Acta Cryst.(2003). E59, o193±o194 DOI: 10.1107/S1600536803001089 Alexander D. Vasilievet al. C3H8N4O2
o193
organic papers
Acta Crystallographica Section E
Structure Reports
Online ISSN 1600-5368
1-Ethyl-2-nitroguanidine
Alexander D. Vasiliev,a*
Alexander M. Astachov,b
Maxim S. Molokeev,a
Ludmila A. Kruglyakovaband
Rudolf S. Stepanovb
aInstitute of Physics, Krasnoyarsk 660036,
Russia, andbSiberian State Technological
University, pr. Mira 82, Krasnoyarsk 660049, Russia
Correspondence e-mail: [email protected]
Key indicators Single-crystal X-ray study
T= 293 K
Mean(C±C) = 0.002 AÊ
Rfactor = 0.036
wRfactor = 0.106
Data-to-parameter ratio = 11.5
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2003 International Union of Crystallography Printed in Great Britain ± all rights reserved
The structure of the title compound, C3H8N4O2, is similar to
other nitrimines. The nitroguanyl group is planar and is stabilized by an intramolecular hydrogen bond. Intermole-cular hydrogen bonds hold the molecules together in the crystal.
Comment
As a continuation of work on the structures of nitrimines (Choi, 1981; Nordenson, 1981a,b; Nordenson & Hvoslef, 1981; Riceet al., 1984; Oyumiet al., 1987; Gaoet al., 1991; Astachov
et al., 2001; Vasiliev et al., 2001), we report here the crystal structure of 1-ethyl-2-nitroguanidine, (I). The molecular structure of (I) corresponds to that of previously investigated nitrimines. Like other nitrimines, the delocalization of -electron density gives values of CÐN, NÐN and NÐO bond lengths intermediate between those characteristic of single and double bonds (Table 1). The formal double bond C1 N2 is, in fact, 0.052±0.057 AÊ longer than the C1ÐN3 and C1ÐN4 bonds. The planar geometry of the nitroguanyl group (r.m.s. deviation 0.065 AÊ and maximum deviation 0.104 AÊ) is stabil-ized by an O1 H1 intramolecular hydrogen bond (Table 2). In other nitrimines, the NÐH O angles are in the range 105± 126, and the O H distance is in the range 1.72±2.24 AÊ (Allen, 2002). The ethyl group does not participate in con-jugation of the nitroguanyl fragment and, as a consequence, the N4ÐC2 bond length is close to those observed in compounds with a single CÐN bond (Allen, 2002).
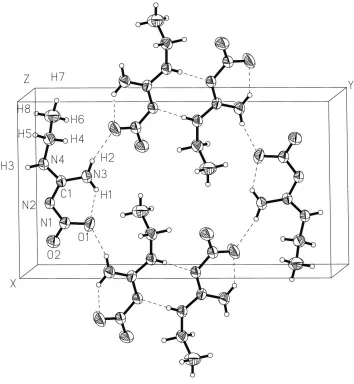
In the crystal structure, each molecule of (I) is connected by three intermolecular hydrogen bonds with two neighbouring molecules (Fig. 1 and Table 2). Atom O1 is involved in intramolecular hydrogen bonding and forms a hydrogen bond with H2 of another molecule. In addition, the molecules are connected, in pairs, by two N2 H3 hydrogen bonds. The second O atom, O2, does not participate in hydrogen bonding and, for this reason, the N1ÐO2 bond is shorter than the N1Ð O1 bond by 0.027 AÊ. This hydrogen-bond network is typical for nitrimines (Allen, 2002).
The structure of (I) agrees well with the methyl derivative of nitroguanidine 1-methyl-2-nitroguanidine, (II) (Nordenson, 1981a). The low-temperature study of (II) exhibits almost identical geometry and, with the exception of N1ÐN2, all
other differences in bond lengths between (I) and (II) do not exceed 2. The molecular packing in (I) and (II) is also very similar: they have the same space-group symmetry and number of molecules in the unit cell, and the same hydrogen bonds with nearly equal geometric parameters.
Experimental
Compound (I) was synthesized as described previously by Fishbein & Gallaghan (1954). Single crystals were obtained by evaporation in air of an aqueous solution of (I).
Crystal data
C3H8N4O2
Mr= 132.13 Monoclinic,P21=n
a= 8.9336 (9) AÊ b= 16.087 (2) AÊ c= 4.3173 (4) AÊ
= 96.119 (8)
V= 616.92 (11) AÊ3
Z= 4
Dx= 1.423 Mg mÿ3 Cu Kradiation Cell parameters from 25
re¯ections
= 20±27
= 1.02 mmÿ1
T= 293 (2) K Lump, colourless 0.320.270.26 mm
Data collection
Kuma KM-4 diffractometer
/2scans
Absorption correction: none 1325 measured re¯ections 1168 independent re¯ections 1049 re¯ections withI> 2(I) Rint= 0.034
max= 69.9
h=ÿ10!10 k=ÿ19!16 l= 0!5
2 standard re¯ections every 50 re¯ections intensity variation: 0.5%
Re®nement
Re®nement onF2
R[F2> 2(F2)] = 0.036
wR(F2) = 0.106
S= 1.04 1168 re¯ections 102 parameters
H atoms treated by a mixture of independent and constrained
w= 1/[2(F
o2) + (0.0632P)2 + 0.1271P]
whereP= (Fo2+ 2Fc2)/3 (/)max< 0.001
max= 0.19 e AÊÿ3
min=ÿ0.16 e AÊÿ3
Extinction correction:SHELXL97 Extinction coef®cient: 0.044 (3)
Table 1
Selected geometric parameters (AÊ,). N2ÐN1 1.3276 (15) N2ÐC1 1.3718 (16) O2ÐN1 1.2303 (15) O1ÐN1 1.2555 (15)
C1ÐN3 1.3164 (17) C1ÐN4 1.3188 (17) N4ÐC2 1.4528 (17) C2ÐC3 1.502 (2)
N1ÐN2ÐC1 119.72 (10) N3ÐC1ÐN4 121.28 (12) N3ÐC1ÐN2 126.41 (12) N4ÐC1ÐN2 112.31 (11) C1ÐN4ÐC2 125.65 (11)
O2ÐN1ÐO1 120.36 (11) O2ÐN1ÐN2 115.99 (11) O1ÐN1ÐN2 123.64 (11) N4ÐC2ÐC3 113.56 (12)
N1ÐN2ÐC1ÐN3 4.4 (2) N1ÐN2ÐC1ÐN4 ÿ176.27 (11) N3ÐC1ÐN4ÐC2 ÿ1.5 (2) N2ÐC1ÐN4ÐC2 179.13 (12)
C1ÐN2ÐN1ÐO2 ÿ176.20 (12) C1ÐN2ÐN1ÐO1 4.9 (2) C1ÐN4ÐC2ÐC3 89.75 (17)
Table 2
Hydrogen-bonding geometry (AÊ,).
DÐH A DÐH H A D A DÐH A
N4ÐH3 N2i 0.82 (2) 2.23 (2) 3.035 (2) 170 (1)
N3ÐH2 O1ii 0.87 (2) 2.04 (2) 2.896 (2) 165 (1)
N3ÐH1 O1 0.85 (2) 1.89 (2) 2.565 (2) 136 (1)
Symmetry codes: (i) 1ÿx;ÿy;ÿz; (ii)xÿ1
2;12ÿy;12z.
H atoms were located in a difference Fourier map and were re®ned as riding atoms for CH2and CH3groups and as free atoms for NH
and NH2groups. Furthermore, the CÐH distance was re®ned as one
parameter for CH2and CH3groups.
Data collection: KM-4 Software (Kuma, 1991); cell re®nement: KM-4 Software; data reduction: DATARED in KM-4 Software; program(s) used to solve structure: SHELXS97 (Sheldrick, 1997); program(s) used to re®ne structure:SHELXL97 (Sheldrick, 1997); molecular graphics:SHELXTL (Sheldrick, 1995); software used to prepare material for publication:SHELXL97.
We are grateful to the Russian Foundation of Fundamental Investigations for partial ®nancial support of the work (grant No. 00-15-96790).
References
Allen, F. H. (2002).Acta Cryst.B58, 380±388.
Astachov, A. M., Gelemurzina, I. V., Vasiliev, A. D., Nefedov, A. A., Kruglyakova, L. A. & Stepanov, R. S. (2001).Energetic Materials ± Ignition, Combustion and Detonation. 32st International ICT Conference, July 3±6, 2001, Karlsruhe, Germany, pp. 139/1±139/10.
Choi, C. S. (1981).Acta Cryst.B37, 1955±1957.
Fishbein, L. & Gallaghan, J. A. (1954).J. Am. Chem. Soc.76, 1877±1879. Gao, A., Rheingold, A. L. & Brill, T. B. (1991).Propel. Explos. Pyrotechnics,
16, 97±104.
Kuma (1991).KM-4Software. Kuma Diffraction, Wrocøaw, Poland. Nordenson, S. (1981a).Acta Cryst.B37, 1543±1547.
Nordenson, S. (1981b).Acta Cryst.B37, 1774±1776. Nordenson, S. & Hvoslef, J. (1981).Acta Cryst.B37, 373±378.
Oyumi, Y., Rheingold, A. L. & Brill, T. B. (1987).Propel. Explos. Pyrotechnics,
12, 46±52.
Rice, S., Cheng, M. Y., Cramer, R. E., Mandel, M., Mower, H. F. & Seff, K. (1984).J. Am. Chem. Soc.106, 239±243.
Sheldrick, G. M. (1995).SHELXTL.Version 5. Siemens Analytical X-ray Instruments Inc., Madison, Wisconsin, USA.
Sheldrick, G. M. (1997). SHELXS97 and SHELXL97. University of GoÈttingen, Germany.
Vasiliev, A. D., Astachov, A. M., Nefedov, A. A., Kruglyakova, L. A. &
Figure 1
supporting information
sup-1
Acta Cryst. (2003). E59, o193–o194supporting information
Acta Cryst. (2003). E59, o193–o194 [doi:10.1107/S1600536803001089]
1-Ethyl-2-nitroguanidine
Alexander D. Vasiliev, Alexander M. Astachov, Maxim S. Molokeev, Ludmila A. Kruglyakova and
Rudolf S. Stepanov
S1. Comment
As a continuation of work on the structures of nitrimines (Choi, 1981; Nordenson, 1981a,b; Nordenson & Hvoslef, 1981;
Rice et al., 1984; Oyumi et al., 1987; Gao et al., 1991; Astachov et al., 2001; Vasiliev et al., 2001), we report here the
structure of crystalline 1-ethyl-2-nitroguanidine, (I). The molecular structure of (I) corresponds to that of previously
investigated nitrimines. Like other nitrimines, the delocalization of π-electron density gives values of C—N, N—N and N
—O bond lengths intermediate between characteristic values of single and double bonds (Table 1). The formally double
bond C1═N2 is in fact longer by 0.052–0.057 Å than the C1—N3 and C1—N4 bonds. The planar geometry of the
nitro-guanile group (r.m.s. deviation 0.065 Å and maximum deviation 0.104 Å) is stabilized by an O1···H1 intramolecular
hydrogen bond (Table 2). In other nitrimines, the N—H···O angles are in the range 105–126° and the O···H distance is in
the range 1.72–2.24 Å (Allen & Kennard, 1993). The ethyl group does not participate in conjugation of the nitroguanyl
fragment and as a consequence the N4—C2 bond length is close to those observed in compounds with single C—N bond
(Allen, 2002).
In the crystal, each molecule of (I) is connected by three intermolecular hydrogen bonds with two neighbouring
molecules (Fig. 1 and Table 2). Atom O1 is involved in intramolecular hydrogen bonding and forms a hydrogen bond
with H2 of another molecule. Besides that the molecules are connected in pairs by two N2···H3 hydrogen bonds. The
second oxygen atom, O2, does not participate in hydrogen bonds and, for this reason, the N1—O2 bond length is shorter
by 0.027 Å than the N1—O1 bond. The similar developed hydrogen-bond net is typical for the nitrimines (Allen, 2002).
The structure of (I) corresponds well with the methyl derivative of nitroguanidine-1-methyl-2-nitroguanidine, (II)
(Nordenson, 1981a). The low-temperature study of (II) exhibits almost equal geometry and, with the exception of N1—
N2, all other differences in bond lengths between (I) and (II) do not exceed 2σ. The molecular packing in (I) and (II) is
also very similar: they have the same space-group symmetry and number of molecules in the unit cell, and the same
hydrogen bonds with nearly equal geometric parameters.
S2. Experimental
Compound (I) was synthesized as described previously by Fishbein & Gallaghan (1954). Single crystals were obtained by
evaporation in air of an aqueous solution of (I).
S3. Refinement
H atoms were found in a difference Fourier map and were refined as riding atoms for CH2 and CH3 groups and as free
Figure 1
The molecular arrangement (I) in a crystal, with the atomic numbering scheme. Dashed lines indicate intra- and
intermolecular hydrogen bonds.
1-Ethyl-2-nitroguanidine
Crystal data
C3H8N4O2
Mr = 132.13 Monoclinic, P21/n
Hall symbol: -P 2yn
a = 8.9336 (9) Å
b = 16.087 (2) Å
c = 4.3173 (4) Å
β = 96.119 (8)°
V = 616.92 (11) Å3
Z = 4
F(000) = 280
Dx = 1.423 Mg m−3
Cu Kα radiation, λ = 1.5418 Å Cell parameters from 25 reflections
θ = 20–27°
µ = 1.02 mm−1
T = 293 K
supporting information
sup-3
Acta Cryst. (2003). E59, o193–o194Data collection
Kuma KM-4 diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
θ/2θ scans
1325 measured reflections 1168 independent reflections 1049 reflections with I > 2σ(I)
Rint = 0.034
θmax = 69.9°, θmin = 5.5°
h = −10→10
k = −19→16
l = 0→5
2 standard reflections every 50 reflections intensity decay: variation 0.5%
Refinement
Refinement on F2
Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.036
wR(F2) = 0.106
S = 1.04 1168 reflections 102 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: difference Fourier map H atoms treated by a mixture of independent
and constrained refinement
w = 1/[σ2(F
o2) + (0.0632P)2 + 0.1271P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001
Δρmax = 0.19 e Å−3
Δρmin = −0.16 e Å−3
Extinction correction: SHELXL97, Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4
Extinction coefficient: 0.044 (3)
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
N2 0.57727 (12) 0.09681 (6) 0.0506 (3) 0.0382 (3)
O2 0.77500 (13) 0.12035 (7) −0.1876 (3) 0.0613 (4)
O1 0.69759 (14) 0.22180 (6) 0.0805 (3) 0.0653 (4)
C1 0.46666 (14) 0.12472 (7) 0.2220 (3) 0.0338 (3)
N4 0.37411 (13) 0.06484 (7) 0.2869 (3) 0.0385 (3)
H3 0.3882 (18) 0.0184 (12) 0.219 (4) 0.048 (4)*
N1 0.68449 (13) 0.14855 (7) −0.0192 (3) 0.0423 (3)
N3 0.44819 (16) 0.20158 (8) 0.3154 (3) 0.0515 (4)
H2 0.367 (2) 0.2150 (12) 0.399 (5) 0.064 (5)*
H1 0.518 (2) 0.2338 (13) 0.271 (5) 0.068 (6)*
C2 0.24698 (15) 0.07412 (8) 0.4678 (3) 0.0425 (4)
H4 0.2700 0.1168 0.622 0.046 (4)*
H5 0.2329 0.0229 0.576 0.060 (5)*
C3 0.10261 (18) 0.09622 (12) 0.2744 (4) 0.0623 (5)
H7 0.0247 0.1029 0.406 0.085 (7)*
H8 0.0761 0.0530 0.128 0.084 (7)*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
N2 0.0375 (6) 0.0255 (6) 0.0531 (7) −0.0023 (4) 0.0122 (5) −0.0029 (4)
O2 0.0527 (7) 0.0493 (7) 0.0879 (8) −0.0023 (5) 0.0354 (6) −0.0077 (6)
O1 0.0680 (8) 0.0315 (6) 0.1025 (9) −0.0163 (5) 0.0380 (7) −0.0124 (5)
C1 0.0350 (6) 0.0260 (6) 0.0400 (6) 0.0019 (5) 0.0027 (5) −0.0004 (4)
N4 0.0413 (6) 0.0268 (6) 0.0490 (7) −0.0002 (4) 0.0119 (5) −0.0024 (4)
N1 0.0405 (6) 0.0292 (5) 0.0589 (7) −0.0016 (4) 0.0125 (5) 0.0005 (5)
N3 0.0491 (8) 0.0283 (6) 0.0813 (9) −0.0009 (5) 0.0262 (7) −0.0089 (6)
C2 0.0452 (8) 0.0369 (7) 0.0475 (7) −0.0008 (6) 0.0150 (6) 0.0017 (5)
C3 0.0420 (9) 0.0712 (12) 0.0736 (11) −0.0015 (8) 0.0054 (8) −0.0116 (9)
Geometric parameters (Å, º)
N2—N1 1.3276 (15) N3—H2 0.87 (2)
N2—C1 1.3718 (16) N3—H1 0.85 (2)
O2—N1 1.2303 (15) C2—C3 1.502 (2)
O1—N1 1.2555 (15) C2—H4 0.9626
C1—N3 1.3164 (17) C2—H5 0.9626
C1—N4 1.3188 (17) C3—H6 0.9515
N4—C2 1.4528 (17) C3—H7 0.9515
N4—H3 0.817 (18) C3—H8 0.9515
N1—N2—C1 119.72 (10) N4—C2—C3 113.56 (12)
N3—C1—N4 121.28 (12) N4—C2—H4 108.9
N3—C1—N2 126.41 (12) C3—C2—H4 108.9
N4—C1—N2 112.31 (11) N4—C2—H5 108.9
C1—N4—C2 125.65 (11) C3—C2—H5 108.9
C1—N4—H3 117.8 (11) H4—C2—H5 107.7
C2—N4—H3 116.5 (11) C2—C3—H6 109.5
O2—N1—O1 120.36 (11) C2—C3—H7 109.5
O2—N1—N2 115.99 (11) H6—C3—H7 109.5
O1—N1—N2 123.64 (11) C2—C3—H8 109.5
C1—N3—H2 119.8 (13) H6—C3—H8 109.5
C1—N3—H1 112.8 (14) H7—C3—H8 109.5
H2—N3—H1 127 (2)
N1—N2—C1—N3 4.4 (2) C1—N2—N1—O2 −176.20 (12)
N1—N2—C1—N4 −176.27 (11) C1—N2—N1—O1 4.9 (2)
N3—C1—N4—C2 −1.5 (2) C1—N4—C2—C3 89.75 (17)
supporting information
sup-5
Acta Cryst. (2003). E59, o193–o194Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
N4—H3···N2i 0.82 (2) 2.23 (2) 3.035 (2) 170 (1)
N3—H2···O1ii 0.87 (2) 2.04 (2) 2.896 (2) 165 (1)
N3—H1···O1 0.85 (2) 1.89 (2) 2.565 (2) 136 (1)