324
means of transport vesiclesthat shuttle between the various organelles. These transport vesicles carry membrane lipids and membrane-bound proteins to their proper destinations in the cell, and they also carry soluble materials destined for secretion. Thus, these organelles and the vesicles connecting them make up a single dynamic system of membranes and internal spaces. Currently, one of the most exciting questions in modern cell biology concerns endomembrane trafficking: How does each of the multitude of proteins and lipids in a cell manage to reach its proper destination at the proper time?
The Endoplasmic Reticulum
The endoplasmic reticulum (ER)is a continuous network of flattened sacs, tubules, and associated vesicles that stretches throughout the cytoplasm of the eukaryotic cell. Although the name sounds formidable, it is actually quite descriptive. Endoplasmicsimply means “within the (cyto)plasm,” and reticulum is a Latin word meaning “network.” The membrane-bounded sacs are called ER cisternae(singular: ER cisterna), and the space enclosed by them is called the ER lumen(Figure 12-1). Of the total membrane in a mammalian cell, up to 50–90% surrounds the ER lumen. Unlike more prominent organelles, such as the mitochondrion or chloroplast, however, the ER is not visible by light microscopy unless one or more of its com-ponents are stained with a dye or labeled with a fluorescent molecule.
The ER was first observed in the late nineteenth century, when it was noted that some eukaryotic cells, particularly those involved in secretion, contained regions that stained intensely with basic dyes. The signif-icance of these regions remained in doubt until the 1950s, when the resolving power of the electron micro-scope was improved dramatically. This allowed cell biologists to visualize for the first time the ER’s elaborate network of intracellular membranes and to investigate the role of the ER in cellular processes. This is a common
A
full appreciation of eukaryotic cells dependson an understanding of the prominent role of intracellular membranes and the com-partmentalization of function within organelles—intracellular membrane-bounded compart-ments that house various cellular activities. Whether we consider the storage and transcription of genetic informa-tion, the biosynthesis of secretory proteins, the breakdown of long-chain fatty acids, or any of the numerous other metabolic processes occurring within eukaryotic cells, many of the reactions of a particular pathway occur within a dis-tinct type of organelle. Also, the movement of molecules between organelles, known as trafficking,must be tightly regulated to ensure that each organelle has the correct com-ponents for its proper structure and function.
We briefly encountered the major organelles found in eukaryotic cells in Chapter 4, and we then learned more about the mitochondrion and chloroplast in Chapters 10 and 11, respectively. We are now ready to consider several other individual organelles in more detail. We will begin with the rough endoplasmic reticulum, thesmooth endo-plasmic reticulum,and the Golgi complex,which are sites for protein synthesis, processing, and sorting. Next, we will look at endosomes,organelles that are important for carry-ing and sortcarry-ing material brought into the cell. Endosomes help to form lysosomes,which are organelles responsible for digestion of both ingested material and unneeded intracel-lular components. We will conclude with a look at peroxisomes,which house hydrogen peroxide–generating reactions and perform diverse metabolic functions.
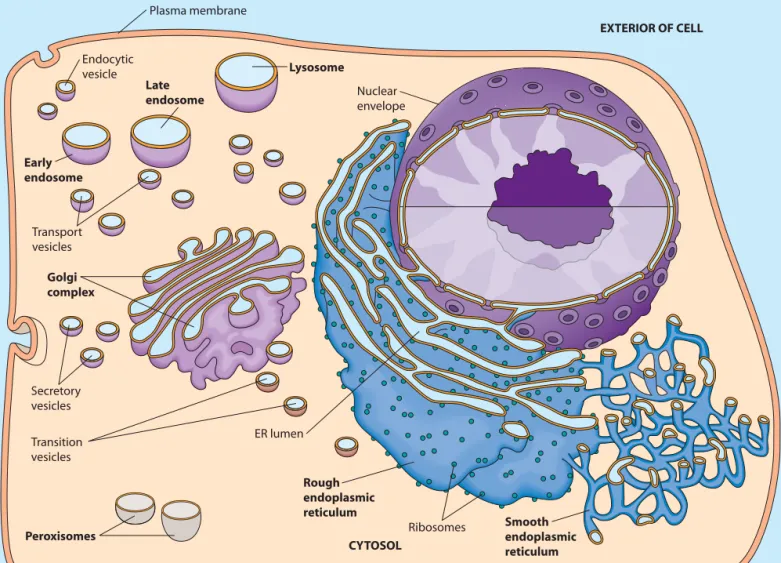
As you study the role of each organelle, keep in mind that the endoplasmic reticulum, the Golgi complex, endo-somes, and lysosomes (but not peroxisomes) comprise the endomembrane systemof the eukaryotic cell, as shown in Figure 12-1. (The nuclear envelope,which we will study in Chapter 18, is closely associated with the endomembrane system.) Material flows from the endoplasmic reticulum to and from the Golgi complex, endosomes, and lysosomes by
12
The Endomembrane System
and Peroxisomes
The Endoplasmic Reticulum 325 theme in scientific discovery—conceptual advances in
one field often follow technological advances in a related (or even an unrelated) field.
We now know that enzymes associated with the ER are responsible for the biosynthesis of proteins destined for incorporation into the plasma membrane or into organelles of the endomembrane system and for synthesis of proteins destined for export from the cell. The ER also plays a central role in the biosynthesis of lipids, including triacylglycerols, cholesterol, and related compounds. The ER is the source of most of the lipids that are assembled to form intracellular membranes and the plasma membrane. The Two Basic Kinds of Endoplasmic Reticulum Differ in Structure and Function
The two basic kinds of endoplasmic reticulum typically found in eukaryotic cells are distinguished from one
another by the presence or absence of ribosomes attached to the ER membrane (Figure 12-2). Rough endoplasmic reticulum (rough ER) is characterized by ribosomes attached to the cytosolic side of the membrane (the side that faces away from the ER lumen; Figure 12-2a). Trans-lation by these ribosomes occurs in the cytosol, but the newly-synthesized proteins will enter the ER lumen shortly. Because ribosomes contain RNA, it was this RNA that reacted strongly with the basic dyes originally used to identify the rough ER. A subdomain of rough ER, the transitional elements (TEs), plays an important role in the formation of transition vesiclesthat shuttle lipids and proteins from the ER to the Golgi complex. In contrast, smooth endoplasmic reticulum (smooth ER) appears smooth due to the absence of ribosomes attached to the membrane (Figure 12-2b) and has other roles in the cell.
Rough and smooth ER are easily distinguished mor-phologically. As illustrated in Figure 12-1, rough ER Plasma membrane
Endocytic vesicle
ER lumen
EXTERIOR OF CELL
Nuclear envelope
Transport vesicles
Transition vesicles
Lysosome
Secretory vesicles
CYTOSOL Late
endosome
Early endosome
Golgi complex
Smooth endoplasmic reticulum Rough
endoplasmic reticulum
Peroxisomes Ribosomes
FIGURE 12-1 The Endomembrane System. The endomembrane system of the eukaryotic cell consists of the endoplasmic reticulum (ER), the Golgi complex, endosomes, and lysosomes (but not peroxisomes). It is associated with both the nuclear envelope and the plasma membrane. The ER lumen is linked to the interiors of the Golgi complex, endosomes, and lysosomes by transport vesicles that shuttle material between organelles, as well as to and from the plasma membrane.
326 Chapter 12 The Endomembrane System and Peroxisomes
(a) Rough endoplasmic reticulum 0.25μm (b) Smooth endoplasmic reticulum 0.25μm FIGURE 12-2 Rough and Smooth Endoplasmic Reticulum. (a)Electron micrograph of endoplasmic
reticulum. The rough ER is studded with ribosomes. (b)Electron micrograph of smooth endoplasmic retic-ulum. The dark spots near the smooth ER appear to be glycogen granules (TEMs).
membranes usually form large flattened sheets, whereas smooth ER membranes generally form tubular structures. The transitional elements of the rough ER are an excep-tion to this rule; they often resemble smooth ER. However, the rough and smooth ER are not separate organelles—electron micrographs and studies in living cells show that their lumenal spaces are continuous. Thus, material can travel between the rough and smooth ER without the aid of vesicles.
Both types of ER are present in most eukaryotic cells, but there is considerable variation in the relative amounts of each type, depending on the activities of the particular cell. Cells involved in the biosynthesis of secretory proteins, such as liver cells and cells producing digestive enzymes, tend to have very prominent rough ER networks. On the other hand, cells producing steroid hormones, such as in the testis or ovary, contain extensive networks of smooth ER.
When tissue is homogenized for subcellular fraction-ation, the ER membranes often break into smaller fragments that spontaneously close to form sealed vesicles known as microsomes.Fractions can be isolated with and without attached ribosomes, depending on whether the membrane originated from rough or smooth ER, respec-tively. Such preparations are tremendously useful for exploring both types of ER. Keep in mind, however, that microsomes do not exist in the cell; they are simply an artifact of the fractionation process. Box 12A presents more detailed information about subcellular fractionation by differential centrifugation.
Rough ER Is Involved in the Biosynthesis and Processing of Proteins
The ribosomes attached to the cytosolic side of the rough ER membrane are responsible for synthesizing both membrane-bound and soluble proteins for the endomem-brane system. So how do proteins enter the ER lumen and
the endomembrane system from their site of synthesis on the opposite (cytosolic) side of the rough ER membrane? Synthesis of proteins destined for the endomembrane system begins on cytoplasmic ribosomes, which attach to the rough ER via receptor proteins in the ER membrane shortly after translation initiation. Newly-synthesized pro-teins enter the endomembrane system cotranslationally— that is, they are inserted through a pore complex in the ER membrane into the rough ER lumen as the polypeptide is synthesized by the ER-bound ribosome (see Figure 22-16). After biosynthesis, membrane-spanning proteins remain anchored to the ER membrane either by hydrophobic regions of the polypeptide or by covalent attachment to membrane lipids. Soluble proteins, including secretory proteins, are released into the ER lumen.
In addition to its role in the biosynthesis of polypep-tide chains, the rough ER is the site for several other processes, including the initial steps of addition and processing of carbohydrate groups to glycoproteins, the folding of polypeptides, the recognition and removal of misfolded polypeptides, and the assembly of multimeric proteins. Thus, ER-specific proteins include a host of enzymes that catalyze cotranslational and posttranslational modifications. These modifications include glycosylation, which is important for sorting of proteins to their proper destinations, and disulfide bond formation, which is essential for proper protein folding. We will discuss the topics of protein biosynthesis, targeting, and folding in more detail in Chapter 22.
The ER is also a site for quality control. In ER-associated degradation (ERAD),proteins improperly modified, folded, or assembled are exported from the ER for degradation by cytosolic proteasomes before they can move on to the Golgi complex. Several human diseases, including cystic fibrosis and familial hyperc-holesterolemia, are associated with defects in these processes.
330 Chapter 12 The Endomembrane System and Peroxisomes Smooth ER Is Involved in Drug Detoxification, Carbohydrate Metabolism, Calcium Storage, and Steroid Biosynthesis
Drug Detoxification.Drug detoxification often involves enzyme-catalyzed hydroxylation because the addition of hydroxyl groups to hydrophobic drugs makes them more soluble and easier to excrete from the body. Hydroxylation of organic acceptor molecules is typically catalyzed by a member of the cytochrome P-450family of proteins. These proteins are especially prevalent in the smooth ER of hepato-cytes (liver cells), in which many drugs are detoxified.
In the hepatocytes, an electron transport system trans-fers electrons from NADPH or NADH to a heme group in a cytochrome P-450 protein, which then donates an electron to molecular oxygen. One atom of molecular oxygen gains two electrons and two , forming . The other oxygen atom is added to the organic substrate molecule as part of a hydroxyl group. Because one of the two oxygen atoms of is incorporated into the reaction product, these cytochrome P-450 enzymes are often called monooxygenases. The net reaction is shown below, where R represents the organic hydroxyl acceptor:
(12-1) The elimination of hydrophobic barbiturate drugs, for example, is enhanced by hydroxylation enzymes in the smooth ER. Injection of the sedative phenobarbital into a rat causes a rapid increase in the level of barbiturate-detoxifying enzymes in the liver, accompanied by a dramatic prolifera-tion of smooth ER. However, this means that increasingly higher doses of the drug are necessary to achieve the same sedative effect, an effect known as tolerancethat is seen in habitual users of phenobarbital. Furthermore, the enzyme induced by phenobarbital can hydroxylate and therefore sol-ubilize a variety of other drugs, including such useful agents as antibiotics, anticoagulants, and steroids. As a result, the chronic use of barbiturates decreases the effectiveness of many other clinically useful drugs.
Another cytochrome P-450 protein found in the smooth ER is part of an enzyme complex called aryl hydrocarbon hydroxylase. This complex is involved in metabolizing polycyclic hydrocarbons, organic molecules composed of two or more linked benzene rings that are often toxic. Hydroxylation of such molecules is important for increasing their solubility in water, but the oxidized products are often more toxic than the original com-pounds. Aryl hydrocarbon hydroxylase converts some potential carcinogens into their chemically active forms. Mice synthesizing high levels of this hydroxylase have a higher incidence of spontaneous cancer than normal mice do, whereas mice treated with an inhibitor of aryl hydrocarbon hydrolase develop few tumors. Significantly, cigarette smoke is a potent inducer of aryl hydrocarbon hydroxylase.
ROH + NAD(P)+ + H2O
RH + NAD(P)H + H+ + O2 ¡
O2
H2O
H+
Recent work shows that differences in activities and side effects of certain medications can result from differ-ences in the presence or activity of particular cytochrome P-450 genes in different patients. This has led to a new field of study known as pharmacogenetics(also called pharmaco-genomics), which investigates how inherited differences in genes (and their resulting protein products) can lead to dif-ferential responses to drugs and medications.
Carbohydrate Metabolism.The smooth ER of hepato-cytes (liver cells) is also involved in the enzymatic breakdown of stored glycogen, as evidenced by the pres-ence of glucose-6-phosphatase,a membrane-bound enzyme that is unique to the ER. Thus, its presence is used as a marker to identify the ER during subcellular fractionation or to visualize the ER using fluorescent antibodies. Glucose-6-phosphatase hydrolyzes the phosphate group from glucose-6-phosphate to form free glucose and inor-ganic phosphate :
(12-2) This enzyme is abundant in the liver because a major role of the liver is to keep the level of glucose in the blood relatively constant. The liver stores glucose as glycogen in granules associated with smooth ER (Figure 12-3a). When glucose is needed by the body, especially between meals and in response to increased muscular activity, liver glycogen is broken down by phosphorolysis (see Figure 9-10), producing glucose-6-phosphate (Figure 12-3b). Because membranes are generally impermeable to phos-phorylated sugars, the glucose-6-phosphate must be converted to free glucose by glucose-6-phosphatase in order to leave the cell and enter the bloodstream. Free glucose then leaves the liver cell via a glucose transporter (GLUT2) and moves into the blood for transport to other cells that need energy. Significantly, glucose-6-phos-phatase activity is present in liver, kidney, and intestinal cells but not in muscle or brain cells. Muscle and brain cells retain glucose-6-phosphate and use it to meet their own substantial energy needs.
Calcium Storage.The sarcoplasmic reticulumfound in muscle cells is an example of smooth ER that specializes in the storage of calcium. In these cells, the ER lumen contains high concentrations of calcium-binding proteins. Calcium ions are pumped into the ER by ATP-dependent calcium ATPasesand are released in response to extracellular signals to aid in muscle contraction (see Figure 14-12). Binding of neurotransmitter molecules to receptors on the surface of the muscle cell triggers a signal cascade that leads to the release of calcium from the sarcoplasmic reticulum and causes the contraction of muscle fibers. We will discuss nerve impulse transmis-sion and muscle contraction in more detail in Chapters 13 and 16.
glucose-6-phosphate +H2O ¡glucose +Pi
(Pi) Functions of Smooth ER
The Endoplasmic Reticulum 331 (a)Proximity of glycogen
to smooth ER
0.5μm
Glycogen granules
Smooth ER
Mitochondrion
Process of glycogen breakdown in liver P
P
Glucose-1-P
Glucose Glycogen phosphorylase
LIVER CELL Glycogen granule
Glucose transporter
Smooth ER
Plasma membrane
In blood Phosphoglucomutase
Glucose-6-P
Glucose Glucose-6-phosphatase
Pi
(b) FIGURE 12-3 The Role of the Smooth ER in the Catabolism
of Liver Glycogen. (a)This electron micrograph of a monkey liver cell shows numerous granules of glycogen closely associated with smooth ER (TEM). (b)The breakdown of liver glycogen involves the stepwise removal of glucose units as glucose-1-phosphate, followed by the conversion of 1-phosphate to glucose-6-phosphate by enzymes in the cytosol. Removal of the phosphate group depends on glucose-6-phosphatase, an enzyme associated with the smooth ER membrane. Free glucose is then transported out of the liver cell into the blood by a glucose transporter in the plasma membrane.
Steroid Biosynthesis.The smooth ER in certain cells is the site of biosynthesis of cholesterol and steroid hor-mones such as cortisol, testosterone, and estrogen. Large amounts of smooth ER are found in the cortisol-producing cells of the adrenal gland; the Leydig cells of the testes, which produce testosterone; the cholesterol-producing cells of the liver; and the follicular cells of the ovary, which produce estrogen. Smooth ER has also been found in close association with plastids in some plants, where it may be involved in phytohormone synthesis.
Cholesterol, cortisol, and the male and female steroid hormones just described share a common four-ring struc-ture but differ in the number and arrangement of carbon side chains and hydroxyl groups (see Figure 3-27e and Figure 3-30). Hydroxymethylglutaryl-CoA reductase (HMG-CoA reductase), the committed step in cholesterol biosynthesis, is present in large amounts in the smooth ER of liver cells. This enzyme is targeted for inhibition by a class of cholesterol-lowering drugs known as statins. In addition, the smooth ER contains a number of P-450 monooxygenases that are important not only in the syn-thesis of cholesterol but also in its conversion into steroid hormones by hydroxlyation.
The ER Plays a Central Role in the Biosynthesis of Membranes
In eukaryotic cells, the ER is the primary source of mem-brane lipids, including phospholipids and cholesterol. Indeed, most of the enzymes required for the biosynthesis of
membrane phospholipids are found nowhere else in the cell. There are, however, important exceptions. Mitochondria synthesize phosphatidylethanolamine by decarboxylating imported phosphatidylserine. Peroxisomes have enzymes to synthesize cholesterol, and chloroplasts contain enzymes for the synthesis of chloroplast-specific lipids.
Biosynthesis of fatty acids for membrane phospholipid molecules occurs in the cytoplasm and incorporation is restricted to the monolayer of the ER membrane facing the cytosol. Cellular membranes, of course, are phospholipid bilayers,with phospholipids distributed to both sides. Thus, there must be a mechanism for transferring phospholipids from one layer of the membrane to the other. Because it is thermodynamically unfavorable for phospholipids to flip spontaneously at a significant rate from one side of a bilayer to the other, transfer depends on phospholipid transloca-tors,also called flippases,which catalyze the translocation of phospholipids through ER membranes (see Figure 7-10). Phospholipid translocators, like other enzymes, are quite specific and affect only the rate of a process. Therefore, the type of phospholipid molecules transferred across a membrane depends on the particular translocators present, contributing to the membrane asymmetry described in Chapter 7. For example, the ER membrane contains a translocator for phosphatidylcholine, and thus it is found in both monolayers of the ER membrane. In contrast, there is no translocator for phosphatidylethanolamine, phos-phatidylinositol, or phosphatidylserine, which are therefore confined to the cytosolic monolayer. When vesicles from the ER membrane fuse with other organelles of the
332 Chapter 12 The Endomembrane System and Peroxisomes endomembrane system, the distinct compositions of the cytosolic and lumenal monolayers established in the ER are transferred to these other cellular membranes.
Movement of phospholipids from the ER to a mito-chondrion, chloroplast, or peroxisome poses a unique problem. Unlike organelles of the endomembrane system, these organelles do not grow by fusion with ER-derived vesi-cles. Instead, cytosolic phospholipid exchange proteins (also called phospholipid transfer proteins) convey phospho-lipid molecules from the ER membrane to the outer mitochondrial and chloroplast membranes. Each exchange protein recognizes a specific phospholipid, removes it from one membrane, and carries it through the cytosol to another membrane. Such transfer proteins also contribute to the movement of phospholipids from the ER to other cellular membranes, including the plasma membrane.
Although the ER is the source of most membrane lipids, the compositions of other cellular membranes vary significantly from the composition of the ER membrane (Table 12-1). A striking feature of the plasma membrane of hepatocytes is the relatively low amount of phosphoglyc-erides and high amounts of cholesterol, sphingomyelin, and glycolipids. Researchers have observed an increasing gradient of cholesterol content from the ER through the compartments of the endomembrane system to the plasma membrane. This correlates with an increasing gradient of membrane thickness. ER membranes are about 5 nm thick, whereas plasma membranes are about 8 nm thick. The observed change in membrane thickness has implica-tions for sorting and targeting integral membrane proteins, which we will discuss after we look at the Golgi complex and its role in protein processing.
The Golgi Complex
We now turn our attention to the Golgi complex, a compo-nent of the endomembrane system that is closely linked, both physically and functionally, to the ER. In the Golgi
complex, glycoproteins from the ER undergo further pro-cessing and, along with membrane lipids, are sorted and packaged for transport to their proper destinations inside or outside the cell. Thus, the Golgi complex plays a central role in membrane andprotein traffickingin eukaryotic cells. The Golgi complex (or Golgi apparatus) derives its name from Camillo Golgi, the Italian biologist who first described it in 1898. He reported that nerve cells soaked in osmium tetroxide showed deposits of osmium in a thread-like network surrounding the nucleus. The same staining reaction was demonstrated with a variety of cell types and other heavy metals. However, no cellular structure could be identified that explained the staining. As a result, the nature—actually, the very existence—of the Golgi complex remained controversial until the 1950s, when its existence was finally confirmed by electron microscopy.
The Golgi Complex Consists of a Series of Membrane-Bounded Cisternae
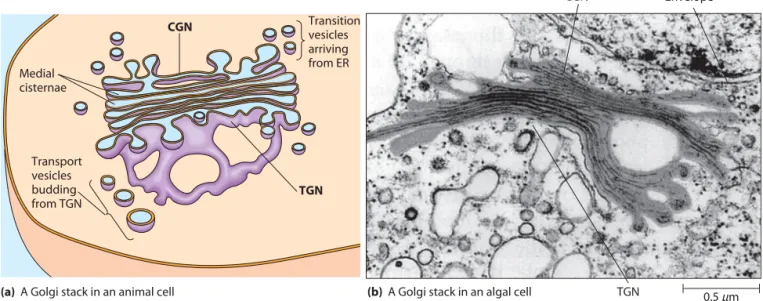
The Golgi complex is a series of flattened membrane-bounded cisternae, disk-shaped sacs that are stacked together as illustrated in Figure 12-4a. A series of such cisternae is called a Golgi stackand can be visualized by electron microscopy (Figure 12-4b). Usually, there are 3–8 cisternae per stack, though the number and size of Golgi stacks vary with the cell type and with the metabolic activity of the cell. Some cells have one large stack, whereas others—especially active secretory cells—have hundreds or even thousands of Golgi stacks.
The static view of the ER and the Golgi complex pre-sented by electron micrographs such as Figure 12-4b can be misleading. These organelles are actually dynamic struc-tures. Both the ER and Golgi complex are typically surrounded by numerous transport vesiclesthat carry lipids and proteins from the ER to the Golgi complex, between the cisternae of a Golgi stack, and from the Golgi complex to various destinations in the cell, including endosomes, lysosomes, and secretory granules. Thus, the Golgi complex lumen, or intracisternal space,is part of the endomembrane system’s network of internal spaces (see Figure 12-1). The Two Faces of the Golgi Stack.Each Golgi stack has two distinct sides, or faces(Figure 12-4). The cis faceis ori-ented toward the ER. The Golgi compartment closest to the ER is a network of flattened, membrane-bounded tubules referred to as the cis-Golgi network (CGN). Vesicles containing newly synthesized lipids and proteins from the ER continuously arrive at the CGN, where they fuse with CGN membranes. The opposite side of the Golgi complex is called the trans face.The compartment on this side of the Golgi complex has similar morphology and is referred to as the trans-Golgi network (TGN).Here, pro-teins and lipids leave the Golgi in transport vesiclesthat continuously bud from the tips of TGN cisternae. These transport vesicles carry lipids and proteins from the Golgi complex to secretory granules, endosomes, lysosomes, and the plasma membrane. The central sacs between the CGN Table 12-1 Composition of the ER and Plasma
Membranes of Rat Liver Cells
Membrane Components ER Membrane MembranePlasma Membrane components as % of membrane by weight
Carbohydrate 10 10
Protein 62 54
Total lipid 27 36
Membrane lipids as % of total lipids by weight
Phosphatidylcholine 40 24
Phosphatidylethanolamine 17 7
Phosphatidylserine 5 4
Cholesterol 6 17
Sphingomyelin 5 19
Glycolipids trace 7
The Golgi Complex 333 and TGN comprise the medial cisternae of the Golgi
stack, in which much of the processing of proteins occurs. The CGN, TGN, and medial cisternae of the Golgi complex are biochemically and functionally distinct. Each compartment contains specific receptor proteins and enzymes necessary for specific steps in protein and mem-brane processing, as shown by immunological and cytochemical staining techniques. This biochemical polarity is illustrated in Figure 12-5, which shows a Golgi stack in a rabbit kidney cell. Staining to detect N-acetylglucosamine transferase I, an enzyme that modifies carbohydrate side chains of glycoproteins, shows that the enzyme is concen-trated in medial cisternae of the Golgi complex.
Two Models Depict the Flow of Lipids and Proteins Through the Golgi Complex
Two models have been proposed to explain the move-ment of lipids and proteins from the CGN to the TGN via the medial cisternae of the Golgi complex. According to the stationary cisternae model,each compartment of the Golgi stack is a stable structure. Trafficking between successive cisternae is mediated by shuttle vesicles that bud from one cisterna and fuse with the next cisterna in the cis-to-transsequence. Proteins destined for the TGN are simply carried forward by shuttle vesicles, while molecules that belong in the ER and successive Golgi compartments are actively retained or retrieved.
According to the second model, known as the cisternal maturation model,the Golgi cisternae are tran-sient compartments that gradually change from CGN cisternae through medial cisternae to TGN cisternae. In this model, transition vesicles from the ER converge to form the CGN, which accumulates specific enzymes for
the early steps of protein processing. Step by step, each cis cisterna is transformed first into an intermediate medial cisterna and then into a transcisterna as it acquires addi-tional enzymes. Enzymes that are no longer needed in late compartments return in vesicles to early compartments. In both models, the TGN forms transport vesicles or Transition
vesicles arriving from ER CGN
Medial cisternae
TGN Transport
vesicles budding from TGN
(a) A Golgi stack in an animal cell (b) A Golgi stack in an algal cell TGN CGN
Nuclear Envelope
0.5μm FIGURE 12-4 Golgi Structure. A Golgi stack consists of a small number of flattened cisternae. (a)At the
cisface, transition vesicles arriving from the ER fuse with membranes of the cis-Golgi network (CGN). At the transface, transport vesicles arise by budding from the trans-Golgi network (TGN). The transport vesicles carry lipids and proteins to other components of the endomembrane system or form secretory vesicles. (b)This electron micrograph shows a Golgi stack lying next to the nuclear envelope of an algal cell (TEM).
CGN CGN
TGN TGN CGN
TGN
FIGURE 12-5 Immunochemical Staining of a Golgi Complex. This electron micrograph shows a Golgi stack in a rabbit kidney cell. The cell section has been stained to detect the enzyme N-acetylglucosamine transferase I,which plays a role in terminal glycosylation of proteins. The arrow and bracket indicate unlabeled cisternae, showing that the enzyme is concentrated in a few medial cisternae close to the cisface of the Golgi stack. This indicates that N-acetylglucosamine is added to existing oligosaccharides on glyco-proteins shortly after the glyco-proteins enter the Golgi stack (TEM).
334 Chapter 12 The Endomembrane System and Peroxisomes secretory granules containing sorted cargo targeted for various destinations beyond the Golgi complex.
Experimental results suggest that these two models are not necessarily mutually exclusive. It is likely that both apply to some degree, depending on the organism and the role of the cell. While the stationary cisternae model is sup-ported by substantial evidence, some cellular components observed in medial compartments of the Golgi complex are clearly too large to travel by the small shuttle vesicles found in cells. For example, polysaccharide scales produced by some algae first appear in early Golgi compartments. Too large to fit inside transport vesicles, the scales nevertheless reach late Golgi compartments on their way to the plasma membrane for incorporation into the cell wall.
Recently, time-lapse fluorescence microscopy has been used in live yeast cells to study individual Golgi cis-ternae in real time. Three-dimensional analysis of images supports the cisternal maturation model and suggests that the cisternae mature at a constant rate. In addition, the rate of movement of labeled secretory proteins through the Golgi complex was measured and was shown to match the rate of cisternal maturation.
Anterograde and Retrograde Transport. The move-ment of material from the ER through the Golgi complex toward the plasma membrane is called anterograde trans-port(anterois derived from a Latin word meaning “front,” and gradeis related to a word meaning “step”). Every time a secretory granule fuses with the plasma membrane and discharges its contents by exocytosis, a bit of membrane that originated in the ER becomes a part of the plasma membrane. To balance the flow of lipids toward the plasma membrane and to ensure a supply of components for forming new vesicles, the cell recycles lipids and proteins no longer needed during the late stages of anterograde transport. This is accomplished by retrograde transport (retrois a Latin word meaning “back”), the flow of vesicles from Golgi cisternae back toward the ER.
In the stationary cisternae model, retrograde flow facilitates both the recovery of ER-specific lipids and proteins that are passed from the ER to the CGN and the transport of compartment-specific proteins back to dis-tinct medial cisternae of the Golgi stack. Material destined for the TGN continues forward. Whether such retrograde traffic occurs directly from all medial cisternae of the Golgi stack back to the ER or by reverse flow through successive cisternae is not yet clear. In the cisternal maturation model, retrograde flow carries material back toward newly forming compartments after receptors and enzymes are no longer needed in the more mature compartments.
Roles of the ER and Golgi Complex
in Protein Glycosylation
Much of the protein processing carried out within the ER and Golgi complex involves glycosylation—the addition of carbohydrate side chains to specific amino acid residues
of proteins, forming glycoproteins.Subsequent enzyme-catalyzed reactions then modify the oligosaccharide side chain that was attached to the protein. Two general kinds of glycosylation are observed in cells (see Figure 7-25). N-linked glycosylation(or N-glycosylation) involves the addition of a specific oligosaccharide unit to the nitrogen atom on the terminal amino group of certain asparagine residues. O-linked glycosylationinvolves addition of an oligosaccharide to the oxygenatom on the hydroxyl group of certain serine or threonine residues. Each step of glyco-sylation is strictly dependent on preceding modifications. An error at one step, perhaps due to a defective enzyme, can block further modification of a carbohydrate side chain and can lead to disease in the organism.
Initial Glycosylation Occurs in the ER
We will focus here on N-glycosylation. Figure 12-6 describes the steps of glycosylation that may occur as a glycoprotein travels from the ER to the CGN and through the Golgi complex to the TGN. Note that specific enzymes that catalyze various steps of glycosylation and subsequent modifications are present in specific compartments of the ER and Golgi complex. The initial steps of N-glycosylation take place on the cytosolic surface of the ER membrane and later steps occur in the ER lumen (Figure 12-7). Despite the variety of oligosaccharides found in mature glyco-proteins, all the carbohydrate side chains added to proteins in the ER initially have a common core oligosaccharide consisting of two units of N-acetylglucosamine (GlcNAc, see Figure 3-26a), nine mannose units, and three glucose units.
Glycosylation begins as dolichol phosphate, an oligo-saccharide carrier, is inserted into the ER membrane (Figure 12-7, step ). GlcNAc and mannose groups are then added to the phosphate group of dolichol phosphate (step ). The growing core oligosaccharide is then translocated from the cytosol to the ER lumen by a flippase(step ). Once inside the ER lumen, more mannose and glucose units are added (step ). The completed core oligosac-charide is then transferred as a single unit from dolichol to an asparagine residue of the recipient protein (step ). Finally, the core oligosaccharide attached to the protein is trimmed and modified (step ).
Usually, the core oligosaccharide is added to the protein as the polypeptide is being synthesized by a ribosome bound to the ER membrane. We know that this cotranslational glycosylation helps to promote proper protein folding because experimental inhibition of glycosylation leads to the appearance of misfolded, aggregated proteins. Addition of a single glucose unit allows other ER proteins to interact with the newly synthesized glycoprotein to ensure its proper folding. One of two ER proteins known as calnexin (CNX, membrane-bound) and calreticulin(CRT, soluble) can bind to the monoglucosylated glycoprotein and promote disulfide bond formation by forming a complex with the glycoprotein and a thiol oxidoreductase known as ERp57, which catalyzes disulfide bond formation. The
6
5 4
3 2
Roles of the ER and Golgi Complex in Protein Trafficking 335 protein complex then dissociates, and the final glucose unit
is removed by an enzyme named glucosidase II.
At this point, a specific glucosyl transferase in the ER known as UGGT (UDP-glucose:glycoprotein glucotransferase) acts as a sensor for proper folding of the newly synthesized glycoprotein. UGGT binds to improperly folded proteins and adds back a single glucose unit, making the protein a substrate for another round of CNX/CRT binding and disul-fide bond formation. Once the proper conformation is achieved, UGGT no longer binds the new glycoprotein, which is then free to exit the ER and move to the Golgi. Further Glycosylation Occurs in
the Golgi Complex
Further processing of N-glycosylated proteins happens in the Golgi complex as the glycoproteins move from the cis face through the medial cisternae to the transface of the Golgi stack. These terminal glycosylations in the Golgi
show remarkable variability among proteins and account for much of the great diversity in structure and function of protein oligosaccharide side chains.
Terminal glycosylation always includes the removal of a few of the carbohydrate units of the core oligosaccha-ride. In some cases, no further processing occurs in the Golgi complex. In other cases, more complex oligosaccha-rides are generated by the further addition of GlcNAc and other monosaccharides, including galactose, sialic acid, and fucose (see Figure 7-26a). Some glycoproteins contain galactose units that are added by galactosyl transferase,a marker enzyme unique to the Golgi.
Given the role of the Golgi complex in glycosylation, it is not surprising that two of the most important cate-gories of enzymes present in Golgi stacks are glucan synthetases,which produce oligosaccharides from mono-saccharides, and glycosyl transferases, which attach carbohydrate groups to proteins. The ER and Golgi complex contain hundreds of different glycosyl trans-ferases, which indicates the potential complexity of oligosaccharide side chains. Within the Golgi stack, each cisterna contains a distinctive set of processing enzymes.
Notice in the preceding discussion that the mature oligosaccharides in glycoproteins are found only on the lumenal side of the ER and Golgi complex membranes and thus contribute to membrane asymmetry. Because the lumenal side of the ER membrane is topologically equiva-lent to the exterior surface of the cell, it is easy to see why all plasma membrane glycoprotein oligosaccharides are found on the extracellular side of the membrane.
Roles of the ER and Golgi
Complex in Protein Trafficking
Membrane-bound and soluble proteins synthesized in the rough ER must be directed to a variety of intracellular locations, including the ER itself, the Golgi complex, endosomes, and lysosomes. Moreover, once a protein reaches an organelle where it is to remain, there must be a mechanism for preventing it from leaving. Other groups of proteins synthesized in the rough ER are destined for incorporation into the plasma membrane or for release to the outside of the cell. Therefore, each protein contains a specific “tag” targeting the protein to a transport vesicle that will carry material from one specific cellular location to another. Depending on the protein and its destination, the tag may be a short amino acid sequence, an oligosac-charide side chain, a hydrophobic domain, or some other structural feature. Tags may also be involved in excluding material from certain vesicles.
Membrane lipids may also be tagged to help vesicles reach their proper destinations. This tag can be one or more phosphate groups attached to positions 3, 4, and/or 5 of a membrane phosphatidylinositol (PI) molecule by a specific kinase. For example, a functional PI 3-kinase is required for proper sorting of vesicles to the vacuole in yeast. In mammalian cells, inhibition of inositol kinases
• Biosynthesis of core oligosaccharide for N-linked glycosylation of certain asparagine residues • Initial processing of core
oligosaccharide
• Identification and removal of misfolded proteins
• Attachment of N-acetylgalactosamine to serine or threonine
• First step of phosphorylation of lysosomal proteins
ER
CGN
Medial cisternae
M
igr
ation thr
ough the G
olgi c
omplex
TGN
• Removal of mannose
• Second step of phosphorylation of lysosomal proteins
• Removal of mannose
• Attachment of N-acetylglucosamine • Addition of galactose
• Addition of sialic acid • Addition of sialic acid
• Attachment of sulfate to tyrosine
FIGURE 12-6 Compartmentalization of the Steps of Glycosylation and Subsequent Modification of Proteins. Enzymes that catalyze specific steps of glycosylation and further modification of proteins reside in different compartments of the ER and Golgi complex. Processing occurs sequentially as proteins travel from compartment to compartment. The steps listed in the figure are examples of potential modifications and do not necessarily occur with all glycoproteins.
352 Chapter 12 The Endomembrane System and Peroxisomes
Lysosome Mitochondria
1μm FIGURE 12-20 Cytochemical Localization of Acid Phosphatase, a Lysosomal Enzyme. Tissue was incubated in a medium containing soluble lead nitrate and b-glycerophosphate, which is cleaved by acid phosphatase, producing free glycerol and phosphate anions. The phosphate anions react with lead ions to form insoluble lead phosphate, which precipitates at the site of enzyme activity and reveals the location of acid phosphatase within the cell. The darkly stained organelles shown here are lysosomes highlighted by deposits of the electron-dense lead phosphate. They are surrounded by mitochondria, which lack acid phosphatase and do not become electron dense (TEM).
system, ER-derived vesicles can attach to Golgi mem-branes without addition of SNARE proteins.
Two main groups of tethering proteins are known at present—coiled-coil proteins and multisubunit complexes. Coiled-coil proteins such as the golginsare important in the initial recognition and binding of COPI- or COPII-coated vesicles to the Golgi. The golgins are anchored by one end to the Golgi membrane and use the other end to contact the appropriate passing vesicle. We know that these proteins are also important in connecting Golgi cis-ternae to each other because antibodies directed against certain golgins block the action of the golgins and disrupt the structure of the Golgi medial cisternae.
The second class of tethering proteins consists of several families of multisubunit protein complexes con-taining four to eight or more individual polypeptides. For example, the exocyst complex of yeast and mammals is important for protein secretion, binding both to the plasma membrane and to vesicles from the TGN whose contents are destined for export. Other types of multi-subunit tethering complexes such as the COG (conserved oligomeric Golgi)complex, the GARP (Golgi-associated retro-grade protein)complex, and the TRAPP (transport protein particle)complex are implicated in the initial recognition and specificity of vesicle–target membrane interaction. Most of the proteins in these complexes are highly con-served among organisms as different as yeast and humans. Identifying the functions of these complexes and the roles of their individual subunits is currently one of the most intriguing frontiers of modern cell biology.
Lysosomes and Cellular Digestion
The lysosomeis an organelle of the endomembrane system that contains digestive enzymes capable of degrading all the major classes of biological macromolecules—lipids, carbo-hydrates, nucleic acids, and proteins. These hydrolytic enzymes degrade extracellular materials brought into the cell by endocytosis and digest intracellular structures and macromolecules that are damaged or no longer needed. We will first look at the organelle itself and then consider lyso-somal digestive processes, as well as some of the diseases that result from lysosomal malfunction.
Lysosomes Isolate Digestive Enzymes from the Rest of the Cell
As we learned in Chapter 4, lysosomes were discovered in the early 1950s by Christian de Duve and his colleagues (see Box 4B, page 92). Differential centrifugation led the researchers to realize that an acid phosphatase initially thought to be located in the mitochondrion was in fact asso-ciated with a class of particles that had never been reported before. Along with the acid phosphatase, the new organelle contained several other hydrolytic enzymes, including b -glucuronidase, a deoxyribonuclease, a ribonuclease, and a protease. Because of its apparent role in cellular lysis, de Duve called this newly discovered organelle a lysosome.
Only after the lysosome’s existence had been predicted, its properties described, and its enzyme content specified was the organelle actually observed by electron microscopy and recognized as a normal constituent of most animal cells. Final confirmation came from cytochemical staining reactions capable of localizing the acid phosphatase and other lysosomal enzymes to specific structures that can be seen by electron microscopy (Figure 12-20).
Lysosomes vary considerably in size and shape but are generally about 0.5 mm in diameter. Like the ER and Golgi complex, the lysosome is bounded by a single membrane. This membrane protects the rest of the cell from the hydrolytic enzymes in the lysosomal lumen. The lumenal side of lysosomal membrane proteins is highly glycosyl-ated, forming a nearly continuous carbohydrate coating that appears to protect membrane proteins from lysosomal proteases. ATP-dependent proton pumps in the membrane maintain an acidic environment (pH 4.0–5.0) within the lysosome. This favors enzymatic digestion of macromole-cules both by activating acid hydrolases and by partially denaturing the macromolecules targeted for degradation. The products of digestion are then transported across the membrane to the cytosol, where they enter various syn-thetic pathways or are exported from the cell.
The list of lysosomal enzymes has expanded consider-ably since de Duve’s original work, but all have the common property of being acid hydrolases—hydrolytic enzymes with a pH optimum around 5.0. The list includes at least 5 phosphatases, 14 proteases and peptidases, 2 nucleases, 6 lipases, 13 glycosidases, and 7 sulfatases. Taken together, these lysosomal enzymes can digest all the major classes of biological molecules. No wonder, then,
Lysosomes and Cellular Digestion 353 A
Exocytosis releasing acid hydrolases to extracellular fluid
Autophagic vacuole forming around mitochondrion Plasma
membrane
Lysosome Residual
body
Release of nutrients as digestion progresses
ER Lysosome
Early endosome Early
endosome
Late endosome
Late endosome Endocytic vesicles
Endocytosis Phagocytosis
Phagocytic vacuole
Vesicles containing acid hydrolase
Golgi complex
D B
C
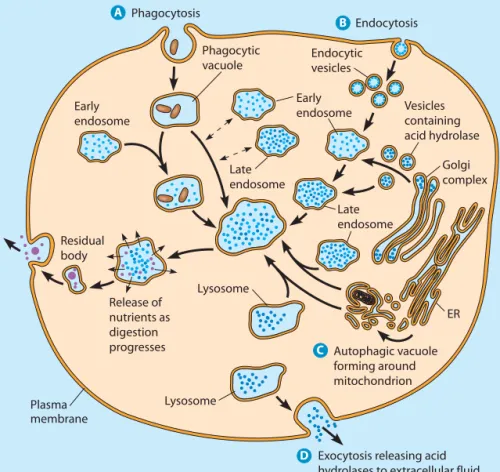
FIGURE 12-21 The Formation of Lysosomes and Their Roles in Cellular Digestive Processes. Illustrated in this composite cell are the major processes in which lysosomes are involved. The pathways depicted are phagocytosis,
receptor-mediated endocytosis,
autophagy, and extracellular digestion. For clarity, the nucleus is not shown.
D C
B
A
that they are sequestered from the rest of the cell, where they cannot quickly destroy the cell itself.
Lysosomes Develop from Endosomes
Lysosomal enzymes are synthesized by ribosomes attached to the rough ER and are translocated through a pore in the ER membrane into the ER lumen before transport to the Golgi complex. After modification and processing in the ER and Golgi complex compartments, the lysosomal enzymes are sorted from other proteins in the TGN. Earlier in the chapter, we described the addition of a unique mannose-6-phosphate tag to soluble lyso-somal enzymes. Distinctive sorting signals are also present on membrane-bound lysosomal proteins. The lysosomal enzymes are packaged in clathrin-coated vesicles that bud from the TGN, lose their protein coats, and travel to one of the endosomal compartments (see Figure 12-9).
Lysosomal enzymes are delivered from the TGN to endosomes in transport vesicles, as shown in Figure 12-21. Recall that early endosomes are formed by the coalescence of vesicles from the TGN and vesicles from the plasma membrane. Over time, the early endosome matures to form a late endosome, an organelle having a full complement of acid hydrolases but not engaged in digestive activity. As the pH of the early endosomal lumen drops from about 6.0 to 5.5, the organelle loses its capacity to fuse with endocytic vesicles. The late endosome is essentially a collection of newly synthesized digestive enzymes as well as extracellular
and intracellular material fated for digestion, packaged in a way that protects the cell from hydrolytic enzymes.
The final step in lysosome development is the activa-tion of the acid hydrolases, which occurs as the enzymes and their substrates encounter a more acidic environment. There are two ways eukaryotic cells accomplish this step. ATP-dependent proton pumps may lower the pH of the late endosomal lumen to 4.0–5.0, transforming the late endosome into a lysosome, thereby generating a new organelle. Alternatively, the late endosome may transfer material to the acidic lumen of an existing lysosome. Lysosomal Enzymes Are Important for Several Different Digestive Processes
Lysosomes are important for cellular activities as diverse as nutrition, defense, recycling of cellular components, and dif-ferentiation. We can distinguish the digestive processes that depend on lysosomal enzymes by the site of their activity and by the origin of the material that is digested, as shown in Figure 12-21. Usually, the site of activity is intracellular. In some cases, though, lysosomes may release their enzymes to the outside of the cell by exocytosis. The materials to be digested are often of extracellular origin, although there are also important processes known to involve lysosomal diges-tion of internal cellular components.
To distinguish between mature lysosomes of different origins, we refer to those containing substances of extracel-lular origin as heterophagic lysosomes,whereas those with