analysis: optim isation and
q uality c rite ria 30
1.3.1 Binary system c rite ria 30
1.3.2 Multicomponent c rite ria 31
1.3.3 C riteria for non-G aussian peaks 35
1.3.4 Column efficiency 37
1.3.4.1 Measurement and calculation methods 38
1.3.4.2 Moment method 39
1.3.4.3 Asymmetric method 39
1.3.4.4 Exponentially modified Gaussian methods 40
1.3.4.5 Choice of method 40
1.3.5 Peak overlap detection by m ultidetector
methods 40
1.3.5.1 D ifference and Ratio methods 41
1.3.5.2 D erivative methods 43
1.3.6 Peak identification 45
1.4 Controllable param eters 48
1.4.1 Column loading 48
1.4.1.1 Flow ra te 48
1.4.1.2 Analytical and p re p a ra tiv e
chrom atography - sample size 48
1.4.1.2.1 Volume overload 49
1.4.1.2.2 C oncentration overload 50
1.4.1.2.3 Comparison of modes 50
1.4.2 Elution 50
1.4.2.1 Iso cratic elution 51
1.4.2.2 G radient elution 51
1.4.2.3 Step change 51
1.4.2.4 Multisegment g ra d ie n ts 52
1.4.2.5 Flow ra te 52
1.4.3 S trateg ies 53
1.5 Theories and Models of
Chrom atographic Separations 53
1.5.1 Plate th eo ry 53
1.5.2 Rate th eo ries 54
1.6 E xpert System s 54
1.6.1 The knowledge base 55
1.6.2 The inference engine 56
1.6.3 E xpert system s fo r chrom atography 57
1.6.3.1 CRISE - CRITeria SElection 57
1.6.3.2 SOS - System optim isation system s 57
1.6.3.3 In te g ra te d packages 59
1.6.3.4 ECAT - E xpert chrom atography a ssista n ce
team 60
1.6.3.5 Method validation 61
1.6.3.6 PREOPT 61
1.6.3.7 Dry-Lab 61
1.6.3.8 MABLAB 62
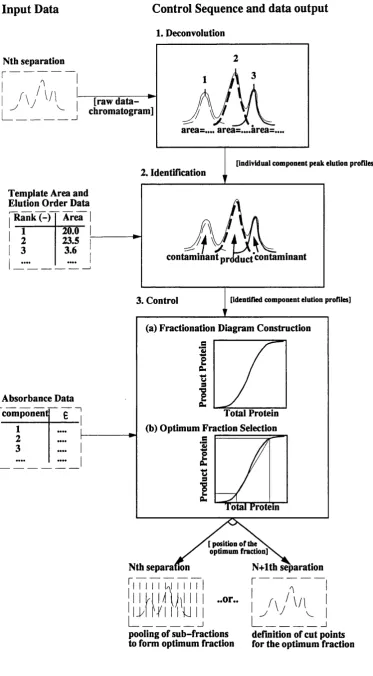
2 Deconvolution 73
2.1 In tro d u ctio n to Deconvolution 74
2.2 C urve fittin g 75
2.2.1 Models 76
2.2.1.1 The exponentially modified Gaussian
function 76
2.2.1.2 The general exponential function 77
2.2.2 O bjective functions 78
2.2.3 C onstraining th e problem 80
2.2.4 Optimisation T echniques 82
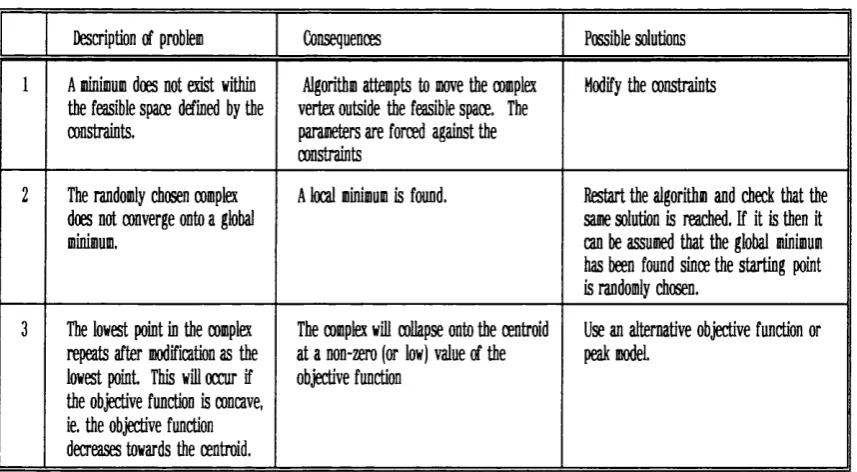
2.2.4.1 Box-Complex optim isation algorithm 82
2.2.4.2 Deconvolution failu re 84
2.2.5 Maximum Acceptable Object Function Size
84 2.2.6 Theoretical Examination of Model
Convergence 86
2.3 Experim ental determ ination of
deconvolution effectiv en ess 87
2.3.1 Choice of system 88
2.3.2 A pparatus 92
2.3.3 Method 92
2.3.4 Chrom atographic method 93
2.3.4.1 Sample p rep aratio n 93
2.3.4.2 Separation conditions 93
2.3.5 Analysis of column fractio n by SDS-PAGE 94
2.3.5.1 Sample p rep aratio n 95
2.3.5.2 Gel p rep aratio n 97
2.3.6 Band quantification by laser densitom etry 97
2.3.7 Experimental R esults 97
2.3.7.1 Chrom atographic Data 97
2.3.7.2 E lectrophoresis Data 98
2.3.8 Deconvolution Analyses of experim ental d ata 98
2.3.8.1 Deconvolution of a single peak 98
2.3.9.1 Conversion of chrom atographic ab so rb an ce profiles to mass based
u n its 101
2.3.9.2 Conversion of electro p h o resis data to
mass based data 103
2.3.10 Comparison of deconvolution re s u lts
with electro p h o resis data 105
2.4 Conclusions 107
3 Product Peak identification 133
3.1 In tro d u ctio n 134
3.2 Fuzzy Logic system param eter selection 135
3.2.1 Reference data s e t selection 135
3.2.2 Membership function selection 136
3.2.3 Matching tech n iq u es 138
3.2.3.1 Matching c riterio n 139
3.2.3.2 Possibility th e o ry 140
3.2.4 Failure modes of fuzzy logic
identification tech n iq u e 140
3.2.4.1 Single Peaks 141
3.2.4.2 O verlapping peaks and combinations of
peaks 142
3.2.4.3 E xtra Peaks 143
3.3 Matching Worked Examples 143
3.4 Egg - white chrom atogram s (deconvolution) 151 3.4.1 Data Handling and Peak In teg ra tio n 152
3.4.2 Fuzzy Logic System P aram eters 152
3.4.3 E gg-w hite S eparation R esults 153
4 Control 164
4.1 Fraction Selection 166
4.1.1 Chromatogram S u b -fractio n s 166
4.1.2 Fractionation Diagrams 167
4.1.2.1 C onstruction of th e Fractionation Diagram 168 4.1.2.2 D isadvantages of th e Fractionation Curve 169 4.1.3 The Fractionation - C oncentration Diagram 169 4.1.4 Selection of th e Optimum Fraction 170
4.2 Effects of deconvolution on fractio n
c u rv es 173
4.2.1 E ffect of deconvolution upon concentration
data 174
4.2.2 Im plications of deconvolution accu racy on
fraction selection 175
4.2.3.1 Selection of c o n stra in ts fo r th e determ ination
of th e optimum fraction 176
4.2.3.2 Selection of th e optimum fractio n 177
4.2.4 Egg-White chromatogram analyses 181
4.3 Conclusions 184
5. Discussion 195
5.1 Separation Perform ance 196
5.2 In teg ratio n of Analysis tech n iq u es 200
5.3 Baseline d rift and noise 203
5.4 Comparison of tech n iq u es developed with o th er c u rre n tly available
softw are 204
5.5 Conclusions 206
6 Conclusions 209
Appendices
Al Computer Programmes and Hardware 216
A2 Deconvolution R esults 243
A3 Fuzzy Logic Matching R esults 250
A4 Nomenclature 255
A5 R eferences 263
L ist of F ig u res Page
F igure 1.1 The Langmuir Isotherm . 66
F igure 1.2 A Typical Chromatogram. 67
Figure 1.3 A schem atic chromatogram showing
th e concept of capacity factor, k. 68
F igure 1.4 The Valley to Peak ratio and Peak
Separation C riterion. 69
F igure 1.5 Two co n g ru en t peaks sep arated by
1.665 sta n d a rd deviations. 69
F igure 1.6 Two in co n g ru en t coincident peaks. 70
F igure 1.7 The A:B ratio. 71
F igure 1.8 The proposed d ata analysis and
control sequence 72
F igure 2.1 A chromatogram co n sistin g of th re e peaks. I t is not possible to determ ine th e peak s ta r ts , e n d s or rete n tio n times because th e peaks
F igure 2.2 The exponentially modified Gaussian
function. 110
F igure 2.3 The General Exponential Function
( a = 1.0 ). I l l
F igure 2.4 The General Exponential Function
( a = 10.0 ). 112
Figure 2.5 The General Exponential Function
( a = 25.0 ). 113
Figure 2.6 Definition of c o n stra in ts for optimisation using th e General
Exponential Functions. 114
F igure 2.7 Box-Comp lex A lgorithm Flow
Diagram. 115
Figure 2.8 T h e f i r s t G a u s s i a n t e s t
chromatogram. 116
Figure 2.9 T h e f i f t h G a u s s i a n t e s t
chromatogram 117
F igure 2.10 The Bio-pilot Chrom atography
System. 118
F igure 2.11 An example of th e complete chromatogram produced from th e
sep aratio n of egg white. 119
F igure 2.12 Two mini gels used to an aly se th e f ir s t egg white sep aratio n (table
F igure 2.13 Deconvolution re s u lts of fittin g a sin g le e x p o n e n tia lly modified Gaussian function and as a single General Exponential Function to
objective function values shown. 121
Figure 2.14 The point d ifferen ces betw een each model peak and th e experim ental
peak shown in fig u re 2.20. 122
Figures 2.15 The f ir s t egg white chromatogram 123 - 2.17 ( Deconvolution re s u lts ). - 125
F igure 2.18 The second egg white chromatogram.
( Deconvolution re s u lts ). 126 F igure 2.19 The th ird egg white chromatogram.
( Deconvolution re s u lts ). 127
F igure 2.20 Densitometry Calibration. 128
F igure 2.21 Extinction coefficients of egg white
components. 129
F igure 2.22 Comparison of th e peaks found by - 2.24 deconvolution and th o se found by
scanning SDS-PAGE gels fo r th e f ir s t to th ird egg white
chromatogram. - 130
132
F ig u re 3.1 The elution o rd e r membership function fo r a refe ren c e s e t peak.
F igure 3.2
(a) The match c riterio n is obtained from th e overlap of a re a membership functions.
(b) A match is considered good if th e match
criterio n is above th e th resh o ld . 159
F igure 3.3 The area data contained in tab le 3.1 may be re p re se n te d by th is
chromatogram. 160
Figure 3.4 Peak a re a membership functions for th e refe ren c e data se t listed in table
3.1. 161
F igure 3.5 The overall match c riterio n fo r th e matching of data in tab le 3.2 with
data in table 3.5. 162
F igure 3.6 The overall match criterio n for th e fo u rth and fifth actual refe ren c e peaks in tab le 3.20 compared with
each refe ren c e peak (table 3.17). 163 F igure 4.1 Flow diagram for th e selection of th e
optimum fractio n based on a
p ro d u ctiv ity th resh o ld . 185
F igure 4.2 The Fractionation Diagram. 186
F ig u re 4.3 The Fractionation - Concentration
Diagram. 187
F ig u re 4.4 A tie line which re p re s e n ts th re e fractio n s with equal purification
fac to rs . 188
F igure 4.6 Fractionation c u rv e s for th e f ir s t
Gaussian te s t chromatogram 190
F igure 4.7 Fractionation c u rv e s for th e fo u rth
Gaussian te s t chromatogram 191
Figure 4.8 The cummulative elution c u rv e for t h e f i r s t G a u s s i a n t e s t
chromatogram 192
Figure 4,9 The cummulative elution c u rv e for th e f o u r t h G a u s s ia n t e s t
chromatogram 193
F igure 4.10 The fractionation diagram s fo r th e firs t, second and th ird egg white
chrom atogram s 194
F igure 5.1 Baseline d rift correction methods 207
F igure A1 The calculation method for th e matching c riterio n used by th e fuzzy logic identification programme ( A 1.3 ). The two portions of th e a re a of in tersectio n a re shown. The in tersectio n point is defined by
List of Tables
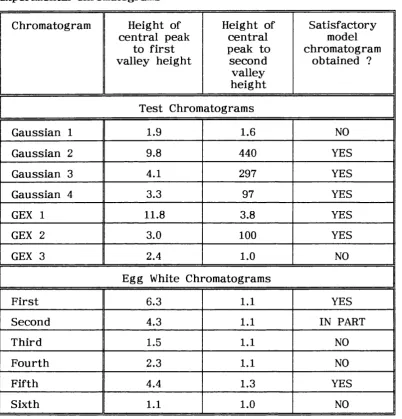
Table 1.1 Quality C riteria for Chromatograms.
Page
32
Table 2.1 Box-Complex Algorithm Failure
Modes. 83
Table 2.2 Physical P ro p e rtie s of Major Egg-
White Components. 91
Table 2.3 G radients used in cation exchange
separation. 94
Table 2.4 Formulation of resolving and
stacking gels. 95
Table 2.5 Formulation of ru n n in g buffer,
staining and destaining solutions. 95 Table 2.6 SDS-PAGE sample ru n n in g b u ffer
composition. 96
Table 2.7 Single Peak Deconvolution R esults. 99
Table 2.8 Egg-White component extinction
coefficients. 103
Table 2.9 Egg-White component dye binding
c h a ra c te ristic s. 104
Table 2.10 Peak to Valley Ratios fo r th e Central peaks in th e te s t and experim ental
chrom atograms. 107
Table 3.1 Reference d ata set: com puter
gen erated chrom atogram s. 144
Table 3.2 Peak data giving a su ccessfu l
Table 3.3 Elution o rd e r matching c rite ria . 145
Table 3.4 Peak a re a matching c riteria. 145
Table 3.5 Overall matching c rite ria . 145
Table 3.6 The transform ed tria l data se t (table
3.2). 146
Tables 3.7 The transform ed d ata se t double
peaks a re a matching c riteria. 147 Table 3.8 Overlapped peaks elution o rd e r
matching c rite ria matched as single
peaks. 147
Table 3.9 Overlapped peaks elution o rd e r
match c rite ria for double peaks. 148 Tables 3.10 Overall match C riteria fo r th e
transform ed d ata set. 148
Table 3.11 Trial d ata se t with an e x tra peak. 149 Table 3.12 Elution O rder Match C riteria. 149
Table 3.13 Peak Area Match C riteria. 149
Table 3.14 Overall Match C riteria. 150
Table 3.15 Expected Egg-W hite Chromatogram Peak Area Data ( R eference Peak
Data ). 153
Table 3.16 D e c o n v o l u t e d E g g - W h i t e Chromatogram Peak Area Data ( tria l Peak data. O bjective function <
Table 4.1 P ro p erties of th e Fractionation -
Concentration Diagram. 170
Table 4.2 Optimum fractio n s for th e te s t
chromatograms. 178
Table 4.3 Actual quality of optimum fractio n s se le c te d f r om d e c o n v o lu tio n
solutions. 179
Table 4.4 Optimum fractio n s for egg white
chrom atograms 182
Table 5.1 Yields for maximum p u rities and P u rities fo r maximum yield fo r te s t chrom atograms
( see section 2.2.6 ) 197
Table 5.2 P u rities a t maximum yields with
differing extinction coefficients. 199
Table 5.3 A typical chrom atographic operation
sequence 200
Table 5.4 Processing times fo r analysis tech n iq u es 201
Tables A2.1 F irst to fifth Gaussian te s t
- A2.5 chromatogrm deconvolution re s u lts .
244 - 245
Tables A2.6 F irst to th ird General Exponential Function te s t chromatogram
- A2.8 deconvolution re s u lts .
245 - 246
Table A2.8 Noisy Gaussian t e s t chromatogram
deconvolution re s u lts . 246
Table A2.9 Noisy General Exponential Function c h r o m a t o g r a m d e c o n v o l u t i o n
Table A2.10 Experimental Deconvolution R esults. 248
Table A3.1 Matching re s u lts fo r th e 1st to 6th 251
A cknow led g e m en ts
I should like to th an k my su p e rv iso rs Dr. N.J. T itch en er- Hooker of U niversity College London and Dr. A.C. Kenney of Drew Scientific, Chiswick, for th e ir advice and encouragem ent d u rin g th e course of th is project.
I am also g ratefu l to th e SERC and OROS In stru m en ts Ltd. for financing th is PhD re se a rc h p ro ject.
I should also like to th an k Mr Martyn Vale and his colleagues in th e E lectronics Workshop for th e ir help with configuration and installation of computer hardw are and softw are, and th e sta ff a t OROS - p articu larly Howard Bates and Penny Vardy for th e ir help with softw are development.
Financial su p p o rt from Biotage UK Ltd. is acknowledged. I am g ratefu l to Dr. P.R. Levison of Whatman Specialty P ro d u cts for helpful discussions and to Pharmacia for loan of equipm ent.
C hapter 1.
1.1 In tro d u ctio n to liquid chrom atography
Liquid chrom atography is a sep aratio n process in which soluble components a re sep arated by ad so rp tio n onto or p a rtitio n with a packed bed of solid material. T here a re sev eral d iffe re n t ty p e s of liquid chrom atography which fall into th e categ o ries - adsorption chrom atography and p artitio n chrom atography. Adsorption chrom atography includes ion exchange and affin ity , while p artitio n chrom atography includes size exclusion ( or gel perm eation ). For each ty p e of chrom atography a d ifferen t ty p e of packing o r media is req u ire d because of th e d ifferen t sep aratio n mechanisms involved. Chrom atography is widely used in th e biotechnology in d u stry . Due to th e relativ ely high cost of chrom atographic media it is generally one of th e last u n it operations. In addition p a rticu lates and o th er m aterials such as lipids, oils and fa ts cause column fouling and in larg e q u an tities blockage of th e packed bed. This th e re fo re means th a t all cells and cell d eb ris must be removed by cen trifu g atio n or filtratio n .
Chrom atography is p articu la rly im portant in th e pharm aceutical in d u stry especially in th e case of protein based d ru g s w here most reg u la to ry a u th o ritie s re q u ire a t least one chrom atographic step to be included in any p rocess ( Sofer and Nystrom, 1989 ), as it is impossible to achieve th e n ecessary p u rity using non-chrom atographic methods.
1.1.1 Ion exchange chrom atography
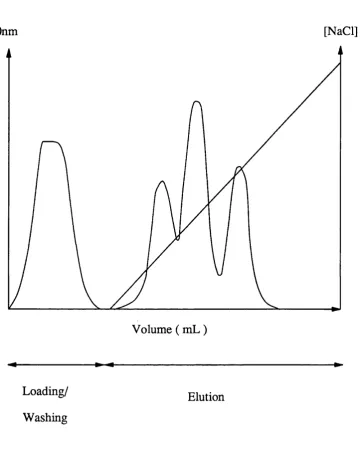
Ion exchange chrom atography is a m ulti-stage process. F irstly th e column is eq u ilib rated to th e loading conditions ie. th e conditions a t which th e functional group and th e protein o r p ro tein s to be bound a re opposite in ch arg e. Next th e mixture is loaded onto th e column and th e column washed with b u ffer to remove any unbound pro tein s. The bound p ro tein s a re th en eluted by changing th e ionic conditions or by changing th e pH. The change in ionic conditions or pH can be c arried out isocratically ie. with a step change to a co n stan t ionic s tre n g th o r by u sin g a g rad ien t ie. a change between two ionic s tre n g th s over a se t time. Each of th ese methods has a d ifferen t effect on th e sep aratio n of components, ie. th e perform ance of th e separation. The o u tp u t resp o n se of th e column will also depend on th e ad so rp tio n isotherm of th e p ro tein s to be sep arated .
1.1.1.1 Adsorption Equilibria
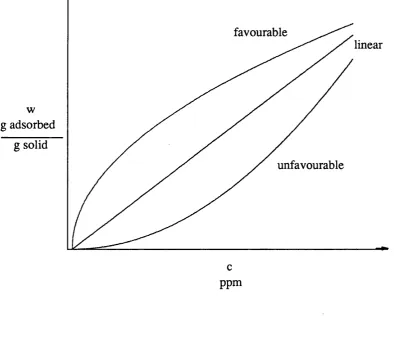
The equilibrium of th e solutes betw een th e mobile and sta tio n a ry phase is often described using th e Langmuir isotherm see equation 1.1
( Langmuir, 1916 ). C ertain assum ptions a re made in th is model: (i) an equilibrium is estab lish ed
(ii) only one p article or species a d so rb s p er site (iii) ad so rp tio n is equally likely on all sites
w = ---— --- (1-1)
( I * K c )
w here w = th e concentration of solute on solid ( g solute ad so rb ed / g solid ) c = concentration of solute in th e liquid ( g solute / g solvent ), b = a c o n stan t related to th e asym ptotic value of th e a d so rb ate
loading ((g solute a d so rb ed .g so lv e n t)/(g so lu te.g ad so rb en t)), and K - 3.n equilibrium co n stan t ( g solvent / g solute ).
When K c » 1, th e isotherm is stro n g ly favourable and when K c < 1 th e isotherm is n early linear. ( See fig u re 1.1. )
In ion exchange chrom atography th e ad so rp tio n isotherm can be considered to be linear over a normal concentration ra n g e used in analytical chrom atography.
The mechanism by which sep aratio n is obtained is one of rev e rsib le adsorption. Separation is possible since su b sta n c es normally have d ifferen t electrical p ro p erties and so a re released a t a d iffe re n t pH or salt concentration. In addition to th e ch arg e effect o th er ty p e s of binding may occur. These a re mainly due to van d e r Waals fo rces and polar in teractio n s. In ion exchange chrom atography most of th e components to be sep arated bind a t th e top of th e column and because of th is columns tend to be sh o rt and have a larg e cro ss sectional area. Once th e desorption step has been completed th e column must be reg e n era ted ie. any rem aining protein must be removed and column r e eq u ilib rated to th e loading conditions. This is usually done by washing with a high salt concentration ( h ig h er th an th a t used in th e elution p rocess ) followed by washing to red u ce th e salt concentration to th e value req u ire d for loading th e column ( re-equiU bration ).
1.1.2 Affinity Chromatography
pH or th e sa lt concentration. Again column reg en eratio n must c arried be out.
1.1.3 Gel perm eation chrom atography
Gel perm eation chrom atography d iffers from both affin ity and ion exchange chrom atography since in th e o ry no chemical or ionic adsorption tak es place. I t relies upon th e size of molecules to be sep arated
( Determann, 1968 ). In p rac tise however some d eg ree of adsorption usually tak e s place. The packing m aterial co n sists of porous beads into which th e species to be sep arated diffuse. The la rg e r molecules a re excluded from th e pores because of th e ir size and so tra v e l down th e column more quickly. Smaller molecules a re retain ed within th e column for a longer period as th e y have a longer path to tra v e l ie. th ey a re held up within th e beads. Molecules which a re medium sized only partially diffuse into th e gel and can be said to be in a sta te of diffusion equilibrium . The components will elu te in o rd e r of decreasing molecular size ie. la rg e st firs t. The param eters which can be varied to improve resolution a re th e bead size, pore size, tem p eratu re ( and hence viscosity ), flow ra te and th e column length. By increasing th e column len g th th e reten tio n time of those molecules which diffuse into th e bead pores is increased in comparison to those molecules which a re not. Reducing th e bead size also improves th e resolution as th e in te r facial a re a of th e gel in creases th u s allowing an equilibrium to be achieved fa s te r b u t causes a h ig h er p re s s u re drop.
1.1.4 H.P.L.C.
The same sep aratio n mechanisms as in low p re s s u re chrom atography may be utilised in HPLC, ie. size exclusion and ion exchange etc. One mechanism which is unique to HPLC is rev e rse d p hase chrom atography w here th e sta tio n ary phase is less polar th an th e mobile phase and is usually only used in HPLC ( Hamilton and Sewell, 1977 ). Chemically bonded octadecylsilane (CDS) is th e most fre q u e n tly used sta tio n ary phase. In adsorption chrom atography w ater is one of th e stro n g e st elution media since it stro n g ly in te ra c ts with th e activ e c e n tre s in silica gel, so th a t adso rp tio n of sample molecules becomes highly re s tric te d and th ey a re rap id ly eluted as a re su lt. The opposite is tr u e of w ater in re v e rse phase chrom atography - w ater cannot wet th e non-polar alkyl g ro u p s and so gives th e slowest elution ra te . Because of th is th e g re a te r th e amount of water in th e elu en t th e longer is th e reten tio n time. A similar list of relativ e affinities to th e su rfa ce as with adsorption to silica may be w ritten b u t with th e d eg ree of affin ity depending on hydrophobicity.
1.2 Chrom atography O utput
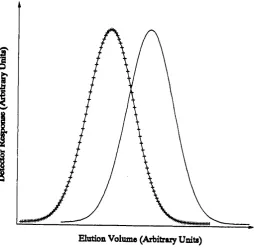
The data produced from a chrom atographic sep aratio n is typically in th e form of a c h a rt known as a chromatogram ( see fig u re 1.2 ). Each peak on a chromatogram re p re s e n ts th e resp o n se of a d etecto r to a change in elu en t composition. This is often th e ab so rb an ce of u ltrav io let radiation b u t o th er means of detection a re available ( see below ). The peak re p re s e n ts a group of molecules which have identical rete n tio n p ro p erties u n d e r th e p a rticu la r sep aratio n mechanism in u se b u t with a v arian ce in accordance with th e peak width. Since th e sep aratio n may be due to one common functional group it is possible fo r each peak to contain a num ber of components ie. more th an one compound may have th e same rete n tio n time. The composition of a peak can only be determ ined by o th e r analytical tech n iq u es eg. fo r p ro tein s gel electro p h o resis.
tailing ^ and anti-Langm uir isotherm s cause fro n tin g ^ ( Meyer, 1988 ). With non-linear isotherm s unlike lin ear isotherm s rete n tio n time will v a ry with sample concentration ( Dose and Guiochon, 1990 ).
An ideal sep aratio n with small sample loadings would produce Gaussian shaped peaks which can be defined mathematically by th e equation :
A; =
a. y 2 %
exp - ( ( V - V i ) 2 a ..
(1.2)
where is th e concentration of component i a t an elution volume V, is th e rete n tio n volume and is th e v arian ce of th e concentration. The param eter is th e a re a of th e peak. Gaussian peaks a re stric tly th eo retical ( Foley and Dorsey, 1983 ) as inefficiencies a re always p re se n t due to e x tra and in tra column so u rces ( see 1.2.1 below ).
A more acc u ra te d escription is th e exponentially modified Gaussian peak, given by equation 1.3 :
A.= X. exp
(
t r f
J
O , X ,/
(1-3)
In th is equation an additional param eter r is in tro d u ced . This param eter d escrib es th e skew of th e peak and is th e decay c o n stan t of an exponential decay function which is convoluted with a p u re Gaussian to produce th e exponentially modified Gaussian function ( Grushka, 1972 ). This model is both th eo retically and experim entally ju stifia b le since peaks a re known to become modified by e x tra column effects, non-equilibrium mass tra n s fe r, and d etecto r resp o n se lag. The u se of th e in co rre ct model may cause an e rr o r in th e v arian ce of up to 50% and an e rr o r in plate count of up to 100% ( Foley and Dorsey, 1983 ).
A num ber of im portant m easurem ents may be tak en from th e chromatogram. The sim plest of th e se is th e rete n tio n volume FJ-, th e volume a t which th e peak maxima occurs.
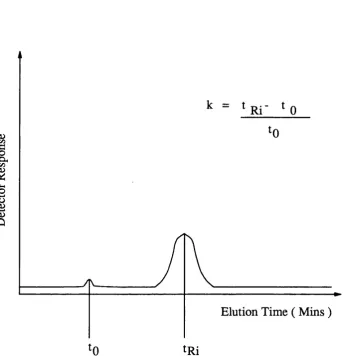
Another m easure of rete n tio n is th e capacity facto r ( see fig u re 1.3 ). This is th e reten tio n volume relativ e to th e reten tio n volume of th e u n retain ed peak {Vg) ( Schoenmakers, 1986 ).
V - V V j
k. = = 1*1 (1.4)
^ Vo
The reten tio n of any p a rticu la r peak may also be m easured relativ e to any o th er peak using th e relativ e rete n tio n param eter.
a.. = ^ (1-5)
" Vni
where th e su b s c rip t j re p re s e n ts th e f ir s t eluting peak and j th e last
eluting peak of a pair. a is th e param eter most d irectly related to selectiv ity and is often itself called selectivity.
It may be ex p ressed in term s of capacity facto r :
=
I
(1.6)1.2.1 Causes of column inefficiency
The next section d escrib es th e causes column inefficiency ie. e ffe cts which give rise to skewed peaks.
1.2.1.1 Extra column effects
= f (1-7)
w here is th e effective total volume and and a re th e volumes due to th e p articles and e x tra column e ffects resp ectiv ely . If th e r e a re o th er co n trib u tio n s to band broadening su ch as an u n su itab le injection valve o r d etecto r th ese may be included in th e equation :
= V„ ): + ( ) +( Ÿ + .... (1.8)
From th e volume, th e peak v arian ce ( tr ) may be calculated which fo r a Gaussian peak is equal to V^/ 4.
1.2.1.2 Intra Column Band Broadening
The v arious so u rces of band broadening which occur within th e column a re discussed in th e following sections. The relativ e im portance of th e se is d ep en d en t upon th e fluid velocity. The concept of red u ced velocity ( defined as v = ud^D^ w here d^ is th e p article diam eter and is th e diffusion coefficient of th e solute in th e mobile phase ) has been used to determ ine th e overall effect of th e se causes of band broadening by relatin g them to th e red u ced plate h eig h t ( red u ced p late h eig h t is defined as h = H/d^. See section 1.3.4 for a definition of p late h eig h t ). Various equations been developed to d escrib e th e relatio n sh ip between red u ced plate h eig h t and red u ced velocity since Van Deemter e t al, 1956 originally su g g ested such a relationship. These in co rp o rate additional term s or tak e into account additional experim ental d ata and th e se have been review ed by Jonsson, 1987. One of th e most widely u sed is th a t of Knox ( Knox, 1986 ) :
h = i4v^ + — + Cv V
1.2.1.2.1 Eddy Diffusion
Eddy diffusion is caused by th e d iffe re n t microscopic flow -stream s th a t flow between th e sta tio n ary phase p articles. As a r e s u lt sample molecules tak e d ifferen t p ath s th ro u g h th e column bed, depending on which flow- stream th ey a re within. The size of band broadening is th u s dep en d en t on th e path len g th and th e width of th e path: a narrow path leads to a high velocity while a wide path leads to a low fluid velocity.
1.2.1.2.2 Mobile Phase Mass Transfer
This re fe rs to band broadening caused by th e flow d istrib u tio n in a flow- stream ie. a sample molecule close to a sta tio n a ry phase p article will tra v e l a s h o rte r d istance compared a t a molecule a t th e c e n tre of th e flow. Again th is re s u lts in band broadening.
1.2.1.2.3 Stagnant Mobile Phase Mass Transfer
With porous packings, th e mobile p hase contained within th e pore is sta g n an t. Sample molecules will d iffuse into and out of th e pores. Due to a v arian ce in th e time sp e n t by sample molecules in th e pores, band broadening will occur since solute molecules of th e same compound will tak e d ifferen t times to p ass th ro u g h th e column.
1.2.1.2.4 Stationary Phase Mass Transfer
If a sample molecule d iffu ses into a pore th e re is a probability th a t th e molecule will p e n e tra te th e sta tio n ary p hase or become attach ed to it in some way. Molecules which do so will move down th e column more slowly in comparison to th o se which diffuse in and o u t of th e pores w ithout becoming attach ed .
1.2.1.2.5 Longitudinal Diffusion
1.2.2 Band-broadening and Scale-up
Typically when a p ro cess chrom atographic sep aratio n is being developed initially lab o rato ry and pilot scale sep aratio n s will be c arried out to determ ine th e c o rre c t o p eratin g conditions for full scale operation. Scaling up th e sep aratio n whilst keeping th e sep aratio n perform ance equivalent to th e smaller scale operation re q u ire s knowledge of th e co n trib u tio n s to band broadening discu ssed in section 1.2.1 above. Generally sep aratio n s a re scaled up by in creasin g th e column c ro ss- sectional area in proportion with th e in crease in flow ra te d esired whilst keeping th e bed h eig h t co n stan t ( providing th a t th e media will not collapse a t wider column diam eters due to lack of wall su p p o rt ). The ra tio of media volume to g rad ien t volume is also k e p t co n stan t ( Sofer and NyStrom, 1989 ). This keeps th e linear flow ra te ( and red u ced fluid velocity if th e same media is used a t both scales ) co n stan t producing th e same num ber of plates available for th e separation. Thus th e sep aratio n within th e column should be equivalent a t both scales.
However o th er facto rs may affect th e perform ance a t th e la rg e r scale due to additional ban d -b ro ad en in g effects. An example of such an effect is th e flow d istrib u tio n system of la rg e r scale equipm ent which is often less efficient in giving even d istrib u tio n acro ss th e bed. G reater axial d isp ersio n will also occur in th e bed and b ran d broadening will also be g re a te r in th e la rg e r column end piece. I t is also im portant to check th a t th e d istan ces between th e column outlet, monitors, and fraction collectors give co n trib u tio n s which a re equivalent, in term s of th e ir band broadening, to th o se a t th e smaller scale.
1.3 Optimisation and Quality Criteria
Many c rite ria have been developed to d escrib e th e quality of a chrom atographic separation. They u se information obtained from a chromatogram such as peak maxima peak minima and points of inflexion to give a numerical value fo r th e quality of th e sep aratio n of mixture components. T here a re two basic ty p e s of c riteria; those which compare th e sep aratio n of a b in ary mixture and th o se which d escrib e th e sep aratio n of a multicomponent mixture.
1.3.1 Binary system criteria
The sim plest and most well known m easure of th e sep aratio n of two peaks in a chromatogram is resolution, R^. This can be defined in two ways ( S nyder and Kirkland, 1979 ):
w here is th e reten tio n volume of component / a n d is th e peak width of th e peak /.
Resolution is a convenient param eter to d escrib e peak sep aratio n since it re la te s th e mean peak width to th e difference in reten tio n volumes - two param eters which determ ine w hether peaks overlap. A resolution, of 2.0, ( ie. Wj + Wj - ~ ) co rresp o n d s to complete peak separation. An a lte rn a tiv e method of calculation is given by :
R , = 1/4 ■ a - 1 ■
f
1
a 1 +
j
. 1 11w here a is th e relativ e reten tio n o r selectiv ity facto r betw een peak i and peak J(k*-/k*^) , ir'j is th e capacity facto r of peak j[(tj-tg )/tg ] and N is th e p late num ber of th e column.
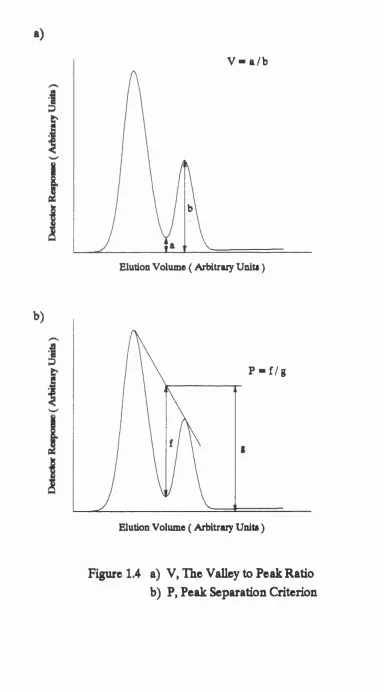
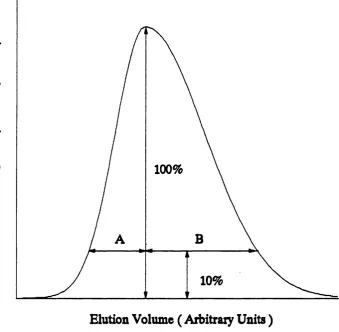
and peak separation P ( Debets, Bajema and Doom bos , 1983 ). These m easures a re not affected by peak asym metry. Peak-to-valley ratio is defined as th e h eig h t of th e valley betw een two peaks divided by th e h eig h t of th e smaller of th e two peaks ( see fig u re 1.4 ). If peaks are completely overlapped th e n V is considered to be u n ity and if th e peaks a re resolved to th e baseline th en V will be zero.
The peak separation, P, is defined as th e d ep th of th e valley between two peaks, below a s tra ig h t line connecting th e two peak maxima, divided by th e h eig h t of th e s tra ig h t line above th e baseline a t th e position of th e valley ( see fig u re 1.4 ). P is said to be zero when th e two peaks overlap completely and u n ity if th e y a re resolved a t th e baseline. All of th e se c rite ria may provide a sa tisfac to ry d escrip tio n of th e separation of two a d ja ce n t peaks. They have however been used, in th e p ast, to describ e multicomponent sep aratio n s. This is achieved by calculating th e value of th e c riterio n fo r each pair of a d ja ce n t peaks. The b est sep aratio n is considered to be th e sep aratio n w here th e most poorly sep arated pair of peaks gives th e h ig h est value of th e criterio n . The u sefu ln ess of th is method is questionable since th e se two component c rite ria only u se information about a d ja ce n t peaks and information about th e d eg ree of separation of o th er peaks in th e r e s t of th e chromatogram is completely ignored ( Debets, Bajema and Doornbos, 1983 ).
1.3.2 Multicomponent C riteria
T a b l e 1. 1 Q u a l i t y C r i t e r i a f o r c h r o m a t o g r a m s
1. Total Overlap
(|) = 2 exp ( -2 )
R- is th e resolution of th e ith p air of peaks
2. Chrom atographic resp o n se function (a)
k
CRF = ( p , ) <=1
(b)
CRF = E P>
p- is th e peak separation of th e ith pair of peaks
(c)
k C/ÎF = 1
* f-1 t is th e sep aratio n time
( d )
k
f-1
(e)
CKF = In
1 = 1
- <%( L )
Pg is th e desired value of peak separation, is th e rete n tio n time of th e la st peak.
(f)
i=l \Po}I + P ( - »i ) is th e maximum allowed rete n tio n time
3. Chrom atographic optim isation function
COF = 5^ W; In i=l
R^ is th e desired resolution 4. Inform ing power
(a)
= E k g ( s , y
i-l
(b)
= t E k g ( a, ) ^ i-l
S- = ( j + w here 0:^ j is th e fractional overlap betw een peak i - l and peak ’ i peak t is th e time or chrom atography
5. Separation number
SN = Y , % ( Pn )' »-l
6. P roduct resolution
Prod,R^ = TÎ R.
is th e resolution of th e ith pair of peaks
Debets e t al (1983) reviewed th ese c rite ria by sim ulating chrom atograms with overlapping Gaussian peaks produced by a com puter program . Each of th e c rite ria were compared with resolution fo r a ran g e of peak sep aratio n s. To find th e peak maxima and minima a peak search ro u tin e was employed to g eth er with a sta n d a rd in teg ratio n package. They found th a t when peaks became resolved to b e tte r th an = 0.5 th e c rite ria improved quickly until baseline resolution was achieved. The c rite ria th en levelled off to a final value. These c rite ria th e re fo re provide a b e tte r estim ate of quality th an does resolution as th e values of th e c rite ria have fin ite limits unlike resolution which may become infinite, with in creasin g peak separation. Once two peaks a re resolved a t th e baseline f u rth e r sep aratio n is to no ad v an tag e and in fact on an economic basis it could be considered th a t f u r th e r sep aratio n is disadvantageous since it will len g th en th e sep aratio n time and so cost more with no increase in p u rity . Debets e t al ( 1983 ) also te ste d multicomponent c riteria. Initially th ey te ste d th ese using a b in ary mixture and found th a t sep aratio n num ber was not a good m easure as th e resp o n se depended stro n g ly on th e way in which th e data was scanned. U ndesired p e rtu rb a tio n s o c cu rred when two maxima were detected on th e chromatogram. Also when two peaks were baseline resolved more irre g u la ritie s occu rred . F u rth e r te s ts were u n d e rta k e n with fo u r components and a v ary in g solvent composition. The re te n tio n time of th e components was assumed to change linearly with changes in solvent composition. They calculated each c riterio n for each selected solvent composition and plotted th e re s u lts . All th e c rite ria found two d ifferen t solvent compositions for which th e sep aratio n was an optimum b u t in th e se c rite ria time was not accounted for. T herefore in all cases th e second of th e two compositions was su p e rio r as it involved th e s h o rte s t time.
co rrectio n s in th e ir calculations. Also when peaks overlap stro n g ly or when peaks become more th an baseline resolved it may actually become impossible to calculate th e c riteria. None of th e c rite ria were able to give an absolute value for th e quality of th e sep aratio n since a good sep aratio n of one pair of peaks may be compensated for by a bad sep aratio n of an o th er pair of peaks. Weighting fac to rs have been applied b u t a re only useful if th e peaks can be identified. When th e reten tio n time of components change with changing solvent composition peaks can only be identified with more complex system s.
In conclusion all th e multicomponent c rite ria rela te d irectly to chrom atographic sep aratio n b u t lack any th eo retical basis. Also if th e elution o rd e r of th e peaks were to change th e n th e re s u lts become in tractab le. Identification of peaks may also be n ecessary to determ ine e ith e r which is th e critical pair of peaks or so th a t th e c o rre c t w eighting may be applied when a multicomponent optim isation criterio n is calculated. 1.3.3 C riteria fo r Non-Gaussian Peaks
proposed, as a general m easure for overlap which is related to th e quantum mechanical overlap in teg ral and can be related to any peak shape. Equations were developed fo r Gaussian, Lorentzian, re c ta n g u la r and tria n g u la r peaks. Chrom atographic d ata was simulated and values of Q co rresponding to d eg rees of peak overlap were calculated for each peak ty p e. Q was found to be zero if th e peaks were not overlapping and u n ity if th e peaks overlapped and were of th e exactly th e same size and shape. Values in between re p re s e n t d iffe re n t d eg rees of overlap. Q was defined by an equation of th e form
[ C ^ C . d t Ÿ
C / dt C / dt
Where and a re th e concentration of th e two components a t a given point on th e chromatogram. The elution time is d escribed by t
P re p ara tiv e chrom atography is often used in a seq u en tial mode with each sta g e providing enrichm ent, ie. p a rtial resolution o r red u ctio n of peak overlap. In analytical chrom atography one is concerned with th e almost to tal resolution of all components for th e p u rp o se of q u an titatio n or identification. P rep arativ e chrom atography th e re fo re re q u ire s a d ifferen t criterio n from resolution since it is possible for a sep aratio n to have zero resolution b u t have a reasonably good fractional overlap. This is dem onstrated by fig u res 1.5 and 1.6 which have equal overlap b u t fig u re 1.6 has a resolution, i?^, of zero. This value of resolution would be of no use in analytical chrom atography since a single peak would be o bserved and th e amounts of th e individual components could not be determ ined. However in p rep a rativ e chrom atography th is may be acceptable since component enrichm ent has o ccu rred . Fractional overlap is difficult to use since th e peak shape must be known before applying th e rele v an t formula and peak shapes a re not available from a chromatogram. A p ro cess known as deconvolution must th e re fo re be c arried out in o rd e r to determ ine peak sh ap es.
Several a ltern a tiv e tech n iq u es have been developed to deconvolute chrom atogram s. The sim plest b u t least a cc u ra te of th e se which a re often used in chrom atographic in teg ratio n packages a re th e p erp en d icu lar drop method ( th e u se of a v e rtica l line draw n from th e valley betw een th e two peaks u n d e r examination to divide th e chromatogram a re a between th e two
components ) and tan g en tial skim method ( w here tan g en tial lines a re draw n on each peak a t th e points of g re a te s t slope to re p re s e n t each peak as a tria n g le ). Papas and Tongas ( 1990 ) in v estig ated th ese tech n iq u es and found th a t in general th e y were in accu rate b u t co n sisten t for an y given se t of peaks. However th e se methods a re not su itab le for determ ination of fractional overlap since th e y do not attem pt to determ ine what peak functions th e chromatogram co n sists of and so a re only useful for estim ating th e amounts of components within th e chromatogram. Several more sophisticated methods have been developed for chromatogram deconvolution. Curve fittin g is mathematically th e sim plest of th ese. Cai and Wu ( 1991 ) and Ju n g and Shin ( 1986 ) used an exponentially modified Gaussian function to d escrib e each peak function within a simulated chromatogram. The param eters fo r th e peak functions were determ ined by minimising th e d ifferen ce between th e chromatogram and th e sum of th e exponentially modified peak functions used to d escrib e th e peak functions using an optim isation algorithm . Vaidya and Hester ( 1984 ) applied a simUar method b u t using a more g eneral empirical peak model ( th e general exponential function - see section 2.2 ). In general c u rv e fittin g has th e ad v an tag e of determ ining both th e am ounts of each component eluted and how th e peaks overlap to produce th e chromatogram. O ther methods exist such as Fourier Transform tech n iq u es ( Nelson, 1991 ), Kalman filte rs ( Hayashi, Shibazaki and Uchiyama, 1987 ) and relaxation based ite ra tiv e methods ( Crilly, 1992 ). In g en eral th ese methods only provide information reg a rd in g th e position ( ie. reten tio n volumes ) and amounts of m aterial eluted r a th e r th an how th e peaks overlap and so a re of no u se in th e determ ination of fractio n al overlap. The overlap of peaks may also be determ ined u sing diode a rr a y d etecto rs ( Fell e t al, 1983 ) which monitor a t sev eral w avelengths sim ultaneously. However th e se re q u ire expensive d e tecto rs in addition to th e com putational requirem ents of c u rv e fittin g . ( Deconvolution of chrom atogram s is d iscussed f u r th e r in C hapter 2. )
1.3.4 Column Efficiency
any indication of w hether th e column is able to se p a ra te a given mixture of compounds and th a t column efficiency is only a m easure of how well th e column has been packed and th e d eg ree of k inetic band broadening. T herefore it can be seen th a t efficiency and perform ance a re two se p ara te term s. Perform ance being th e ability to c a rry o u t a p articu la r separation - high efficiency alone will not g u aran tee th is.
Several param eters affect th e determ ination of column perform ance. These include elu en t composition, viscosity, velocity, tem perature, column length, packing ty p e, packing size and m easurement and calculation method chosen. I t is th e re fo re v ital to s ta te u n d e r what conditions efficiency has been calculated, as well as what method has been used. Many methods make no e ffo rt to remove any co n trib u tio n obtained from th e liquid chrom atography system and so th e value fo r efficiency obtained is th e to tal efficiency for th e whole system . One m easure of p articu lar in te re s t is H or HETP ( h eig h t equivalent to a th eo retical plate ) as it a d ju s ts for th e len g th of th e column. HETP { or H ) refle cts th e ratio of column efficiency to column len g th whilst th e o th er u ses th e in v erse ratio. ( Generally it is not used as chrom atographers a re conditioned to p re fe r a high value of N ra th e r th an a low value of H \ )
1.3.4.1 Measurement and calculation methods
In analytical chrom atography peaks a re often assum ed to be Gaussian in shape and because of th is N is defined as follows
N = (1-13)
o2
Where PJ. is th e reten tio n volume and é is th e v arian ce in volume u n its. (} can be w ritten in term s of th e peak width divided by a constant. The value of th e co n stan t depends upon w here th e w idth is m easured on th e peak, ie. 4%, 50%, or 100% of peak height. This can be incorporated into th e above equation
a
Where a is a co n stan t, th e value of which depends on th e p ercen tag e of th e peak h eig h t a t which th e width, is m easured.
Several approaches a re available fo r th e calculation of peak variance. The following is a list of d ifferen t calculation methods: inflection method, width a t half height, ta n g e n t method, h e ig h t/a re a method, moment method and asym m etry-based methods. Biddlingm eyer and Warren (1984) teste d each of th e calculation methods for accu racy and consistency. The moments and asym metric methods were found to be th e most accu rate. The sim plest methods, ie. m easuring th e width a t half h eig h t and m easuring th e width using tan g en tial lines p ro jected to th e base line, were found to be th e least accu rate, b u t w ere th e easie st to calculate.
1.3.4.2 Moment Method
In th is method no assum ptions a re made about th e peak shape. The c h a ra c te ristic s of th e peak a re ex p ressed in term s of sta tistic a l moments. Peak area is th e zero th moment, th e f ir s t moment is th e mean (for Gaussian peaks th is is th e peak maximum) and th is o ccurs a t th e c en tre of mass. The second moment is th e peak v arian ce and th e th ird and fo u rth moments a re m easures of skew ness. The second moment may th e re fo re be used in place of peak v arian ce to calculate N.
1.3.4.3 Asymmetric Methods
Two ty p e s of asym metric methods a re available. The sim pler of th e two u se s an empirical ratio of th e peak w idths e ith e r side of th e peak maxima m easured a t 10% peak h eight. This ratio has th e a d v an tag e of satisfy in g th e ch ro m ato g rap h er’s in tu itiv e idea of peak asym m etry and is easy to m easure. I t however lacks any th eo retical basis, b u t th is should not preclu d e its use.
The o th er method u se s an exponentially modified Gaussian peak to r e p re s e n t peak skew ness. This function c o n sists of a Gaussian function combined with an exponential decay function. The to tal v arian ce is;
= " 'o + T'
1.3.4.4 Exponentially Modified Gaussian Methods
The u se of exponentially modified Gaussian peaks has been investigated as a more accu rate descrip tio n of chrom atographic peaks ( Jeansonne and Foley, 1992, Foley and Dorsey, 1983, Grushka, 1972, and Yau, 1977 ). The exponentially modified Gaussian model may be ju stifie d since it is known th a t in tra and ex tra column effects have th e effect of skewing peaks causing th e changes in v arian ce produced by exponentially modified Gaussian peak functions. The u se of th e c o rre c t model is im portant - th e u se of th e in co rrect model may lead to g re a t inaccuracies in plate counts and variances. The most fre q u e n tly used formula fo r th e num ber of plates for real peaks is :
BIA + 1.25
This formula includes th e asym m etry ( using th e B/A asym m etry ratio as shown in fig u re 1.7 ) of th e peak in th e calculation of th e num ber of th eo retical equilibrium sta g es ( ie. plates ) co rresponding to th e operation of th e column.
1.3.4.5 Choice of Method
Since th e re a re many d ifferen t methods of calculating efficiency which do not always give equivalent values it is im portant to decide why efficiency is to be m easured before choosing which method to use. Generally m ethods which a re least sen sitiv e to peak asym m etry a re th e b e st choice b u t if th e only objective is to monitor th e efficiency of a column over a period of time th e n any of th e methods may be useful. If comparisons a re to be made with columns of d ifferen t size or d ifferen t packings th en a more acc u ra te method which gives a c o n sisten t r e s u lt is req u ire d . With any evaluation of column efficiency th e capabilities and limitations of th e calculation methods used must be tak en into account. When evaluating re p o rte d efficiencies contained within lite ra tu re or commercial information it is difficult to make any d irec t com parisons due to th e lack of any sta n d ard isa tio n of experim ental conditions.
1.3.5 Peak overlap detection by multi detector methods
The most fre q u e n tly used d etecto r in chrom atography is th e u ltrav io let d etecto r. Most of th ese d e tecto rs o p erate a t fixed w avelength and
m easure th e absorbance of th e eluent. O ther more advanced UV d etecto rs can d etect a t more th an one w avelength - both dual and m ultiwavelength a re available, th e most sophisticated being th o se capable of scanning over a spectrum of w avelengths. These d etecto rs a re known as photodiode a rra y s . UV d etecto rs a re relativ ely in sen sitiv e to flow ra te or tem p eratu re changes b u t th e choice of mobile phase is re s tric te d as th ey must be tra n s p a re n t a t th e w avelength a t which th e d etecto r is operating ( Hamilton and Sewell 1977 ).
Another ty p e of d etecto r is th e re fra c tiv e index d etecto r. These work using a differential technique in which th e sample’s re fra c tiv e index is compared with th a t of th e eluent. Any su b sta n c e which has a re fra c tiv e index significantly d ifferen t from th e elu en t may be d etected b u t th e method is v e ry sen sitiv e to changes in elu en t and so is u n su itab le for g rad ie n t elution unless solvents a re chosen with identical re fra c tiv e indices. I t is also v e ry tem p eratu re d ep en d en t ( E n g elh ard t 1979 ). Compounds may be d etected using fluorescence. The p ro cess stream containing th e su b stan ce to be d etected is f ir s t excited by UV radiation and monitored for emission a t an o th er w avelength. Fluorescence detection is v e ry specific b u t fluorescence may be su p p re sse d or quenched by th e p resen ce of contam inants. The se v ere effect th a t su ch contam inants have on th is detection system means th a t carefu l selection of column conditions must be c arried out.
1.3.5.1 Difference and Ratio Methods
More th an one d etecto r may be used fo r detection. This has th e ad v an tag e of providing more information about th e sep aratio n eg. if th e sep aratio n contains one su b stan ce which flu o resces th en th e d eg ree of overlap may be m easured by comparing th e chromatogram obtained from UV with th a t from fluorescence.
RAT = (A1/A2) when A2 > Al > th resh o ld RAT = 2 - (A2/A1) when A1 > A2 > th resh o ld RAT = 0.0 when A1 and A2 < th resh o ld
RAT = -0.1 when A1 < th resh o ld and A2 > th resh o ld or when A2 < th resh o ld and A1 > th resh o ld .
Where A1 and A2 a re th e ab so rb an ces m easured a t th e two d ifferen t w avelengths a t any given point in th e chromatogram.
Initially th ey teste d th e ratio method using simulated fully-resolved peaks. They found th a t th e ratio produced block shaped resp o n ses when th e ratio was plotted ag ain st th e elution time. They also showed th e ratiogram provided information when th e simulated peaks were overlapped. When th e two peaks were fully resolved two blocks were shown on th e ratiogram co rresponding to each of th e peaks. Once peaks were overlapped th e blocks combined with a ste p change from th e fir s t value to th e second. As th e d eg ree of overlap increased th e change from th e f ir s t to th e second became smoother un til th e two peaks coincided exactly when a single block o ccu rred . These re s u lts were fo r p e rfe ct data ie. no noise, or baseline offset, or time delay between signals. Baseline offsets may occur because of incompletely co rrected background signal and may be variable or constant. They have th e effect of d isto rtin g th e blocks into asym metrical shapes. This effect can be red u ced by increasing th e th resh o ld value b u t th is has th e effect of making th e p u rity check of th e peak more difficult. Any d isto rtio n of th e ratiogram caused by time delays may be removed by a d ju stin g th e d etecto rs so th a t th ey a re m easuring a t th e same w avelength and determ ining th e time delay between th e two by comparing chrom atograms. This time delay can th en be tak en into account. Tailing may also d isto rt th e ratiogram and lead to m isin terp retatio n s eg. more components ap p ear th a n a re actually p resen t. Again c le are r data may be gained by in creasin g th e th resh o ld value b u t th is red u c es th e amount of information available. The ability of th is method to identify overlapping peaks also depends on th e difference between th e RAT of th e two d ifferen t peaks. The g re a te r th e difference th e sim pler th e recognition of overlap. Drouen, Billiet and de Galan (1984) found th a t if th e difference in th e RAT of two components was less th an 0.05 th en th e peak sep aratio n must be g re a te r th an 2 o f o r overlap to be identified. This is still b e tte r th an single w avelength detection. The
selection of w avelengths for m ultichannel monitoring is im portant and it should be made to gain as much information as possible. Mathematical tech n iq u es ( key se t facto r analysis (KSFA) ) have been developed to determ ine th e optimum w avelength se ts ( Warren, Bidlingmeyer and Delaney, 1987a and Warren, Bidlingmeyer, and Delaney, 1987b ).
Drouen, Billiet, and de Galan (1985) extended th e ir previous work on dual w avelength d etecto rs to m ultiwavelength d etecto rs such as diode a rra y d e tecto rs which allow rap id scanning and a more detailed analysis of overlapped peaks to be obtained by m easuring th e ab so rb an ce spectrum of th e eluent. This in tu r n gives more a cc u ra te analysis and identification of sev erely overlapping components. However with proteins, since th e ir absorbance spectrum s a re often v e ry similar, diode a rra y method would be essen tial ( C arr e t al, 1988 ).
A facto r analysis method has been developed for th e resolution of unresolved peaks ( Sakema, e t al, 1990 ). Using th is method estim ates can be made of th e elution profiles of components eluting a t th e risin g or tailing edge of a peak. With unresolved th re e component peaks, peak a re a of th e th re e components were determ ined to within 10%. Methods have been developed ( Marr, Seaton, Clark and Fell, 1990 ) using modern m utliw avelength d e tecto rs for examining th e homogeneity, id en tity and p u rity of peaks, based on work for stopped flow conditions ( Ostojic, 1974 ).
Krstulovic, Rosie and Brown (1976) used th e ratio of peak a re a s to id en tify chrom atographic peaks which were fully resolved b u t p u rity was not considered as th e method was p u rely for identification p u rp o ses. O ther methods have been devised fo r analysis of chrom atogram s m easured on two w avelengths. Li and A rrington (1979) used th e difference betw een chrom atogram s for th e identification of components as well as q uantification of components in overlapped peaks. They were able to find a relatio n sh ip between th e amount of su b sta n c e p re s e n t and th e d ep th of th e valley caused by th e su b tra ctio n of th e chromatograms.
1.3.5.2 D erivative Methods
d eriv ativ e of th e function has two maxima and one minimum which a re positioned a t ( th e maxima ) and x=x^ ( th e minimum ). Where x^ is th e Gaussian peak’s c e n tre of mass and o is th e sta n d ard deviation. The ratio s of th e maxima and minimum a re in d ep en d en t of th e peak height. The ratio s of th e second d eriv ativ e maxima and minimums were defined as follows:
R| maximum on fro n t of peak / minimum Rg maximum on tail of peak /minimum
Rj maximum on fro n t of peak / maximum on tail of peak
A g rap h of R^ and Rg was obtained for v arious peak h eig h t ratio s of overlapping peaks onto which co n to u rs of resolution were draw n. Once c u rv e s such as th ese a re produced it is th en possible to id en tify peaks if th e peak width a re known since th e second d eriv ativ e ratio s will give information about th e peak h eig h t and resolution. This method has th e ad v an tag e of not being too d ep en d en t upon th e exact sh ap e of th e chrom atographic peaks b u t it is n e ce ssa ry th a t th e data is of a good and rep ro d u cib le quality and so may not be of g eneral use. The main disadvantage of th is method is th a t calibration sep aratio n s must be c arried out to d etect overlapped peaks and th e extension of th is method to multicomponent system s would re q u ire a larg e database of information about overlap of each peak with each o th er peak, eg. a combination of four peaks would re q u ire information about six perm utations of overlapping peaks. Measurement of ab so rb an ce ratio s would re q u ire much less information in comparison - th e value of th e ratio for each p u re peak which could be gained from one sep aratio n with all peaks fully resolved. A nother disad v an tag e is th a t a num erical d ifferen tiatio n m ust be c arried out which may not be a cc u ra te depending upon th e quality of th e data. D ifferentiation also causes magnification of noise in th e signal. The limits of th is method have been found to be d ep en d en t on th e peak h eig h t ratio and th e relativ e sep aratio n ( G rushka and Israeli, 1990 ).
1.3.5.3 Methods suitable for general use
Ideally th e method chosen for ju d g in g th e quality of th e sep aratio n should be g eneral enough to be applied to sep aratio n s based on th e same mechanism and on o th er mechanisms. Fluorescence detection is heavily d ep en d en t upon p ro p erties of th e components within th e m ixture to be sep ara ted and th e re fo re will not be considered. I t is also su b je c t to