ACKNQMLEDGEflENTS
I would like to thank my wife and son for their patience
and cooperation. Dr. Singer for being a very helpful and
understanding advisor. Dr. Digiano and Dr. Leith for their beneficial suggestions, and Jose Felix—Filho and Ning—Wu
TABLE OF CONTENTS
page l.O INTRODUCTION... 1-1 2.0 THEORY AND LITERATURE REVIEW...___... 2-1
2.1 PARTICLES AND PARTICLE AGGREGATION...2-1
2.1.1 PARTICLES IN WATER... 2-1 2.1-2 FACTORS AFFECTING PARTICLE AGGREGATION 2-2
2.1.3 THE EFFECT OF DISSOLVED ORGANIC
MATERIAL ON PARTICLE STABILITY... 2-3 2-1-4 THE EFFECT OF Ca^"- ON PARTICLE
STABILITY... 2-5 2.2 OZONE APPLICATIONS IN WATER TREATMENT... 2-7 2.2.1 BENEFICIAL EFFECTS OF OZONE ____.____. 2-7 2.2.2 ADVERSE EFFECTS OF OZONE... 2-3 2.3 OZONE EFFECTS ON PARTICLE STABILITY... 2-11
2.3.1 OZONE EFFECTS WITHOUT COAGULANT
ADDITION... 2-12 2.3-1.1 NATURAL WATERS... 2-12 2.3.1.2 MODEL WATERS... 2-14 2.3-2 OZONE EFFECTS WITH COAGULANT ADDITION. 2-15 2.3.2.1 NATURAL WATERS... 2-15 2.3.2.2 MODEL WATERS... 2-10 2.3.3 TRENDS IN OZONE EFFECTS... 2-18 2.3.4 OZONATION OF HUMIC SUBSTANCES... 2-20 2.3.5 POTENTIAL MECHANISMS OF OZONE-INDUCED
MICROFLOCCULATION... 2-22 3.0 MATERIALS AND METHODS. ... 3-1
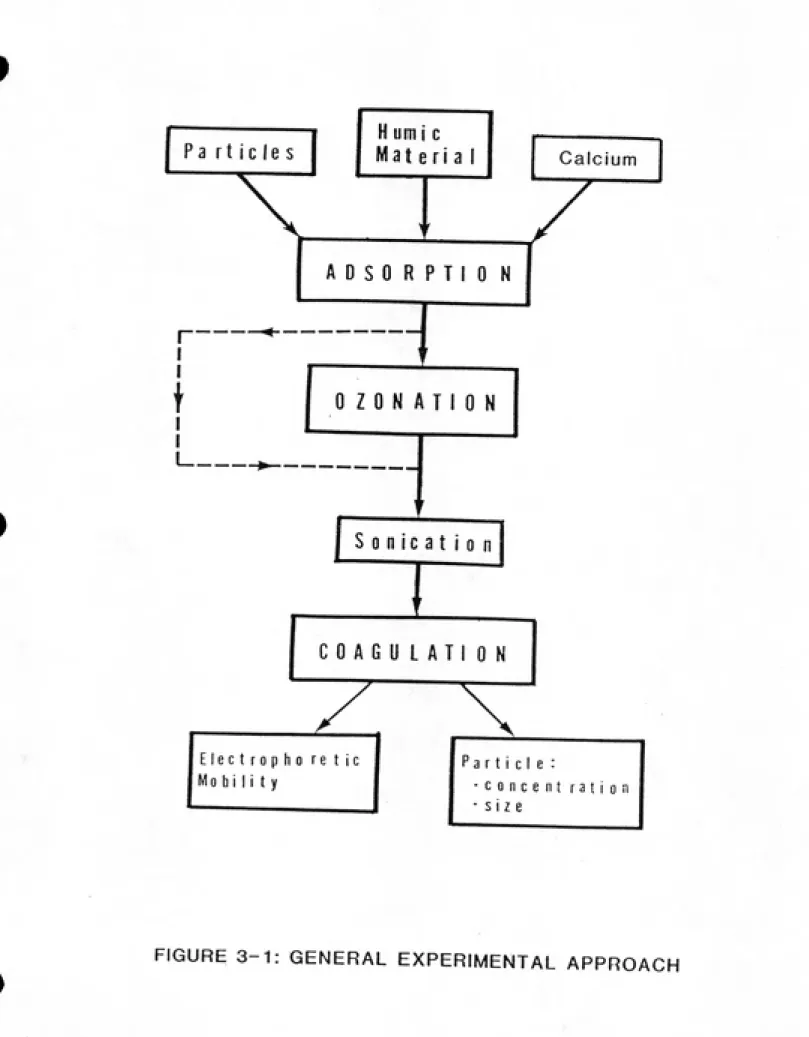
3.1 GENERAL EXPERIMENTAL APPROACH... 3-1 3.E SOLID PHASE____... 3-3 3.2.1 RATIONALE FOR USING a-AlaOg...3-3 3.2.2 CLEANING...____.____3-4
3.2.3 SIZE FRACTIONATION... 3-5
3.2.4 PREPARATION AND HANDLING OF STOCK
SUSPENSIONS... 3-5 3.3 HUMIC MATERIAL...- ... 3-h
3.4 OZONE...____3-7 3.4.1 PREPARATION... 3-7 3.4.2 MEASUREMENT. ...3-9
3.5 DETAILED EXPERIMENTAL PROCEDURE____...3-10 3-5.1 CALCIUM / HUMIC MATERIAL / OZONE
3.5.2 ALUM / HUMIC MATERIAL / OZONE
EXPERIMENTS... 3-15
3.6 EXPERIMENTAL REPRODUCIBILITY AND ACCURACY--- 3-16 3.7 ADSORPTION PROCEDURE... 3-18 3.8 SETTLING AND FILTERING TESTS... 3-19 3.9 DATA HANDLING... 3-EO 4.0 EXPERIMENTAL RESULTS AND DISCUSSION... 4-1
4.1 INTRODUCTION... 4-1 4.e CHARACTERIZATION OF THE SOLID PHASE...--- 4-E
4.3 THE STABILIZING EFFECT OF AQUATIC HUMIC
MATERIAL... 4-S 4.4 DESTABILIZATION BY ALUM...--- 4-7
4.5 THE EFFECT OF CALCIUM ON THE STABILITY OF
PARTICLES... 4-10 4.6 ELECTROPHORETIC MOBILITY... 4-14 4.7 OZONATION EFFECTS... 4-15 4.8 IMPLICATIONS OF EXPERIMENTAL RESULTS... 4-ai
5.0 MODELING OF EXPERIMENTAL RESULTS... 5-1
5.1 INTRODUCTION... 5-1 5.S MODEL DEVELOPMENT...__ 5-1
5.3 SENSITIVITY TESTING OF THE MODEL... 5-10 5.4 MODEL INPUTS... 5-12 5.5 COMPARISONS OF EXPERIMENTAL RESULTS AND MODEL
PREDICTIONS... 5-lS 5.6 IMPLICATIONS OF MODELING RESULTS... 5-18
REFERENCES... ref-1
APPENDIX A : PARTICLE SIZE ANALYSIS AND
ELECTROPHORETIC MOBILITY... A-1
A.l BASICS OF RESISTIVITY BASED PARTICLE SIZE
ANALYSIS... A-1 A.e BASICS OF ELECTROPHORETIC MOBILITY
MEASUREMENT... A-^
LIST OF FIGURES
FIGURE FOLLOWS PAGE 3-1 GENERAL EXPERIMENTAL APPROACH...3-1
3-a INITIAL VOLUME DISTRIBUTION...-... 3-3
4-1 CHANGES IN THE TOTAL NUMBER OF PARTICLES WITH
TIME : EFFECTS OF HUMIC ACID... 4-2 4-2 EFFECT OF FULVIC AND HUMIC ACID ON THE STABILITY
OF ALUMINA PARTICLES... 4-4
4-3 IMPACT OF FULVIC AND HUMIC ACID ON
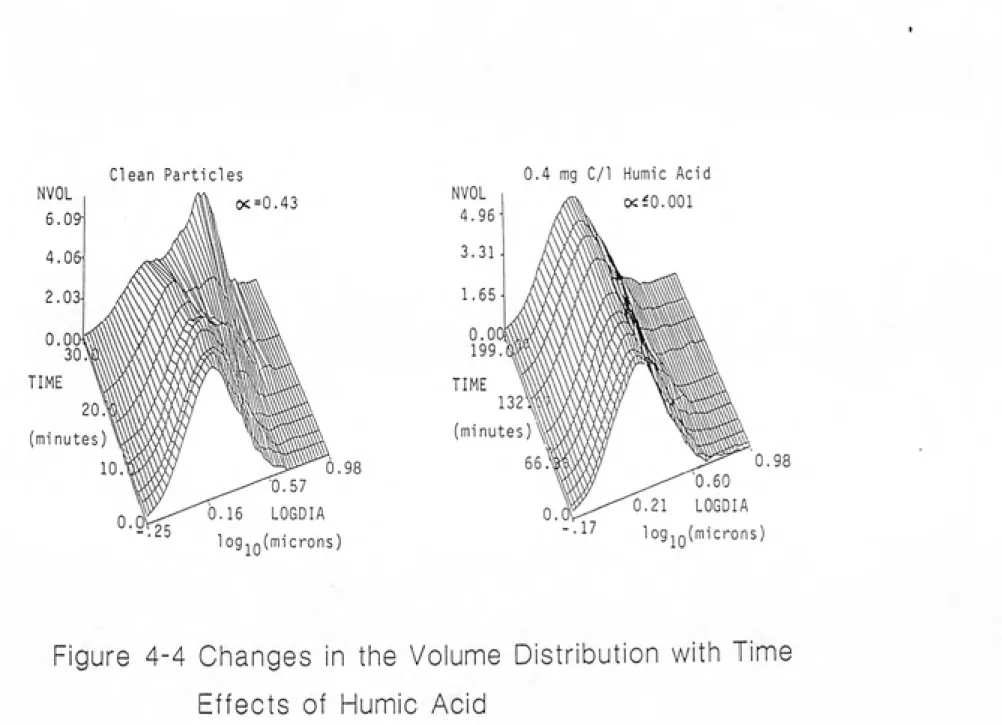
ELECTROPHORETIC MOBILITY OF ALUMINA PARTICLES .. 4-5 4-4 CHANGES IN THE VOLUME DISTRIBUTION WITH TIME
EFFECTS OF HUMIC ACID... 4-6 4-5 PARTICLE DESTABILIZATION BY ALUM IN
ADSORPTION-CHARGE NEUTRAL I ZAT I ON...4-8 4-6 CHANGES IN THE VOLUME DISTRIBUTION WITH TIME
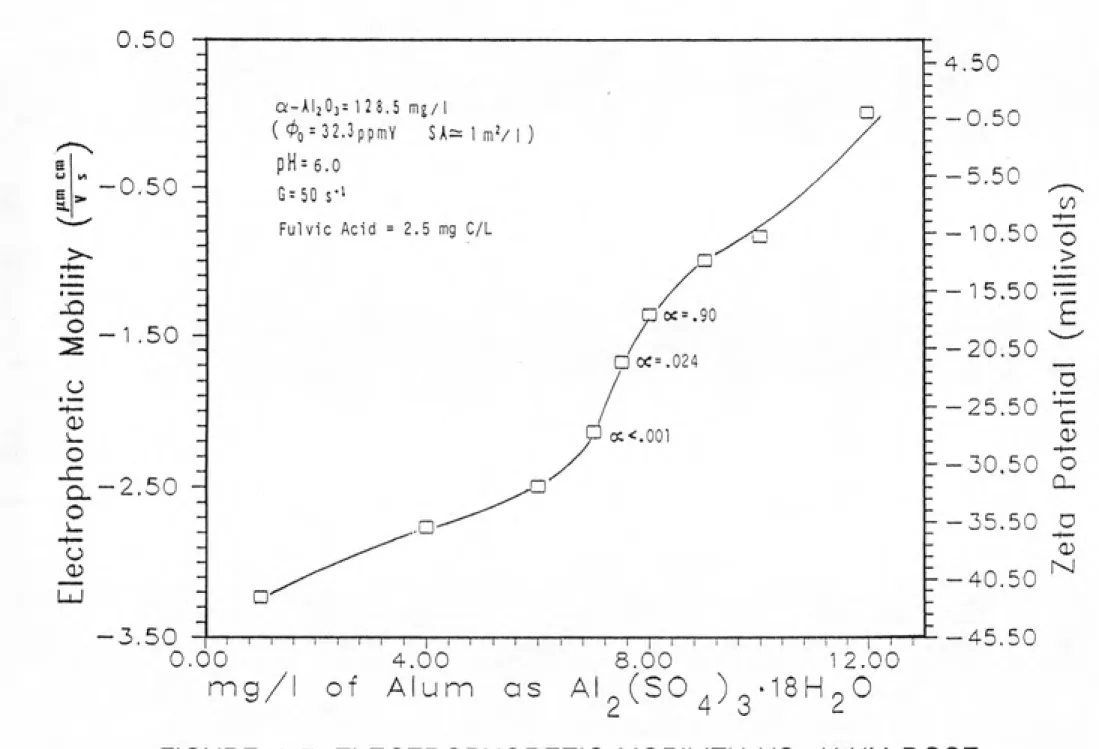
DESTABILIZATION WITH ALUM...4-8 4-7 ELECTROPHORETIC MOBILITY VS. ALUM DOSE ... 4-9 4-8 CHANGE IN THE TOTAL NUMBER OF PARTICLES WITH TIME
EFFECTS OF CALCIUM WITH 0.4 mg C/1 HUMIC ACID .. 4-10
4-9 EFFECTS OF CALCIUM ON THE STABILITY OF ALUMINA
WITH 0.4 mg C/1 HUMIC ACID... 4-10
4-10 CHANGES IN THE VOLUME DISTRIBUTION WITH TIME
EFFECTS OF CALCIUM WITH 0.4 mg C/1 HUMIC ACID.. 4-10
4-11 EFFECTS OF CALCIUM ON THE ELECTROPHORETIC
MOBILITY OF ALUMINA PARTICLES WITH 0.4 mg C/1
HUMIC ACID... 4-10 4-IE EFFECTS OF CALCIUM ON THE STABILITY OF ALUMINA
WITH 5.0 mg C/1 FULVIC ACID ...4-11
4-13 EFFECTS OF CALCIUM ON THE ELECTROPHORETIC
MOBILITY OF ALUMINA PARTICLES WITH 5.0 mg C/1
FULVIC ACID... 4-11
4-14 THE RELATIONSHIP BETWEEN ELECTROPHORETIC MOBILITY
AND THE COLLISION EFFICIENCY FACTOR OF ALUMINA
^-15 CHANGES IN THE VOLUME DISTRIBUTION WITH TIME
THE INABILITY OF OZONE TO DESTABILIZE ALUMINA
PARTICLES EQUILIBRATED WITH HUMIC MATERIAL ... h-15 4-16 CHANGES IN THE TOTAL NUMBER OF PARTICLES VS. TIME
EFFECTS OF OZONE WITH 1-O mM Ca and 0.4 mg C/1
HUMIC ACID...4-16
4-17 THE EFFECT OF OZONE AND CALCIUM ON PARTICLE
STABILITY AT 0.4 mg C/1 HUMIC ACID--...4-17 4-18 CHANGES IN THE VOLUME DISTRIBUTION WITH TIME
THE EFFECTS OF OZONE AT 1.0 mM Ca and 0.4 mg C/1
HUMIC ACID... 4-17
4-19 THE EFFECT OF OZONE AND CALCIUM ON PARTICLE
STABILITY...___... 4-17
4-20 THE EFFECT OF OZONE ON THE RELATIONSHIP BETWEEN
ELECTROPHORETIC MOBILITY AND THE COLLISION
EFFICIENCY FACTOR ... 4-18
5-1 SIMILAR PATHS OF AGGREGATION FOR DIFFERENT
VALUES OF ALPHA... 5-8 5-2 TEST OF INTEGRATION TIME STEP AND DISCRETIZATION
OF THE VOLUME DISTRIBUTION ... 5-11 5-3 CHANGES IN THE TOTAL NUMBER OF PARTICLES WITH TIME
PREDICTION CURVES VS. EXPERIMENTAL OBSERVATIONS. 5-14 5-4 CHANGES IN THE VOLUME DISTRIBUTION WITH TIME
PREDICTION CURVES VS. EXPERIMENTAL DATA POINTS.. 5-14 5-5 CHANGES IN THE VOLUME DISTRIBUTION OVER TIME
PREDICTION CURVES VS. EXPERIMENTAL OBSERVATIONS. 5-15
5-6 CHANGES IN THE VOLUME MEAN DIAMETER WITH TIME
PREDICTION CURVES VS. EXPERIMENTAL OBSERVATIONS. 5-15 5-7 CHANGES IN THE TOTAL NUMBER OF PARTICLES WITH TIME
EFFECT OF A SETTLING TERM___... 5-16 5-8 CHANGES IN THE VOLUME DISTRIBUTION WITH TIME
WITHOUT THE SETTLING TERM... 5-16
5-9 CHANGES IN THE VOLUME DISTRIBUTION WITH TIME
WITH THE SETTLING TERM ... 5-16
5-10 CHANGES IN THE VOLUME MEAN DIAMETER WITH TIME
EFFECT OF A SETTLING TERM ... 5-I6
A-1 DIAGRAM OF THE ELZONE PARTICLE SIZE ANALYZER ... A-1
F-1 CHANGES IN THE VOLUME DISTRIBUTION WITH TIME
LIST OF TABLES
TABLE PAGE
^-1 THE EFFECT OF AQUATIC HUMIC MATERIAL ON THE
STABILITY OF ALUMINA PARTICLES (pH=7.5> ... ^-3
4-E THE EXTENT OF ADSORPTION OF NATURAL ORGANIC
MATERIAL ON a-AlsO^ and y-AleOg ... 4-7 ^-3 ADSORPTION-CHARGE NEUTRALIZATION WITH ALUM
(2.5 mg C/1 of FULVIC ACID, pH=6.0) ...4-9
4-4 EFFECTS OF Ca^* ON THE STABILITY OF ALUMINA... 4-lS 4-5 OZONATION OF ALUMINA PARTICLES EQUILIBRATED WITH
HUMIC MATERIAL... 4-16 4-6 THE CORRELATION BETWEEN ALPHA AND PARTICLE /
TURBIDITY REMOVAL ... follows pg. 4-18
4-7 PRE-OZONATION PRIOR TO DOSING WITH ALUM
(S.5 mg C/1 FULVIC ACID, pH=6.0)...4-19 4-8 RAW WATERS SUSCEPTIBLE TO THE COAGULATING EFFECTS
OF OZONE ... follows pg. 4-EO
5-1 SENSITIVITY OF THE MODEL TO THE TIME STEP... 5-11 D-1 SUMMARY TABLE : HUMIC ACID, CALCIUM AND ALUMINA
EXPERIMENTAL RESULTS ... D-1
D-S SUMMARY TABLE : FULVIC ACID, CALCIUM, AND ALUMINA
EXPERIMENTAL RESULTS ... ... D-S D-3 SUMMARY TABLE : FULVIC ACID, ALUMINA, AND ALUM
INTRODUCTION
"Most pollutants in water are particles or are
associated with particles" <0'Melia, 1980; Lawler et al, 1980). Particles in water include inorganic solids such as
clays, metal oxides and asbestos -fibers, and organic
particles such as humic substances? algae? protozoan cysts,
bacteria? and viruses. The health significance of particles is amplified by the adsorption? complexation? and
bioaccumulation of toxic metals and synthetic organic
chemicals. Consequently? it is desirable in water treatment?
wastewater treatment? and in polluted surface waters to
maximize particle aggregation and hence sedimentation and removal.
In this research? some of the factors which control
particle aggregation in natural waters and water treatment
plants are evaluated through a review of the literature?
mathematical modeling? and controlled laboratory experiments.
Orthokinetic coagulation rate experiments? coupled with particle size distribution measurements? were used to
evaluate the impact of humic material? calcium? and ozone on
the colloidal stability of a-AlaCfc,. Electrophoretic mobility (EPM) was also measured to allow a comparison between EPM and the particle collision efficiency factor («), which is the fraction of collisions which result in aggregation.
The experimental conditions were selected to simulate
natural waters and water treatment conditions. The alumina
particles such as clays, which are composed o-f aluminum and
silicon oxides.
Aquatic humic material was used to evaluate the extent o-f particle stabilization associated with various
concentrations of dissolved organic material. A number of investigators have suggested that natural organic material controls the stability, and to some extent, the surface
chemistry of aquatic particles, but much of this evidence is
based solely on EPM measurements without evaluating the amount of adsorbed organic material necessary to stabilize the particles.
Several investigators have shown that high levels of
calcium hardness correspond to greater rates of aggregation
in model and natural waters. The extent of destabi1ization
observed for various levels of calcium hardness is quite
varied in the literature (Eppler et al, 1975; Jekel, 19SO;
Ali and O'Melia, 1984). Experiments were performed in this
research to evaluate the effect of calcium hardness on the
stability of alumina particles at a low concentration of
humic acid <0.4 mg C/L) and at a high concentration of fulvic acid (5 mg C/L).
Approximately 1,000 water treatment plants in Europe use
ozone- The principal uses of ozone are oxidation,
particularly for taste and odor removal, disinfection, and to
increase biodegradabi1ity of the organic material. The use
of ozone in the U.S. is presently small but rapidly
increasing. Approximately 8 U.S. water treatment plants were
using ozone in 19SO (Rice et al, 1981) and S4 plants in 1985
(Rice, 1985) . The primary use o-f ozone in the U.S. is as a
pre—oxidant, particularly -for taste and odor control. Due to
the present and future regulations on organic halides and
synthetic organic chemicals, and to the rising expectations
o-f pure, odor—free water by the U.S. consumer (Prendivi 1 le,
1985), the use o-f ozone in the U.S. is anticipated to
significantly increase. Ozone has not yet been adopted widely in the United States because of its relatively high
cost (Singer, 1935). However, recent applications of ozone
as a microflocculant in the United States have demonstrated
substantial savings in overall treatment cost by allowing higher filtration rates, decreased coagulant requirements,
and subsequent decreases in solids handling costs
(Prendiville, 1985).
The factors causing certain raw waters to be
susceptible to ozone—induced microflocculation are not well established. Hypotheses relating ozone benefits to algal content, metal cations and/or levels of organic carbon have
been proposed (Reckhow et al, 1986). Felix-Filho (1985)
showed that increased levels of iron, added as Fe(II),
increased the susceptibility to ozone-induced
microflocculation for suspensions of alumina particles
equilibrated with solutions of iron and humic material at pH=7.5. This research extends the work of Felix-Filho
(1985), by testing the effect of calcium on the
susceptibility to ozone—induced microflocculation.
Suspensions tested consist of alumina particles equilibrated
concentrations of aquatic humic material <0.4 mg C/L humic
acid and 5.0 mg C/L fulvic acid).
Experiments with alum under adsorption—charge
neutralization destabi1ization conditions (pH=6.0) were conducted to investigate ozone trends under water treatment conditions.
The objectives of this research 3ire as follow:
• Quantify the effect of various levels of humic material on the stability of alumina particles at pH=7.5. This
extends the work of Felix—Filho (1985) and tests the
hypothesis of Davis (1980> that under conditions typical
of natural waters, only small amounts of dissolved organic carbon adsorb to suspended particles, yet this organic material may significantly influence surface properties of particles, such as surface charge and colloidal stability.
• Evaluate the effect of Ca^"^ on the stability of alumina
particles equilibrated with solutions of calcium and humic material. This extends the work of Ali et al (198'^), who
demonstrated that higher levels of calcium in lakes may
result in increased rates of particle aggregation.
• Test the effect of calcium and humic material on the
susceptibility of alumina particles to ozone-induced microflocculation. This extends the work of Felix-Filho
(1985) and provides for the laboratory testing of an
apparent trend in the literature of raw waters of higher
calcium hardness demonstrating a higher susceptibility to ozone—induced
microflocculation-• Measure the electrophoretic mobility (EPM) of all
suspensions to allow a comparison of this surrogate
measure of particle stability to rigorous measurements of
changes in the particle size distribution with time.
• Test a mathematical model for particle aggregation
using the Smoluchowski equation. Test the ability of this
CHAPTER 2
THEORY AND LITERATURE REVIEW
2.1 Particles and Particle Aggregation.
2.1.1 Particles in Water.
Particles in water include inorganic solids such as
clays, metal oxides? and asbestos -fibers? and organic
particles such as humic substances? algae? protozoan cysts?
bacteria? and viruses. The health signi-ficance of particles
is ampli-fied by their ability to adsorb? complex? and bio-accumulate toxic metals and synthetic organic chemicals.
Consequently? it is desirable in water treatment? wastewater
treatment? and in polluted surface waters to maximize
particle removal. Stokes law -for particle settling predicts
faster settling and hence increased removal for larger
particles. The Kozeny equation -for -filtration predicts less
headloss for flow through filters of larger particles. By
combining the theoretical equations for particle settling and
filtration with a numerical aggregation model using
Smoluchowski's equations for the aggregation of particles?
Lawler et al <1980> and O'Melia (1980) have shown that
particle aggregation results in greater removal of particles
by sedimentation and filtration? and that larger aggregates
create less headloss when filtered through a packed-bed
E.l.E Factors Affecting Particle Aqgreqation.
The theoretical equation for predicting the rate of particle aggregation is Smoluchowski's equation. A detailed
discussion of Smoluchowski's equation and other complexities
of modeling particle aggregation is provided in Chapter 5.
Despite the complexities of particle aggregation? the
major factors influencing aggregation may be generalized as follow:
MAJOR FACTORS INFLUENCING THE RATE OF AGGREGATION
Velocity gradient
Number and size distribution of the particles
Shape of the particles (porous floe rather than coalesced spheres)
Density of the particles
Temperature and viscosity of the solution
Surface chemistry of the particles (sorbed material) Dissolved organic material
Natural polymers, biopolymers
Ionic Strength
Major Divalent Cations (Ca^^.MgS*)
Iron and Manganese / Other multivalent cations
Chemicals added in treatment plants (oxidants and coagulants)
The term "particle stability" is often used to describe the
resistance of particles to aggregation. Particles of high
stability aggregate slowly or not at all. Much of the
literature stresses the importance of adsorbed organic
material in controlling particle stability in natural waters-Howe ver , some evidence indicates that increased levels of
calcium and other cations in natural waters may somewhat
offset the stabilizing effect of natural organic material. Due to their importance in natural waters and this research,
the effects of dissolved organic material and calcium are
discussed further.
2.1.3 The Effect of Dissolved Organic Material on Particle
Stabi1ity.
An abundance of literature exists which indicates that dissolved organic material adsorbs onto particles and to a considerable extent controls their stability and surface chemistry. References will be presented to support this
statement and address its important but uncertain corollary
that all solid phases in a given solution will have similar
stability and surface chemistry due to the dominating role
of the dissolved organic material.
Davis and Gloor (1981) showed that when
positively-charged alumina particles were suspended in filtered lake water with a dissolved organic carbon (DOC) concentration of
3.3 mg C/Lj the zeta potential of the particles dropped from initially positive values over a pH range of 4-8.5 to quite negative values over the same pH range. When the lake water
was uv-oxidized to lower the DOC to a concentration of 0.5 mg
C/L before adding the particles, the zeta potential of the
particles was only slightly decreased, remaining positive for
Hunter (1980) found that the electrophoretic mobility
<EPM) of alumina and silica particles shifted to negative and
more negative values respectively, upon addition to filtered
seawater unless the seawater was exhaustively UV—oxidized
prior to adding the particles.
Tipping (1981), Davis <1982) and Tipping and Cooke
<19SH) have shown that the greater the organics coverage, the
more negative the EPM of coated oxide particles.
Numerous investigators (Kavanagh, Posner, and Quirk,
1977: Parfitt, Farmer, and Russell, 1977; Davis and Leckie,
1978; Kummert and Stumm, 1980) have studied the adsorption of
model organic compounds on metal oxide surfaces, and have
shown that adsorption occurs through ligand exchange as a
result of the types of functional groups associated with the
organic molecule.
Investigators have shown that in natural aquatic
systems, the DOC is typically in such excess that only a
small percentage will adsorb. Davis (198E) showed that the
percent of humic material adsorbing onto Y~AleQ3 dropped from
30% to EB% for a decrease in the solid phase concentration
from lO m^/mg C to S.5 m^/mg C. In natural systems, the solid phase concentration is typically much lower than even
S.5 m^/mg C; consequently one may expect very low percentages
of carbon adsorption. Felix-Filho (1985) showed that the
addition of only 0.33 mg C/L of humic acid was sufficient to
stabilize 1S8 mg/L of «-alumina particles (1 m^/L surface
area). Davis (1980) proposed that while adsorption processes
do not control the concentration of dissolved organic carbon
in lakes and rivers, adsorbed organic compounds may influence
surface properties of suspended particles, such as surface
charge and colloidal stability. Numerous investigators
<Kavanagh, Posner, and Quirk, 1977; Felix-Filho, 1985;
Logtenberg and Stein, 1986) have shown that organics cause
metal oxide particles to have more negative electrophoretic
mobilities and correspondingly lower particle aggregation
rates. The hypothesis that all particles in a given body of
water may have similar colloidal stability is due to
observations that for a given natural water, most particles
typically fall in a narrow range of electrophoretic
mobilities <Hunter, 19S2 with estuary samples) and to the ability to fit aggregation models to natural waters (Ali,
1984).
£.1.4 The Effect of Ca^"^ on particle stability.
It has been shown by All <1984) with lake sediment, and
Jekel <1980) with polystyrene latex particles that increasing
levels of calcium can offset the stabilizing effect of humic
material and result in increased rates of aggregation. Yet,
Eppler, Neis, and Hahn (1975) found very little effect of
calcium on the stability of several solid phases and on
kaolinite equilibrated with natural organic material.
Black, Birkner, and Morgan (1965) demonstrated that
coagulation of negatively-charged kaolinite particles by
anionic polyelectrolytes was due to the presence of
counterions such as Ca^*.
The destabilizing effect of calcium could be attributed
to several phenomena. Adsorption—charge neutralization is
feasible since calcium is known to complex both metal—oxide
surface functional groups (sMe-OH) and the natural organic
materials which coat particles. Double-layer compression may¬
be significant in natural waters as predicted by double-layer
theory. A third possibility is that of a calcium cation bridge between negatively charged humic substances and/or negatively charged solid surfaces as suggested by Black? Birkner, and Morgan (1965). Sreenland (1971) indicated that
adsorption of humic and fulvic acids on clay surfaces such as
montmoriilonite is due to the presence of polyvalent
exchangeable metal cations which form cation bridges between the negatively—charged humic substances and the negatively-charged clay particles. Stumm and Morgan <1981) state that Ca^* and Mg^* increase the collision efficiency factor "not
only by their contribution to ionic strength but also because of their tendency to coordinate with the carboxyl and OH~
functional groups of the huraic substances and of the hydrous
oxides and clays". Tipping and Cooke (1981) showed that
addition of goethite particles into surface water samples
from four lakes all caused the electrophoretic mobilities to drop from positive to negative. This was attributed to the
large adsorbed humic molecules shifting the shear plane from
the oxide surface outward into the humic material. The
electrophoretic mobilities were less negative for higher
concentrations of spiked Ca^* or Mg^*. It was proposed that
the divalent metal offset the stabilizing effect of the
adsorbed organics by bringing their divalent positive charge
into the shear plane through interactions with the adsorbed
humics. The major -functional groups governing the
interactions of Ca^* or Mg="*" with the humic material was thought to be the various carboxyl groups since they are
deprotonated at neutral pH.
The destabilizing effect reported for Ca^-*- may be
observed with many other metal cations based on the results
of Hunter (1980) who measured electrophoretic mobilities of
anion exchange resin equilibrated with seawater and a wide
variety of spiked metal cations and found that all of the
metal cations could reverse the charge of the particles if added in high enough concentrations. Charge reversal is a characteristic of complexing/adsorbing species and cannot be
explained by double—layer theory.
S.2 Ozone Applications in Mater Treatment.
E.S.I Beneficial Effects of Ozone.
Approximately 1,000 water treatment plants in Europe and
approximately 30 water treatment plants in the U.S. use
ozone. Costs of ozone have been reported to range from 0.45 to 4 cents/ljOOO gallons of water treated (Rice et al 1981).
The beneficial effects of ozone in water treatment are
listed belowi
ͣ
Disinfection with ozone is practiced in France by
satisfying the initial ozone demand then providing contact
with 0.4 mg O^/L for at least 4 minutes, typically ranging from 4 to 12 minutes (Rice et al 1981).
-Oxidation of Iron and Manganese.
-Enhanced Nitrification was noted by Sontheimer et al (1978)
for biological nitrification and total organic carbon (TOO
removal on GAC^ in a slow sand filter and in ground passage.
-Color Removal is one of the highest reported uses of ozone
(Rice et al, 1981; O'Donovan, 1965).
-Taste and Odor Control is the highest reported use of ozone
in the U.S. <Rice et al, 1981). Sontheimer (1985) reports taste and odor control to be the primary use of ozone
worldwide.
-Increased Biodeqradabi1ity is evident in reservoirs, on
GAC, and on slow sand filters. Pre-ozonation followed by
reservoir storage precedes coagulation at the SIO mgd Choisy—Le—Roi plant at Paris, France and at the 317 mgd plant at Moscow, Russia. "Schalekamp states that in
Switzerland the use of ozonation before GAC adsorption
increases the operating life of the GAC before regeneration is required by five to six times." (Rice et al, 1981).
-Destruction of TOC, THIiFP, TOXFP, and chlorine demand. (Where: TOC=total organic carbon, THMFP=trihalDmethane
formation potential, and TOXFP=total organic halogen
formation potential). Reckhow and Singer (1984) found that
TOC destruction for fulvic acid is negligible at
typical ozone doses (up to 5 mg Os/L). However, ozone
was found to appreciably decrease THMFP, TOXFP, and
chlorine demand of the fulvic acid and of Chapel Hill, NC raw water, particularly in the presence of added
b i carbonate.
-Synthetic Organic Chemicals may be partially oxidized
-Coagulating Effects reported with ozone are discussed in detail in Section S.3.
2-2.S Adverse Effects of Ozone
Reckhow and Singer (1984) report decreased coagulatability of TOC, THMFP, and TOXFP with sweep
flocculation doses of alum for a 4.1 mg C/L fulvic acid
solution in the absence of natural particles after
pre-ozonation with 5 mg Og/L. Similar results are presented by
Reckhow and Singer (1984) for THMFP and TOXFP and by Reckhow
(1984) for TOC using Chapel Hill, NC raw water (high TOC, low
hardness). The decreased coagulatability slightly offset the
initial decreases in TOC, THMFP and TOXFP by the ozone for
alum doses greater than between lO and 20 mg/L. Jekel
(1983b) also observed decreased removal oT humic TOC by alum
a-fter ozonation at higher doses in the absence of particles.
Scrivner et al <1980) showed no impact on THMFP removal and a
small decrease in the overall removal o-f TOC -for ozone doses
over 4 mg/L with three North Carolina waters (high TOCj low
hardness) using alum doses o-f at least 30 mg/L as
Als(SQ*)a.iaHEO.
It is also noteworthy that Scrivner et al (1980) and
Johnson and Randtke (1983) showed similar decreases in alum
coagulatabi 1 ity o-f TOC -following pre—oxidation with KMnO*,
and chlorine? respectively, at high oxidant doses. High
doses o-f all o-f the oxidants tested (ozone, KMnO^, and
chlorine) decreased the coagulatabi 1 ity o-f TOC. This is
further supported by Van Breeman et al (1979) who tested a
pre—ozonation dose of 40 mg/L and a pre-chlorination dose of
lO mg/L and found both to significantly decrease the
coagulatabi1ity of a fulvic acid solution containing 31 mg
C/L (no particles) using both alum and iron coagulants.
In the results of Van Breeman et al (1979), Scrivner et al (1980), Jekel (1983b), and Reckhow (1984) ozone was not
demonstrated to significantly decrease the coagulatibi1ity of
THMFP, TOXFP or even TOC for lower more typical pre-ozonation
doses (1 to 2 mg Og/L), Jekel (19e3b) concluded his article
by noting that "lower ozone doses may improve the organics
removal by rendering the substances to become betteradsorbable on hydroxide floes, without the detrimental
solubilization of flocculant-organic complexes" which is
presumed to have dominated at high ozone doses.
More research is needed on TOC removal at low ozone doses and on TOC removal with and without ozone in the
presence of various solid phases since most of the literature
on TOC removal involves sweep coagulation in the unrealistic absence of natural particles.
The pilot plant studies of Saunier et al <1983) and the
present operation of the SIO mgd Choisy-Le-Roi plant in Paris
(Reckhow et al, 1986) do not reveal an adverse effect of
ozone on TOC removal. Richard (1978) even found improvements
in TOC removal in jar tests with alum coagulation of Moulle
River water (France).
Ozone increases the biodegradabi1ity of natural
organic material. Consequently, it may be deemed necessary to provide sand or SAC filtration? or reservoir storage, to give microorganisms opportunity to degrade any biodegradable products and hence prevent aftergrowth in the distribution
system. This may be considered an undesirable effect of
ozone though the increased biodegradabi1ity has been used in
several cases to enhance TOC removal.
Veenstra <19S3> and Reckhow and Singer (1984) found
that ozone alters the type of chlorinated organic compunds
formed by subsequent chlorination. Unfortunately, there is
insufficient toxicological/ epidemiological evidence to
conclude how these compounds differ in carcinogenicity (i.e.,
whether this is a beneficial or an adverse health effect). A few investigators have found adverse effects of ozone
on the coagulatability of raw water particulates. This is
seemingly contradictory to most of the literature on
ozone-induced micro-flocculation, but perhaps can be partly
explained by considering the constituents of the raw water
and the type of coagulant used as discussed in section 2.3.3.
2.3 Ozone Effects on Particle Stability.
Very high oxidant doses have been shown to decrease
colloidal stability presumably by oxidation/alteration of
organics (Gibbs, 1983 with HOCLj Felix-Filho, 1985 with
ozone). However, it has also been found with specific
solutions or raw waters that low doses of ozone can
considerably decrease colloidal stability or the coagulant
dose necessary to destabilize particles (Kurz, 1977; Bryant
and Yapijakis, 1977; Haier, 1979; Schalekamp, 1979; Richard,
1981; Brown and Caldwell/ Camp Dresser and McKee, 1982;
Gurol and Pitadella, 1983; Jekel, 1983; Saunier, 1983;
Felix-Filho, 1985). Other observed ozone benefits include
increased removal of particulates and turbidity and longer
filter runs. These benefits have occasionally been referred to
as "microflocculation" and this term is used throughout this
research to generally describe a coagulation benefit
attributed to ozonation.
The microflocculation effect of ozone on particle
stability is often attributed to some impact of ozone on
natural organic material (Maier, 1979; Jekel, 1983b). This
is because ozone has been found to significantly alter
aquatic humic material in ways which might affect particle
stability (see Section 2.3.4). The following discussion
focuses on the constituents of raw waters and model waters
tested for ozone—induced coagulation benefits. This provides
some insight to the factors which may influence ozone-induced microflocculation. The observations from the literature Are grouped bys 1> whether or not a coagulant was added and; S)
whether the system examined was a model aquatic system or natural raw water. This is because the impacts on a raw
water may differ depending on the type and dose of coagulant
added and because natural raw waters are not as
well-characterized as those simulated in the laboratory. The terra coagulant here refers to those added in treatment plants
(polymers, iron and aluminum coagulants).
E-3.1 Ozone Effects Without Coagulant Addition. S.3.1.1 Natural Maters.
A number of investigators have ozonated natural waters then measured the change in turbidity or particle
distribution. Schalekamp <1979> found that ozone decreased
the turbidity of Lake Zurich (Switzerland) water by EO to 40 percent. He also found that doses of 1 and E.5 mg Og/L
decreased the total number of particles and shifted the
particle distribution to larger particles indicating that
coagulation did occur. However, a dose of 7 mg Og/L was found to increase the number of small particles. The Lake
Zurich water had a TOC of 1,E5 mg/L, calcium hardness of
S^O mg/L as CaCOg, moderate particle content (90
particles/ML) and low algae content (E-A^ algal cells/HL;
Reckhow et al, 1986).
eurol and Pidatella (19S3) obtained similar results with Delaware River (Philadelphia) water. Ozone doses up to lO
mg/L reduced the total number of particles and shifted the
distribution toward larger particles. Ozone doses over lO
mg/L caused the opposite effect, an increase in the total
number of particles with apparent disaggregation of larger
particles, Delaware River water samples demonstrated to have
a positive ozone effect had a TDC of 3-^mg/Lj turbidity of
10—20 IMTU, pH of 7.0-7.Sj and were winter samples low in
algae (organic tubidity). Hardness was not reported.
O'Donovan (1965) reported that the turbidity of the
ozonated water was usually less than that of the raw water
for three Irish Lakes (all high in organic content, hardness
values of lOj lOO, and 150 mg/Lj and ozone doses of 4, 5j and
3 mg/L, respectively).
These results demonstrate a microflocculation effect of
ozonation at low to moderate doses with perhaps an adverse
effect at ozone doses much higher than those typically used
for pre-o>;idation in water treatment (typically under about 5
mg Og/L). However, a few investigators have found
conflicting results.
Sommerville and Rempel (197S), Fiessinger et al (1979),
and Maier (1979) reported that ozone can cause increases in
the turbidity of raw waters. Fiessinger et al also reported
an increase in the number of particles. Investigators have
explained this increase in turbidity as being caused by the
polymerization of dissolved metabolites of biological
activity (Maier, 1979; Gurol and Pitadella, 1983). Since it
may accompany a marked decrease in the number of algae (Sommerville and Rempel, 197E and Pak et al, 19S1>, the
increased turbidity may also be due to lysis of the algae into "fragments as well as aggregation of algal metabolites.
Maier (1979) observed that during the summer months, the
turbidity of filtered Lake Constance (West Germany) water increased significantly upon the addition of 1-S mg/L Og. However J during the winter months when the water is high in inorganic turbidity, the initial turbidity decreased with ozone doses up to 1.5 mg/L.
Since natural waters may contain many particles less than 1 Hm diameter, it is also possible that aggregation of
these particles could explain the slight increase in
turbidity and number of particles counted. This is because the measured turbidity is greatest for particles about 0.5 to
1 Hm in diameter and the lower limit of particle counters is
about 1 H-ro.
5.3.1.g Model Waters.
Kurz (1977) ran jar tests with a solution of lOO mg/L kaolin, lOOO mg/L Ca hardness as CaCOa, and lO and 20 mg/L
humic substances. Kurz found that lower turbidities were
achieved when ozone was added, the benefit leveling
off for an Og/C mass ratio of about O.5. Kurz observed a
greater ozone benefit for the higher humic concentration (20
mg/L humic substance).
Jekel (19S3b) demonstrated that pre-ozonation of humic
material before equilibrating with the solid phase decreased
the ability of the humic material to stabilize silica particles.
Felix—Filho and Singer (1985) have shown that doses of ozone from 0.6 to over 5 mg/L have little effect on the
stability of colloidal alumina previously equilibrated with
dissolved humic material? but in the presence of iron (O.OIO and 0.015 mli Fe) j ozone doses as low as 1 mg/L greatly
increased the rate of particle aggregation <the Fe
concentration was below the dose required for coagulation in
the absence of ozone).
2-3.S Ozone Effects With Coagulant Addition.
S.3.a.1 Natural Waters.
Saunier (1983) performed the first of severail pilot
plant studies leading to the design of the SIO mgd
Choisy-Le-Roi water treatment plant in Paris, France. Saunier found
that ozone doses of 0.3 to l.S mg/L greatly improved particle
and turbidity removals and decreased the required dosage of
polyaluminum chloride by almost 50% for an ozone dose of 1.2 mg/L. The raw water was that of the Seine River (median
values of TOC was 4.0 mg/L, calcium hardness was 243 mg/L as
CaCOa, O.OO'^ mM Fe(III), 0.007 mM Al , turbidity of 15 Jtu,
seasonal algae peaks; Saunier, 1983).
Gerval et al (1985) found coagulant aid benefits of
ozone to be evident with the full scale Choisy-Le-Roi 210 mgd
plant.
Fiessinger et al (1979) also observed improved
coagulation with polyaluminum chloride of Seine River water
that was pre—ozonated (1.O mg/L Da).
Richard (1931) per-formed laboratory studies with water
ͣ
from the Moulle River, France. Richard found that pre—
ozonation enhanced the removal of turbidity and particles
and lowered the alum coagulant demand in jar test
flocculation/sedimentation with 50 to S50 mg/L aluminum
sulphate. Richard noted no effect of ozone on
electrophoretic mobility, though ozone did increase the number
of negatively charged groups as determined by colloidal
charge titration. One might presume that this reflects an
increase in solution-phase carboxyl and hydroxy1 groups. Chlorine and chlorine dioxide did not produce this change in
the colloidal titration. The constituents of the Moulle
River stre TDC of 10 mg/L, Hardness of 3^0 mg/L as CaCOa,
relatively high algae concentration (^7 algae/P-L); LReckhow et al <1986)3.
Jekel (1933b) reported on a pilot plant study with Ruhr River water (Federal Republic of Germany) in a direct
filtration scheme. In pilot plant runs, it was found that 1.6
mg Oa/L pre—ozonation in the absence of coagulant gave
filtered water particle concentrations 2.3 times lower than the control (no ozone-no coagulant) and even a little lower
than obtained with polyaluminum chloride coagulant at 0.009 mli Al=3* (0.25 mg Al^*) without ozone. The combination of 1.6 mg Oa/L and 0.009 mM Al=^* provided filtered particle
concentrations 3.5 times lower than were obtained with 0.009
mM Al=3* dose without ozone. The Ruhr river water has high
hardness and a TDC concentration of about ^ mg/L.
Prendiville and McBride <1983) reported on one of
several sets of pilot tests which led to the incorporation of pre—ozonation in the design for the 580 mgd Los Angeles <CA)
direct filtration plant. Prendiville and McBride found that
pre—ozonation allowed an increase in the filtration rate from
*? to 13.5 gpm/ft^. This increased filtration rate was
accompanied by a decrease of 50% in the ferric chloride
coagulant requirement and 25% in the cationic polymer
requirement (Brown and Caldwell/ Camp Dresser and McKee. 198S). The Los Angeles water supply has a TOC concentration
of 3 mg/Ly hardness of SO mg/L as CaCOs, high total dissolved
solids and high Al (0.019 mM Al) (Reckhow et al, 1986). Maier (1979) reported on a study of filtration of Lake Constance water (West Germany) following ozonation. It was found that throughout the dose range studied (0.5 to 2.3 mg
Os/L), ozone improved the fi1terabi1ity of suspended matter,
independent of the turbidity of the raw water. It was also
observed that ozone lowered the aluminum sulphate requirement
to achieve the same degree of removal of suspended matter.
Several investigators have found no effect or an adverse effect on turbidity or particle removal using ozone with a
coagulant. Bcrivner (1980) showed little impact of ozone on
the removal of turbidity and TOC in jar tests for three North
Carolina waters of high TOC (3.4-6.7 mg C/L) and low hardness
at high alum doses, 30-40 mg/L as Ale(SO^)3-ISHeO,
sampled in the
winter-Reckhow (1984) also observed a negative impact of ozone
on turbidity removal with a winter sample of University Lake
(NO water (TOC = 6.8 mg C/L, Calcium Hardness =: 20 mg/L as
CaCOs, and a high turbidity of 60 NTU). The sample was pre—
ozonated with 6 mg/L O^, then coagulated with 0-40 mg/L alum.
Mallevialle (1979) reported a slight decrease in
turbidity removal and a slight increase in TOC removal in jar tests o-f Seine River water with aluminum sulphate
following a dose of 1 mg/L ozone.
a.3.a.2 Model Waters.
The only model waters where coagulant was added were those of humic material solutions without particles or calcium (Van Breeman et al, 1979; Larose, 19Sa; Reckhow,
1994). In all cases, ozone at high doses was found to
hinder sweep coagulation of the humic material in the absence
of natural particles and calcium. This disparity with what
has been observed for natural waters might be attributed to
the lack of calcium? natural particles, algae, or other
constituents, and to the high ozone doses
tested-Experiments at lower, more typical, ozone doses and with model particles and varied hardness concentrations are greatly needed to understand the effects of ozone on both
turbidity and TOC removal.
a.3.3 Trends in Ozone Effects
Considering the raw-water characteristics in which ozone
has been tested for a microflocculation effect, it is
immediately apparent that many of these waters are of
moderately high to very high hardness. However, the effect
of varied calcium doses has not been systematically studied.
Other factors such as increased dissolved organic carbon and
algal content have also been related to increased microflocculation (Reckhow et al, 1986). It has been
suggested that there is a minimum concentration of organics
below which microflocculation will not be observed. The work of Felix-Filho C1985) showed ozone benefits can occur at very
low organic concentrations- The writer suggests that this disparity may be due to several different mechanisms
predominating under different conditions. When ozone benefits are reported to require a minimum organic carbon
concentration, the benefits slvs also typically greatest in
the summer suggesting an algae—related mechanism such as the agglomeration of algal metabolites. It could also be that the benefit of ozone is just more apparent for high levels of TOC as one might expect for a mechanism involving increased associations of Ca^"*" with organics following ozonation.
Another clear trend is that high doses of pre—oxidants (ozone, KMnO^, and chlorine) hinder sweep flocculation of humic material in the absence of natural particles and cations. In most cases, the use of ozone corresponds to
fairly low doses of coagulant, but Richard <19S1) notes ozone
benefits at high, sweep flocculation doses of aluminum
sulphate. Ozone-microflocculation benefits have been
observed with alum, ferric chloride, and cationic polymer
with no clear evidence, at present, of a dependence on the type
of coagulant used.
2.3.4 Ozonation o"f Humic Substances
The e-f-fects o-f ozone on colloidal stability are o-ften attributed to an e-f^fect of" ozone on the natural organic material present in the solution. Consequently? an
understanding af the e-ffects o-f ozone on humic sustances is
necessary be-fore exploring the mechanisms by which ozone
might induce micro-flocculation.
The reactions o-f ozone with constituents in water can be
described by two pathways? one being that Cff direct molecular oxidation, the other being a radical indirect reaction.
In the molecular oxidation pathway, ozone reacts directly with constituents in the water. These direct reactions are highly selective and o-ften slower than the
indirect reactions (Hoigne, 198S)- In molecular oxidation, it is the electrophi 1 ic character o-f ozone that governs the reactions with organic molecules. Consequently, double and
triple bonds, aromatic, and heterocyclic sites are among the preferred targets for an ozone attack (Bailey, 1972).
In the radical pathway, the dissolved ozone decomposes before it reacts with solutes. This decomposition leads to the -formation o-f hydroxyl (H0-> and hydroperoxyl (HOe" )
radicals, as well as hydrogen peroxide (h^Cfe). The
decomposition is accelerated by a radical chain reaction in
which hydroxide ions act as initiators, and the -free radicals
produced act as chain carriers (Hoigne and Bader, 1978;
Hoigne, 1982). When organics are present, the chain reaction
may lead to the formation o-f intermediate organic radicals,
including peroxy radicals (R00-> (Hoigne and Bader, 1978;
Hoigne, 1982). Reactions ot these radicals with organic
substrates is relatively rapid (second order rate constants
on the order of lO'' 1 i ter/mole-second > and non—selective,compared to the direct oxidation pathway. Hence, the free
radical reaction pathway leads to greater destruction of
total organic carbon (TOO.
A dependence of the ozone—induced microflocculation
phenomenon on pH and alkalinity is logical from a chemical
point of view, although it has not yet been tested.
Hydroxide ions catalyze ozone decomposition which should
cause a pH effect. Competition between H"*" and metal cations,
such as Ca^"^, for COO~ sites and other functional groups on
the humic material also suggest a pH effect. An alkalinity
effect is also suspected because the carbonate and
bicarbonate ions Are radical scavengers. Consequently, the
molecular ozone pathway is likely to predominate over the
radical pathway in solutions of high alkalinity.
Mallevialle (1979) indicated that some depolymerization
of humic macromolecules occurs with small doses of ozone,
accompanied by the liberation of phenolic or quinonic
molecules. With larger quantities of ozone, aromatic rings
Btre broken and aliphatic aldehydes and acids are produced.
Upon ozonation, color readily disappears, chemical oxygen
demand and total oxygen demand decrease, and TOC decreases,
but to a much lesser degree. The concentration of carboxylic
acid groups was reported to go through a maximum and then
diminish, while the concentration of polyhydroxyaromatic
substances decreases uniformly, but at a slower rate than the
decrease in color.
Schalekamp <1979), Gilbert (1979), Richard (1981), and
Reckhow and Singer (1984), are among others who found little
destruction of TOC at typical ozone doses (less than 5
mg Oa/L)- Rather than completely mineralizing the humic
material to CQs, it seems that typical ozone doses simply
alter the humic material by reducing the apparent molecular
weight of the humic substances (Gilbert, 1979? Maier, 1979;
Anderson, 1983; Brunet et al, 1983; Veenstra et al, 1983),
and by increasing the number of carboxyl and other oxygenated
functional groups (Mallevialle et al, 1978; Maier, 1979;
Schalekamp, 1979; Jekel, 19S3b; Reckhow and Singer, 1984).
E.3.5 Potential Mechanisms of Ozone—Induced Microflocculation.
Reckhow et al (1986) list the mechanisms which
have been proposed for the influence of ozone on coagulation.
It is important to note that several of these mechanisms
could occur in the same water, and different raw waters may
cause different mechanisms to predominate.
Some of the mechanisms imply increased removal
of the dissolved organic carbon (DOC) and some imply hindered
removal of DOC. Consequently, measuring the removal of DOC
is an important consideration in understanding how ozone
behaves. However, there is presently insufficient evidence
as to the effect of ozone on DOC removal, particularly in the
presence of natural particles.
The mechanisms listed by Reckhow et al are discussed
below with several hypothesized explanations.
Due to the importance o-f carboxyl groups in the
adsorption and complexing behavior o-f humic material, the
increase in the number o-f carbo>:yl groups brought about by
ozonation is o-ften attributed to be responsible -for the
micro-flocculation ef-fect o-f ozone (Maier, 1979; Reckhow et
al. 1986), Reckhow et al (1986) note that an increase in
the carboxyl content ma-/ lead to increases in the association
between the organics and metal cations. It is unknown to
what extent additional metal—organic complexes which may
result from ozonation are soluble or may directly
precipitate? thereby enhancing DOC removal. Kurz (1977)
showed that increased calcium levels increase the turbidity
0"f a solution o-f calcium and humic material presumably
indicating the -formation o-f insoluble complexes. The
metal-organic complexes (soluble or insoluble) may serve as bridges
between particles which may help explain the anomaly o-f
enhanced coagulation observed with natural waters but
hindered coagulation of solutions of humic material without
natural particles. In the absence of natural particles, the
metal-organic complexes have no solid phase on which to sorb,
other than the coagulant floe which may be diminished due to
the binding of coagulant in more organic complexes. An
increase in the carboxyl content may increase adsorption to
alum floe which may hinder floe formation and cause a greater
coagulant requirement but perhaps increase the ultimate
removal of TOC given efficient solid-liquid separation.
Ozonation also decreases the molecular weight of the
humic material which may offset the increase in carboxyl
content and perhaps even result in decreased adsorption onto
alum floe. Although it is not in the list of mechanisms
summarized by Reckhow et al <19S6), the increased carboxyl
content may also cause increased complexation/adsorption of
C3i^* with adsorbed humic material and hence decreased
particle stability? regardless of the extent of adsorption or
precipitation of the solution phase complexes.
The hypothesis of a loss of organics from the solid
surface because of decreased adsorption due to ozonation was
given by Jekel, 19S3b with silica particles (negative EPM)
and high Ca hardness. However, it has not been
experimentally verified that ozone decreases adsorption of
humics onto silica or other natural particles.
The hypothesis of the formation of meta-stable organics
which may polymerize and serve as a polyelectrolyte bridge
fell short of predicting the lack of destabi1ization of alumina particles equilibrated with humic acid following
ozonation in the absence of metal cations (Felix—FiIho,
1985). However, this explanation may still be valid if one
considers the polymerized organics as neutral or oppositely
charged to the particle surface and hence requiring a metal
cation bridge between the polymer and the particle. This is
possible even with positively charged alumina particles since
they are coated with organics making them negatively charged
as is the anionic polyelectrolyte humic material.
The hypothesis that ozone breaks up metal-organic
(1985) with FeiI but would not explain the work of several
others (Kurz, 1977; Jekel, 1983b). Cromley and O'Connor
(1976) have shown that ozone can oxidize -ferrous iron despite
organic complexes which prevented oxidation by aeration
alone.
Pak et al (1981) noted that ozone causes lysis of algae.
This could lead to the release o-f biopolymers which could
enhance coagulation. However, this explanation does not
account "for the marked ozone benefits observed by Felix—Filho
(1985) with a solution of humic acid and iron equilibrated
with alumina particles prior to ozonations or of Kurz (1977)
with kaolin, humic substance and a high concentration of
c a 1 c i urn.
Several of the mechanisms seem appropriate to explain
the results of isolated experiments without being able to
explain results of some other experiments. Consequently, it
is emphasized here that several of the mechanisms may
actually occur and the predominant mechanism if any may be
specific to the constituents of the water tested. Maier
(1979) also stated this possibility by noting that there is
some evidence that ozone-induced microflocculation is
algae-dependent and there is also sound evidence that ozone-induced
microflocculation can occur in solutions void of algae: Kurz
(1977). The two mechanisms discussed by Maier sire:
1: Ozone causes the products of phytoplankton to precipitate,
causing the benefit of ozone to depend on the algae
concentration, showing the greatest benefits in the summer
months.
2: Ozone increases the polarity o-f the organic matter,
mani-fested particularly in a considerable increase in the
number of carboxyl groups. The increase in the number of carboKyl groups causes the organic matter to adsorb more strongly on the turbidity material and cross-link
particles together through a bridge formation. Because increased ozone doses decrease the size of the organic
molecules? there is an optimum ozone dose where the organic
material is left sufficiently large but also has a large number of carboxyl groups to allow it to bridge particles
similar to the action of polyelectrolytes in water treatment,
It is interesting to note that anionic polymers are large aliphatic molecules containing many carboxyl groups and that it has been observed that the coagulation of negatively-charged kaolinite particles by anionic polyelectrolytes was
found to require the presence of a counterion such as Ca^*.
A number of mechanisms have been proposed for
ozone-induced microflocculation, yet none has been experimentally
verified. The research performed for this report is not a
mechanistic study, but observations are made regarding the
effects of calcium and humic material on the susceptibility
of particles to ozone-induced microflocculation.
CHAPTER 3
MATERIALS AND METHODS
3.1. General Experimental Approach
In this research? orthokinetic coagulation rate
experiments? coupled with particle size distribution
measurements? were used to evaluate the impact of humic
material? calcium? and ozone on the colloidal stability of
ot-AlEOa.
The alumina particles were cleaned of organic and
inorganic impurities by heat treatment <50C>° C for 24 h) and
washing with O.IN NaOH. The alumina was size—fractionated to
produce a material of sufficiently narrow size distribution
such that the material could be described as monodisperse
with regard to the rate of particle collisions (Fel i>;—Fi Iho ?
19B5>.
The alumina was suspended in solutions of fixed ionic
strength (3 or 30 mM> and constant pH (7.5) containing
various concentrations of humic material and calcium (see
Figure 3-1). This is the same general experimental approach
as that used by Felix-Filho (1985). The suspensions were
sonicated to disperse the particles then placed on a
reciprocating shaker to achieve adsorption equilibrium. A
second sonication preceded the dosing of certain suspensions
with ozone. Suspensions were then placed back on the
reciprocating shaker overnight to re-establish adsorption
equilibrium. The suspension was sonicated again, then mixed
H umi c
Mat eria Caicium
ADSORPTION
OZONATION
I Sonication
COAGULATION
Electrophoretic
Mobility
Particle:
•concent ration
•size
in a A^OO—ml beaker with a stainless steel impeller at a mean
velocity gradient o-f 50 sec"*.
At di-f-ferent times during the coagulation run, a sample
was carefully withdrawn from the beaker and diluted with electrolyte (2% NaCl> for particle size analysis. The concentration and size distribution of the particles was measured using an Elzone IIS LSD/ADC-80XY resistivity-based particle size analyzer (Particle Data Inc., Elmhurst, ID. A 30 Hm orifice was employed to allow the measurement and sizing
of particles ranging from 0.6 to 9.5 Hm in diameter.
For relatively monodisperse suspensions under the type of turbulent flow conditions developed in the beaker, the
orthokinetic coagulation rate can be expressed as
dN/dt = -^f a§GN/TT (1 )
where N = number concentration of particles at time t
a = collision efficiency factor <a measure of particle stabi1ity)
$ = volume fraction of particles per unit volume of suspension <irdp,=^/6)
dp = particle diameter G = velocity gradient
Integration of equation 1 yields
In <N/No) = -4 a$Gt/Tr (a)
Hence, if particle volume is conserved and $ and G are known,
a semilog plot of the number concentration of particles as a
function of time allows for the calculation of alpha («>, the
collision efficiency factor, from the initial slope of the
semilog plot. For particles which are very unstable and have
a tendency to coagulate rapidly, alpha approaches one. For
very stable particles, alpha approaches zero.
Electrophoretic mobility (EPM) is often used as a surrogate
measure of particle stability. In this research, EPM is
measured in addition to the determination of <x to provide a
comparison of « and EPM, and to give insight into the factors
affecting ex.
3.S. Solid Phase
The alumina solid phase selected for this research was
Linde SF—6, a finely—divided crystalline aluminum oxide
(«—AleD^a, Union Carbide, Indianapolis, IN) with a number mode
diameter of 1.S3 Mm and a volume mode diameter of 1.85 pm.
The volume distribution of the sonicated alumina particles is
shown in Figure 3-E. According to the manufacturer, the
material is 99.955i AleOa, has a density of 3.98 g/cm^, and a
specific surface between 5 and S m^/g.
3.g.l. Rationale for using «—AlgQp^
Several factors contributed to the desirability of using
DIAMETER (microns)
1 2 3 4 5 6 7 8
80.00 ____ I I I 1 I 1 I 1
A
^-s
** ^r
'w
ͣfr ^c
o
* *
o
;S 60,00
/ %
>*• -fr Tir
O)
* %
_o
^ <r£
Q.
-it *
%
a or
^ 40,00 •ft' •jV
^i^. it ͣ*
ͣ
§
ͣ
itT5r
*
i? T^
o ^r ͣsir
^
%
g5 20.00
—>< <r si/ ͣ a >
-/
0.00-—i , , r—,—|—p-—i -r-
-i—i—i—i—i—i—i—i—i—i—|—i—i—1—i—i i9lliHi&&il&&it#tik *
0.30 0.10 0.50
LOG10 (DIAMETER in microns)
FIGURE 3-2: INITIAL VOLUME DISTRIBUTION
1. clays are composed of aluminum and silicon oxides;
S. the surface of clays is thought to be covered by a layer
of simple oxides <Breenland, 1971). When hydrous iron and
aluminum oxides are present as precipitated coatings on
the clay surface? very strong associations between the
clay and humic substances can develop. These associations
involve ligand exchange as well as simple anion exchange reactions. Consequently, these hydrous metal oxides may
play a large role in determining the extent of adsorption
of organic matter by clays;
3. the clean alumina particles sre slightly positively-charged at pH 7,5, but sufficiently close to the iso¬ electric point <pH 8.0) for particle aggregation to be very rapid. This allows the stabilizing power of humic material to be distinctive with regard to decreased rate of
aggregation and decreased electrophoretic mobility
<Felix-Filho, 1985);
^. alumina crystals can be rigorously cleaned of organic and
inorganic impurities and offer a well defined solid surface, whereas kaolinite has been found to experience
structural damage upon rigorous cleaning (Felix-FiIho,
19S5>, perhaps due to a dependence on natural organic
material and/or various cations for binding of the lattice
structure.
3.E.g. Cleaning
Organic material and metal cations present on the
stability and complicate the interpretation of experimental
results. Consequently, the alumina particles were rigorously
cleaned (after Pel i >:-Fi Iho , 1985).
The alumina particles were cleaned of organic and
inorganic impurities by heat treatment <500*^ C for 24 h)
followed by a procedure of Hohl and Stumm (1976) which
consisted of washing the material with O.1 N NaOH for about
lO minutes, followed by repeated centrifugation and dispersion in distilled, deionized water.
3.g.3. Size Fractionation
The alumina was size-fractionated by repeated settling
overnight, siphoning of the supernatant, and redispersion in
an ultrasonic bath to produce a material of sufficiently
narrow size distribution such that the material could be
described as monodisperse with regard to the rate of particle
collisions (Pelix-FiIho, 1985).
3.5.4. Preparation and Handling of Stock Suspensions
Following fractionation, the alumina particles were again
heated at 5000C for 24 h. The alumina particles were then
stored in dry form. Alumina stock suspensions were prepared
by adding a known amount of dry material to a known volume of
suspensions <4.S83 g/L AleOa) were used within three weeks to
avoid complications with slow dissolution of alumina.
3.3. Humic Material
The humic material used in this research was aquatic humic acid extracted from Bay—Tree Lake water (formerly Black
Lake), NC, and aquatic fulvic acid extracted from Lake
DrummondJ VA. The extraction procedure used was that
developed by Thurman and Malcolm (1981), with the slight
modifications introduced by Christman et al (1981). Previous
applications of this method for extracting humic acid from
Black Lake resulted in the following elemental composition:
Table 3-1: Elemental Composition of Black Lake Humic Acid
(in percent by weight)* C H N
^5 if.5 2.6 50.71 7.05 5.84
O Ash Reference
- 4.6 Christman et al (1981)
12.03 Steel ink et al (1983)
* totals do not add up to 100% because percentages of O, S;
P, or moisture were not indicated or were not shown
The extraction of fulvic acid from Lake Drummond performed as
part of this research resulted in the following elemental
composition:
Table 3-2: Elemental Composition of Lake Drummond Fulvic Acid
(in percent by weight)
C H N O Ash 55,73 4.33 0.85 - 1.49
The isolated humic and fulvic acids were stored in dry
form in a dessicator in the dark. Stock solutions of fulvic
material into a known volume of O.S P-m—f i 1 tered, distilled,
deionized water, with pH adjusted to 7.0 with NaOH before and
after addition of the fulvic acid. The mixture was thenstirred for S hours in the dark and then stored at h**C in the
dark. Stock solutions of humic acid were prepared by raising
the pH of O.S >im—fi 1 tered, distilled, deionized water to 12
with NaOH, adding the humic acid, stirring S hours in the
dark, neutralizing to pH 7 with HNOg, aging overnight at 4*^
in the dark, then centrifuging <2000rpm, ~600 g for 2 hours)
and decanting the top S/3 of the humic acid solution for
storage at ^**C in the dark until use. The total organic
carbon (TOO content of humic acid and fulvic acid stock
solutions was typically measured the day after preparation
and checked periodically (Beckman Carbon Analyzer Model 915B
Tocamaster, Beckman Instruments Inc., Fullerton,
CA)-Solutions of potassium hydrogen phthalate were used as TOC
calibration standards. The samples were acidified with HNOs
to pH~3, and bubbled with purified nitrogen gas for at least
20 minutes for the removal of inorganic carbon before
injection into the carbon analyzer. Stock concentrations
were typically SO to S5 mg C/1 humic acid and lOO to 200 mg
C/1 fulvic acid. Humic and fulvic acid stock solutions were
used between 1 day and 3 weeks after preparation and control
experiments revealed no change as a result of the storage.
3.4. Ozone
3.4.1. Preparation
The production of ozone gas was achieved by passing
U.S.p. grade oxygen through a Hydropurge purifying column
(Coast Engineering Laboratories? Gardena? CA), and then
through a Grace LG-2-LI laboratory ozone generator <Union
Carbide? South Plain-field, NJ). The output -from the ozone
generator was controlled by varying the gas flow rate and by
adjusting the power input to the generator. Felix-Filho
(1985) reported that ozone gas phase concentrations as high as
3.554 by weight could be achieved with this system. All materials in contact with the ozone gas or ozonated water were made of stainless steel, glass? or teflon.
All dosing with ozone was by batch addition of ozone stock solution. Felix—Filho (1985) performed similar
experiments with both batch and semi—continuous ozonation.
Similar results were achieved and the batch ozonation was
reported to be simpler and more reproducible.
Ozone stock solutions were prepared and calibrated
immediately prior to dosing. Ozone stock solutions were made
with ozone—demand-free water (distilled? deionized water
ozonated overnight? then oxygenated overnight to remove the remaining ozone). Two ml of O.l N HNOa was added to 500 ml
of ozone-demand-free water which was then refrigerated
overnight (decreased pH and decreased temperature allow the
production of higher stock concentrations of ozone). The
following day, ozone was bubbled into the solution for approximately 20 minutes? the ozone concentration was
determined? and the test suspension was dosed with ozone by
slow addition with the pipette tip submerged and the solution
All glassware used with the ozone stock solution or -for
ozone analysis was cleaned as follows: soaked at least 30
minutes in acid dichromate, rinsed lO times with distilled, deionized water, soaked at least 1 night with ozonated
ozone-demand—free water, then rinsed ten times with
ozone—demand-free water.
3.4.5. Measurement
Three different methods were used to measure the ozone
concentration in the stock solution immediately prior to
dosing:
1. ultraviolet absorption at 260 nm, using a molar
absorptivity of 3,000 L-mol"*-cm-^ (Reckhow, 1984);
S. the indigo method of Bader and Hoigne <19Sl)j and
3. the iodometric method (Standard Methods, 16th edition,
1985), without purging the ozone into a KI solution as
there were no interfering chemicals in the stock ozone
solution (i.e., the iodometric procedure as it is described
for the determination for residual chlorine).
The agreement between these three methods was good. The UV—
absorption and iodometric method typically agreed within 55i
of each other and the indigo method typically gave
concentrations 7—lB% lower than the other two. This could have been due to aging of the indigo reagent, presence of
other ozone-demanding impurities (unlikely), or a systematic
error such as more loss of ozone by volatilization or a