Copyright
q
1997, American Society for Microbiology
Evaluation of a Quantitative Competitive PCR Assay for
Measuring Herpes Simplex Virus DNA Content
in Genital Tract Secretions
ANN HOBSON, ANNA WALD, NANCY WRIGHT,
ANDLAWRENCE COREY*
Departments of Laboratory Medicine and Medicine, University of Washington, and the Program in Infectious Diseases,
Fred Hutchinson Cancer Research Center, Seattle, Washington
Received 9 August 1996/Returned for modification 17 September 1996/Accepted 2 December 1996
Previous studies have shown an association between the approximate titer of herpes simplex virus (HSV)
DNA in clinical specimens and the ability to isolate HSV from genital secretions. To control for variance in
amplification conditions, we developed a competitive quantitative PCR (QC PCR) for the detection of HSV
DNA. The assay accurately measured from 10 to 10
6copies of HSV DNA. We compared the QC PCR with our
previous semiquantitative detection method and found concordance for 61 of 63 positive specimens. We also
evaluated the HSV DNA content from individual swabs of genital secretions obtained from individual sites of
the genital tract (cervix, vulva, and rectum) with that from one swab with secretions from all three sites. The
concordance for detecting HSV DNA was 91%; for only 4 of 143 collection days was there a >1 log difference
between the two collection methods. A single swab with secretions from all three genital sites and evaluated in
a QC PCR format can accurately measure the frequency of subclinical and clinical shedding of HSV and the
titer of HSV shed from the genital region. Such an approach should be very useful in the evaluation of antiviral
chemotherapy for HSV.
Detection of viral nucleic acids in clinical specimens has
been shown to be associated with replicating virus, and in vivo
and in vitro inhibition of viral replication by an antiviral
com-pound can be monitored by quantitation of the nucleic acids (5,
7, 11). We have previously shown the association between a
semiquantitative measurement of herpes simplex virus (HSV)
DNA levels and the ability to isolate HSV in genital lesions (4).
More recently, we have shown the association between the
approximate titer of HSV in clinical specimens and the
isola-tion of HSV in tissue culture (11). To control for variance in
amplification conditions on a run-to-run and sample-to-sample
basis, most authorities have recommended that quantitative
PCR-based assays be performed in a competitive format (1, 6).
Competitive strategies involve coamplification of a range of
concentrations of a modified PCR product with the target
DNA and determination of the point at which the modified
DNA and the target DNA give the same amount of PCR
product. Ramakrishnan et al. (10) have described a
quantita-tive competiquantita-tive PCR method for HSV type 1 (HSV-1) DNA
detection involving a modified PCR product with a single base
change that produces a restriction enzyme site, which allows
for the differentiation between products on the basis of the
product size after digestion.
We describe a competitive quantitative (QC) PCR strategy
for HSV-2 DNA detection in which a PCR product from which
82 bp have been deleted is used as a competitor. The assay
accurately measured amounts of HSV-2 DNA from 10 to 10
6copies. We compared the results obtained by this assay with
those obtained by our previously published method (4), in
which dilutions of positive specimens are run by PCR and the
results are visually compared with the results from separate
PCR assays with known amounts of HSV-2 DNA. During this
study, we also compared the results obtained with swabs of
genital secretions from individual sites of the genital tract
(cervix, vulva, and rectum) with those obtained with a single
swab containing secretions from all three sites.
MATERIALS AND METHODS
Collection of specimens.We selected one woman who, in previous studies, had been shown to have both frequent clinical and subclinical genital herpes (12). She gave informed consent for a protocol approved by the University of Washington Human Subjects Review Committee. We used a prospective crossover design in which the patient obtained specimens on a daily basis for 174 consecutive days. During the initial 80 days she was treated with acyclovir at 800 mg three times a day, and during the subsequent 94 days she received no antiviral therapy. This study design was selected to evaluate the specificity and utility of the QC PCR assay during a period of limited viral shedding compared to those during active viral shedding. On each day, the woman collected samples on four separate swabs upon awakening. Swabs were taken first from individual sites: the cervicovaginal, the vulvar, and the perianal areas, in that order. Then, a single swab was used to collect secretions from all three sites in the same order (a “mixed” swab). The swabs were placed in digestion buffer and were sent to the PCR laboratory, where they were stored at2208C until they were processed. DNA was prepared as described previously with phenol and phenol-chloroform extraction and eth-anol precipitation (2). Samples were obtained on 143 of the 173 study days.
PCR methods.The PCR amplified part of the HSV-2 glycoprotein B gene, as described previously (3). Briefly, the primers used were HSV2a-1 (59-CTGGTC AGCTTTCGGTACGA) and HSV2a-2 (59-CAGGTCGTGCAGCTGGTTGC). Amplification was for 35 cycles, and 20% glycerol was included in each reaction mixture. Products were detected by liquid hybridization, gel electrophoresis (on 2% agarose for standard noncompetitive PCR or 3% Nusieve–1% agarose for QC PCR) and autoradiography. In select experiments with high DNA input (103 copies or greater), liquid hybridization was not performed and gels were stained with ethidium bromide.
Synthesis of competitor DNA.For the QC PCR method, a shortened version of the HSV2 PCR product was made. The modified product had the HSV2a-1 and HSV2a-2 primer sequences at each end but was 82 bp shorter (260 bp compared with 342 bp for the HSV2 PCR product). To synthesize this product, a new primer was used in place of HSV2a-1. This primer (HSVQC) contained 18 bases of the internal HSV2a PCR product sequence along with the HSV2a-1 primer sequence and 10 bases adjacent to it, attached to the 59end (Fig. 1). Amplification of HSV-2 DNA (105copies) with this modified primer and the HSV2a-2 primer generated a 260-bp product. This product was separated on 3% Nusieve–1% SeaPlaque and was visualized by ethidium bromide staining. The band was cut out, melted at 658C, and diluted 53104-fold in 10 mM Tris (pH 8.0). A total of 5ml of the diluted DNA was amplified with the HSV2a-1 and HSV2a-2 primers to give the final 260-bp competitor DNA. This product was again gel purified to remove the primers.
* Corresponding author. Mailing address: 1124 Columbia St.,
M-115, Seattle, WA 98104. Phone: (206) 6702. Fax: (206)
667-4411.
548
on May 15, 2020 by guest
http://jcm.asm.org/
FIG. 1. Construction of the competitor DNA. HSV-2 DNA was amplified with the HSV QC primer and the HSV2a-2 primer to give the 260-bp competitor DNA.
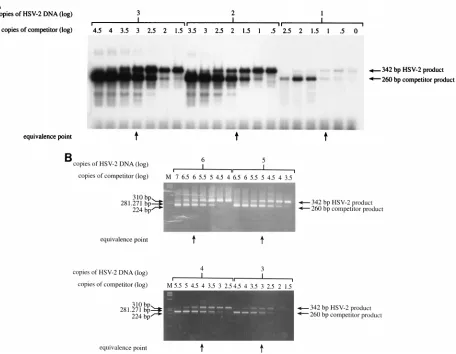
FIG. 2. Serial 10-fold dilutions of HSV-2 DNA from 106to 10 copies were each amplified with 7 different 3.16-fold (half-log) dilutions of competitor DNA. Products were detected by liquid hybridization, gel electrophoresis, and autoradiography (A) or by gel electrophoresis and ethidium bromide staining (B). The equivalence points (arrows), at which the 342-bp band and the 260-bp band have equal intensities, were determined by visual inspection. For panel A, the equivalence point in one dilution set was between the two competitor concentrations.
V
OL. 35, 1997
QUANTITATIVE COMPETITIVE PCR FOR HSV DNA
549
on May 15, 2020 by guest
http://jcm.asm.org/
[image:2.612.83.539.338.692.2]The purified competitor DNA was then quantitated by PCR. Dilutions of this DNA were amplified with the HSV2a-1 and HSV2a-2 primers, and the products were detected by liquid hybridization. An estimate of the copy number was made by comparison of the intensity of the bands obtained with those generated by amplification of known amounts of HSV-2 DNA. The precise copy number was then determined by coamplification of serial twofold dilutions of the DNA with 1,000 copies of linearized plasmid pHS108, which carries the entire glycoprotein B gene (8). Products were detected by liquid hybridization. The dilution giving equivalent intensities of the two bands (the 342-bp HSV2a product from the plasmid and the 260-bp product from the competitor) was then adjusted to contain 1,000 copies in 5ml (the volume used in the PCR).
HSV DNA PCR of specimen DNA.Purified DNA from each specimen was first run in a noncompetitive HSV PCR to check for inhibition of the reaction by the specimen and to obtain an estimate of the HSV copy number. To detect any inhibition in the clinical sample, 50 copies of a modified PCR product, HSV2a-Dros, with aDrosophilaprobe region in place of the HSV probe region, were added to each reaction mixture to monitor for inhibition of the reaction by the DNA preparation (3). All specimens in this study were positive for the Drosoph-ilaprobe, indicating no inhibition of the reaction. Copy number was determined by comparing the intensity of the band with the intensities of bands from known amounts of HSV-2 DNA (10 to 104copies) run in separate tubes. Specimens with copy numbers of greater than 103per reaction (63 specimens) were diluted in serial 10-fold dilutions and were rerun. In addition, for these 63 specimens, a dilution of the DNA estimated to contain from 102to 105copies was run in a QC PCR assay with four different 10-fold dilutions of competitor with from 102to 105 copies. The copy number of the competitor giving a 260-bp band with an intensity equal to that of the 342-bp band was determined, and this was considered to be the copy number of the diluted target DNA.
RESULTS
QC PCR method validation.
We initially evaluated the
lin-earity of the QC reaction by coamplifying the competitor with
known copy numbers of authentic HSV-2 DNA. This HSV-2
DNA had been purified by phenol and phenol-chloroform
extraction and ethanol precipitation, as described previously
(2), from human diploid fibroblasts infected with a laboratory
strain of HSV-2. The DNA level was quantitated by repeated
comparison with known amounts of pHS108 DNA by PCR. In
the validation experiment, 10-fold dilutions containing from 10
to 10
6copies of HSV-2 DNA were coamplified with 7 half-log
(3.16-fold) dilutions of the competitor DNA (Fig. 2). Products
were detected either by ethidium bromide staining of gels or,
for lower copy numbers, by liquid hybridization. In each case,
the copy number of the competitor giving equivalent intensities
of the 342- and 260-bp bands was determined. In each case it
was found to equal the known copy number of the added
HSV-2 DNA.
Detection of HSV DNA by noncompetitive PCR.
Figure 3
illustrates the detection of HSV DNA in the genital tract by
anatomic site, therapy, and day of observation. As described in
Materials and Methods, specimens with copy numbers greater
than 10
3were diluted serially and rerun to get an estimate of
the copy number. HSV was isolated in tissue culture on 2 of 72
days while the study subject was on acyclovir therapy, whereas
it was isolated on 21 of 71 days while she was on no therapy.
One clinical recurrence lasting 4 days was noted while she was
on acyclovir therapy, whereas three recurrences, lasting a total
of 33 days, occurred while she was on no therapy. HSV DNA
was detected by standard PCR on 7 of 72 days while the subject
was on acyclovir therapy, whereas it was detected on 51 of 71
days while she was on no therapy.
Comparison of QC PCR with dilution titers of HSV DNA.
We then evaluated all 63 of the 137 separate samples in which
HSV DNA was detected in the noncompetitive assay at
$
10
3copies per reaction (after dilution of the specimen DNA when
necessary). A comparison of the QC PCR with the dilution
method is presented in Table 1. There was excellent
concor-dance between the two methods, with identical titers being
present in 36 of the 63 clinical specimens and titers within 1
FIG. 3. Detection of HSV by culture and HSV DNA PCR in a woman initially treated with acyclovir and then followed without antiviral therapy. Bars indicate the amount of HSV DNA detected in cervical, vulvar, and rectal samples (specimens with 1 to 10 copies of HSV DNA were plotted here as 10 copies).1, positive culture; the presence of genital lesions is also indicated.3, days on which a specimen was not available.
on May 15, 2020 by guest
http://jcm.asm.org/
dilution of each other (1 log) being present in 61 of 63
speci-mens. For both of the other two specimens, QC PCR showed
the copy number to be 2 logs higher than that by the dilution
method.
Comparison of single-site versus multiple-site swab
speci-mens.
We then compared the accuracy of detecting HSV DNA
from a swab containing secretions from all three anatomic sites
with that of detecting HSV DNA from the swabs containing
secretions from the individual sites. The results for the
detec-tion of HSV DNA were concordant between the two collecdetec-tion
methods on 91% of the days (Table 2). Both collection
meth-ods were negative for HSV DNA on 80 days and positive on 50
days. On 8 days, swabs with specimens from individual sites
contained HSV DNA, while the swab with specimens from all
three sites did not, and on 5 days the swab with specimens from
all three sites contained HSV DNA, while the swabs with
specimens from individual sites were negative for HSV DNA.
Of the 13 days on which discrepant results were obtained, for
4 days PCR product at
,
10 copies in the positive sample was
involved.
Table 3 evaluates the titers of HSV in samples from 143 days
in which both mixed and individual swab specimens were
col-lected. The concordance between the two collection methods
was again excellent. On 112 of 143 collection days the titers for
the swabs containing specimens from all three sites were
iden-tical to the highest titer for a swab specimen from an individual
site, and on only 5 of 143 collection days was there
.
1 log
difference between the titers obtained by the two collection
methods.
DISCUSSION
This study extends current methods of accurately
quantitat-ing HSV DNA levels in swab specimens. We have developed a
QC PCR method which can be used to quantitate HSV levels
over a large linear range and which more completely controls
for the difference in amplification variability seen with diverse
clinical specimens. Quantitation of HSV DNA levels in clinical
specimens is particularly helpful in the evaluation of the
re-sponse to antiviral chemotherapy (5). When subjects have
clin-ical lesions, HSV DNA is detected by PCR in high copy
num-bers. In this setting, the QC PCR offers advantages because of
the large linear range and could be a useful tool for evaluating
the rapidity with which antiviral agents influence HSV
clear-ance from mucosal sites.
Another point of interest in our study was the high
concor-dance of the results for swabs with secretions collected from
several anatomic areas compared to those for swabs with
se-cretions taken from individual sites. Our subject collected
sam-ples very systematically. Our subject obtained all samsam-ples upon
awakening in an established order and after a detailed
instruc-tion period. Sample collecinstruc-tion is an important aspect in
eval-uating any diagnostic assay for HSV infection (9). Both the
frequency of detection and the titer of virus obtained were
similar between swabs containing specimens from all three
sites and those containing specimens from a single site.
Dif-ferences of
.
1 log in the amounts of virus were seen in only 5
of 143 specimens. Thus, it appears that sampling of sites
col-lectively may be as accurate a means of detecting the presence
of HSV DNA in the genital tract on a single day compared to
sampling of individual sites. This is an important advance for
studies of systemic antiviral agents.
In summary, we have described a QC PCR assay which
accurately quantitates HSV DNA levels. While this assay is
labor-intensive, it may be useful in research settings.
More-over, it appears that a single swab specimen with secretions
taken from all three genital sites and evaluated in a QC PCR
format can qualitatively and quantitatively establish the
fre-quency and titer of subclinical and clinical shedding of HSV in
the genital tract.
ACKNOWLEDGMENTS
This work was supported by National Institute of Health grant
30371. Anna Wald is the recipient of a 1994–1996 American Social
Health Association Postdoctoral Research Fellowship in Sexually
Transmitted Diseases.
REFERENCES
[image:4.612.56.298.90.189.2]1.Becker-Andre, M., and K. Hahlbrock.1989. Absolute mRNA quantification using the polymerase chain reaction (PCR). A novel approach by a PCR aided transcript titration assay (PATTY). Nucleic Acids Res.17:9437–9446. 2.Cone, R. W., A. C. Hobson, Z. Brown, R. Ashley, S. Berry, C. Winter, and L. Corey.1994. Frequent detection of genital herpes simplex virus DNA by polymerase chain reaction among pregnant women. JAMA272:792–796.
TABLE 1. Comparison of QC PCR with dilution titers for HSV-2
DNA in the genital tract
Titer by QC PCR
No. of samples with the following titers by dilutiona:
103 104 105 106 107 Total
10
31
0
0
0
0
1
10
40
7
3
1
0
11
10
50
6
21
12
1
40
10
60
0
2
7
2
11
10
70
0
0
0
0
0
Total
1
13
26
20
3
63
aBoldface numbers indicate equivalence.
TABLE 2. Comparison of samples containing secretions from all
three sites and samples containing secretions from individual sites
for detection of HSV-2 DNA in the genital region
Result for specimen
No. of days with the following result for
individual specimens: % Discrepant results All negative Positive at oneor more site Total
Negative
80
8
a88
9
Positive
5
a50
55
9
[image:4.612.313.555.93.216.2]aOn 4 of these 13 days, trace amounts of PCR product which do not amplify reproducibly were involved.
TABLE 3. Comparison by PCR of HSV titers in swabs with
secretions from multiple and individual genital sites
Titer of HSVDNA in swabs from
individual sites
Highest HSV DNA titer in mixed swab specimensa
Negative ,10 10 102 103 104 105 106 Total
Negative
81
2
2
0
1
0
0
0
86
,
10
3
2
1
0
0
0
0
0
6
10
3
2
3
1
0
0
0
0
9
10
21
1
2
7
1
0
0
0
12
10
30
1
1
0
1
0
1
0
4
10
40
0
0
0
0
2
3
0
5
10
50
0
0
0
0
1
12
1
14
10
60
0
0
0
0
1
4
12
7
aHighest titer of HSV DNA in a swab from any one site (vulvar, cervicovagi-nal, or rectal site). Boldface numbers indicate equivalence.
V
OL. 35, 1997
QUANTITATIVE COMPETITIVE PCR FOR HSV DNA
551
on May 15, 2020 by guest
http://jcm.asm.org/
[image:4.612.59.299.640.708.2]3.Cone, R. W., A. C. Hobson, and M.-L. W. Huang.1992. Coamplified positive control detects inhibition of polymerase chain reactions. J. Clin. Microbiol. 30:3185–3189.
4.Cone, R. W., A. C. Hobson, J. Palmer, M. Remington, and L. Corey.1991. Extended duration of herpes simplex virus DNA in genital lesions detected by polymerase chain reaction. J. Infect. Dis.164:757–760.
5.Diaz-Mitoma, F., M. Ruben, S. Sacks, P. MacPherson, and G. Caissie.1996. Detection of viral DNA to evaluate outcome of antiviral treatment of pa-tients with recurrent genital herpes. J. Clin. Microbiol.34:657–663. 6.Galliland, G., S. Perrin, and H. F. Bunn.1990. Competitive PCR for
quan-titation of mRNA, p. 60–69.InM. A. Innis, D. H. Gelfand, J. J. Sninsky, and T. J. White (ed.), PCR protocols: a guide to methods and applications. Academic Press, Inc., New York, N.Y.
7.Hardy, D. A., A. M. Arvin, L. L. Yasukawa, R. N. Bronzan, D. M. Lewinsohn, P. A. Hansleigh, and C. G. Prober.1990. Use of the polymerase chain reaction for successful identification of asymptomatic infection with herpes simplex virus in pregnant women. J. Infect. Dis.162:1031–1035.
8.Pachl, C., R. L. Burke, L. Stuve, L. Sanchez-Pescador, G. Van Nest, F.
Masiarz, and D. Dina.1987. Expression of cell-associated and secreted forms of herpes simplex virus type 1 glycoprotein gB in mammalian cells. J. Virol.61:315–325.
9.Peterson, E. P., O. W. Schmidt, L. C. Goldstein, R. C. Nowinski, and L. Corey.1983. Typing of clinical HSV isolates using mouse monoclonal anti-bodies to HSV-1 and HSV-2: comparison with type-specific rabbit antisera and restriction endonuclease analysis of viral DNA. J. Clin. Microbiol.17: 92–96.
10. Ramakrishnan, R., D. J. Fink, G. Jiang, P. Desai, J. C. Glorioso, and M. Levine.1994. Competitive quantitative PCR analysis of herpes simplex virus type 1 DNA and latency associated transcript RNA in latently infected cells of the rat brain. J. Virol.68:1864–1873.
11. Wald, A., L. Corey, R. Cone, A. Hobson, G. Davis, and J. Zeh.Frequent genital herpes simplex virus 2 shedding in immunocompetent women. Sub-mitted for publication.
12. Wald, A., J. Zeh, G. Barnum, L. G. Davis, and L. Corey.1996. Suppression of subclinical shedding of herpes simplex virus type 2 with acyclovir. Ann. Intern. Med.124:8–15.