0095-1137/06/$08.00⫹0 doi:10.1128/JCM.02291-05
Copyright © 2006, American Society for Microbiology. All Rights Reserved.
Identification and Characterization of Bacterial Pathogens Causing
Bloodstream Infections by DNA Microarray‡
Berit E. E. Cleven,
1† Maria Palka-Santini,
1† Jo
¨rg Gielen,
1Salima Meembor,
1Martin Kro
¨nke,
1,2and Oleg Krut
1*
Institute for Medical Microbiology, Immunology, and Hygiene,1and Center of Molecular Medicine Cologne, Medical Center,2 University of Cologne, Goldenfelsstr. 19-21, 50935 Cologne, Germany
Received 2 November 2005/Returned for modification 11 January 2006/Accepted 1 May 2006
Bloodstream infections are potentially life-threatening and require rapid identification and antibiotic sus-ceptibility testing of the causative pathogen in order to facilitate specific antimicrobial therapy. We developed a prototype DNA microarray for the identification and characterization of three important bacteremia-causing
species: Staphylococcus aureus, Escherichia coli, and Pseudomonas aeruginosa. The array consisted of 120
species-specific gene probes 200 to 800 bp in length that were amplified from recombinant plasmids. These probes represented genes encoding housekeeping proteins, virulence factors, and antibiotic resistance deter-minants. Evaluation with 42 clinical isolates, 3 reference strains, and 13 positive blood cultures revealed that
the DNA microarray was highly specific in identifying S. aureus, E. coli, and P. aeruginosa strains and in
discriminating them from closely related gram-positive and gram-negative bacterial strains also known to be etiological agents of bacteremia. We found a nearly perfect correlation between phenotypic antibiotic resistance
determined by conventional susceptibility testing and genotypic antibiotic resistance by hybridization to theS.
aureusresistance gene probesmecA(oxacillin-methicillin resistance),aacA-aphD(gentamicin resistance),ermA
(erythromycin resistance), andblaZ(penicillin resistance) and theE. coli resistance gene probesblaTEM-106
(penicillin resistance) andaacC2(aminoglycoside resistance). Furthermore, antibiotic resistance and virulence
gene probes permitted genotypic discrimination within a species. This novel DNA microarray demonstrates the feasibility of simultaneously identifying and characterizing bacteria in blood cultures without prior amplifi-cation of target DNA or preidentifiamplifi-cation of the pathogen.
The presence of living microorganisms in the blood of a patient is usually indicative of a serious invasive infection re-quiring urgent antimicrobial therapy (30). The mortality asso-ciated with bloodstream infections may range from 20 to 50% and depends on several factors, including the pathogen and host (30). Many septic episodes are nosocomial and may be due to microorganisms with increased antimicrobial resistance.
Staphylococcus aureus, Escherichia coli, coagulase-negative staphylococci (CoNS), Klebsiella pneumoniae, Pseudomonas aeruginosa,Enterococcusspp.,Streptococcusspp.,Candida al-bicans, andEnterobacter cloacaeare the most frequent etiolog-ical agents of bacteremia and fungemia in Europe (10, 20, 29) and the United States (4, 30). Rapid and reliable detection of bloodstream infections, including characterization of the pathogen to the species level and determination of its anti-biotic susceptibility pattern, is crucial for several reasons: (i) appropriate antimicrobial agents can be selected, and thus, unnecessary treatment with ineffective antibiotics can be avoid-ed; (ii) the prognosis of the patients can be improvavoid-ed; (iii) the acquisition of resistance in pathogens may be decelerated; and (iv) expenditure on antimicrobials and overall hospital costs can be reduced (2, 12).
Routine microbiological detection of bacteremia relies on enrichment of the causative pathogen using automated contin-uous-monitoring blood culture systems followed by Gram stain, subculture on agar, and subsequent biochemical identi-fication and susceptibility testing. When the blood culture is noted to be positive, definitive identification and antibiotic susceptibilities are usually not available earlier than 24 to 72 h. In general, automated identification systems type pathogens to the species level; additional strain-specific information (e.g., virulence factors) require additional time-consuming and ex-pensive phenotypic and genotypic tests and are not performed routinely (18).
In recent years, numerous studies have demonstrated the value of molecular techniques in order to identify and geno-type bacteria or fungi in blood specimens. Assays using rRNA-based oligonucleotide probes such as fluorescence in situ hy-bridization (16, 17, 24) or microarrays (1, 22) have been shown to allow rapid species identification in blood cultures. How-ever, methods solely based on rRNA probes allow species identification only and do not provide information on antibi-otic susceptibility and other strain-specific characteristics (e.g., virulence genes). For the molecular detection of antibiotic resistance in staphylococci, several multiplex PCR-based as-says have been described (23, 34, 36). The major drawback of multiplex PCR is the limited number of genes that can be analyzed in one reaction and that a preidentification to the species level is required.
A promising genotyping method that allows the simulta-neous identification of a wide variety of genes is provided by the DNA microarray technology (43). DNA probes specific to
* Corresponding author. Mailing address: Institute for Medical Mi-crobiology, Immunology and Hygiene, Medical Center, University of Cologne, Goldenfelsstr. 19-21, 50935 Cologne, Germany. Phone: 49-221-47832103. Fax: 49-221-47832134. E-mail: [email protected].
† B.E.E.C. and M.P.-S. contributed equally to this study.
‡ Supplemental material for this article may be found at http://jcm .asm.org/.
2389
on May 16, 2020 by guest
http://jcm.asm.org/
selected genes are spotted on a solid substrate (usually glass) in a lattice pattern. Target DNA to be analyzed is then labeled with a reporter molecule (e.g., fluorescent dye) and hybridized to the array, and specific target-probe duplexes are detected by measuring the fluorescent signals associated with each spot. There are two types of DNA microarrays: one is the oligonu-cleotide-based array and the other is the PCR product-based array (43). DNA microarrays of both formats have been ap-plied successfully either to the detection of genes encoding resistance to-lactam (14, 19, 25), erythromicin-macrolide (25, 38), tetracycline (6, 25), and gentamicin-aminoglycoside (25) antibiotics or to the analysis of virulence factors (3, 11, 39, 42). The aim of the present study was to establish a DNA-chip (microarray) using gene-specific PCR products as capture probes, which allow both the identification of bacterial species and their further characterization in regard to antibiotic resis-tance and virulence. The practicability and specificity of the DNA microarray for the identification and characterization of
Staphylococcus aureus, Escherichia coli, and Pseudomonas aeruginosa grown in blood culture specimens was evaluated with clinical isolates and positive blood cultures. We demon-strate here its high degree of specificity, its applicability to blood cultures, and its suitability for detecting resistance genes.
MATERIALS AND METHODS
Reference strains, clinical isolates, and culture conditions.Bacterial reference strains were obtained from the American Type Culture Collection (ATCC; Manassas, Va.), the Deutsche Sammlung von Mikroorganismen und Zellkul-turen (Braunschweig, Germany), or the Network on Antimicrobial Resistance in
Staphylococcus aureus(Herndon, Va.). Clinical isolates were obtained from our routine microbiology laboratory. The following bacteria were used for evaluation of the specificity of the microarray: Staphylococcus aureus (ATCC 29213, NRS123 alias MW2, five clinical isolates),Staphylococcus epidermidis(five clin-ical isolates),Staphylococcus capitis(clinical isolate),Staphylococcus haemolyticus
(clinical isolate),Staphylococcus hominis(clinical isolate),Staphylococcus warneri
(clinical isolate),Staphylococcus auricularis(clinical isolate),Micrococcusspp. (clin-ical isolate),Escherichia coli(ATCC 25922, six clinical isolates),Pseudomonas aerugi-nosa(ATCC 27853, five clinical isolates),Klebsiella pneumoniae(three clinical iso-lates),Proteus mirabilis (two clinical isolates), Serratia marcescens (two clinical isolates),Enterobacter cloacae(clinical isolate),Enterobacter aerogenes(clinical iso-late),Acinetobacter baumannii(clinical isolate),Stenotrophomonas maltophilia (clin-ical isolate),Enterococcusspp. (clinical isolate),Enterococcus faecalis(clinical iso-late), andStreptococcus pneumoniae(clinical isolate).
Bacterial strains and clinical isolates were grown overnight at 37°C with con-stant shaking in 5 ml of Luria-Bertani broth or tryptic soy broth (30 g/liter; Merck) containing 3 g of yeast extract/liter. Enterococci and streptococci were grown in 10 ml of tryptic soy broth plus yeast without agitation under 5% CO2. Overnight cultures were harvested after centrifugation at 2,560⫻gfor 10 min. After the supernatant was discarded, the pellet was washed in 1 ml of TE (10 mM Tris-HCl [pH 7.5], 1 mM EDTA) and recovered by centrifugation at 17,900⫻g
for 10 min. Cell pellets were used for DNA preparation.
Blood cultures.Positive blood cultures were used for microarray validation as they were encountered in the routine laboratory. Aerobic and anaerobic blood culture bottles (BACTEC; Becton Dickinson, Heidelberg, Germany) were inoc-ulated with blood from patients with suspected septicemia and placed in a BACTEC 9240 blood culture system (Becton Dickinson), a continuous-reading, automated, and computed blood culture system that detects the growth of mi-croorganisms by monitoring CO2production. Incubation was performed accord-ing to the manufacturer’s recommendations. Bottles with a positive growth index were removed from the incubator, and aliquots of 1 ml of the blood culture suspensions were taken aseptically with a needle syringe. One 1-ml aliquot of the blood culture suspensions was mixed with 1 ml of 0.1% Triton X-100 and kept at room temperature for 5 min in order to disrupt human blood cells. Bacterial cells were then harvested after centrifugation at 17,900⫻gfor 10 min, and the pellets were washed in 1 ml of TE, recovered by centrifugation, and used for DNA preparation. A second 1 ml-aliquot was examined by Gram stain and subcultured on agar plates. The organisms grown on agar plates were characterized and
tested for susceptibility using a VITEK-2 system (bioMe´rieux, Inc., Nu¨rtingen, Germany), Etest strips (AB Biodisk, Solna, Sweden) or disk diffusion tests following the method recommended by the Clinical and Laboratory Standards Institute (9). For microarray hybridization experiments, DNA was prepared from 13 blood cultures positive forS. aureus(n⫽4),S. epidermidis(n⫽3),S. pneumoniae(n⫽2),P. aeruginosa(n⫽1),E. coli(n⫽2), andP. mirabilis(n⫽
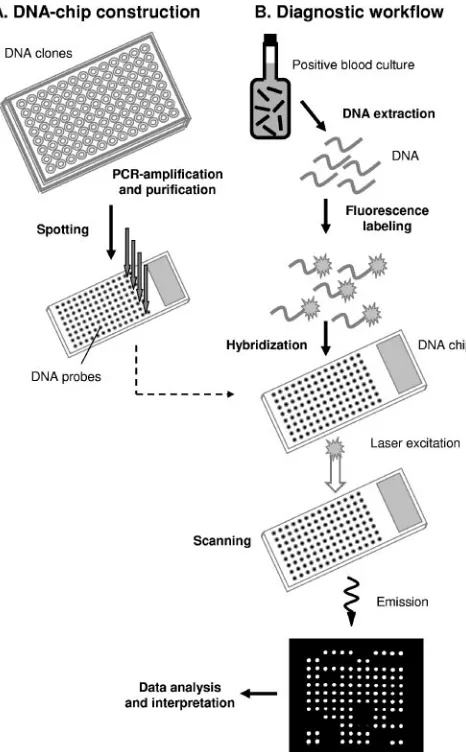
1). The workflow of the microarray assay is outlined in Fig. 1.
[image:2.585.305.538.68.444.2]DNA preparation.Total cellular DNA was extracted and purified either by using the First-DNA All-Tissue kit (GEN-IAL GmbH, Troisdorf, Germany) following the instructions of the supplier or by enzymatic lysis followed by phenol-chloroform extraction. For the latter protocol, cell pellets were resus-pended in 500l of lysis buffer (20 mM Tris-HCl [pH 8.0], 2 mM EDTA [pH 8.0], 1.2% Triton X-100), and lysozyme (Sigma, Taufkirchen, Germany) was added to reach a final concentration of 0.8 mg/ml. In addition, lysostaphin (Sigma) was added to a final concentration of 0.2 mg/ml to promote staphylo-coccal lysis or mutanolysin (0.5 U/l; Sigma) was added to lyse streptococci and enterococci. After incubation at 37°C for 1 h, cell lysates were treated with proteinase K (1 mg/ml; Sigma) for 1 h at 55°C and then with RNase A (0.2 FIG. 1. Workflow for hybridization assay used with the prototype microarray. (A) For DNA-chip construction, capture probes were pro-duced by PCR amplification of plasmid-cloned gene segments, fol-lowed by ethanol precipitation. Purified probes were deposited onto glass slides by robotic printing. (B) For hybridization assays, bacterial target DNA was extracted from positive blood cultures, clinical iso-lates, or reference strains and then labeled with fluorescent dyes and hybridized to the spotted DNA capture probes. Images of fluorescent, hybridized probes were acquired by using a laser scanner and pro-cessed by computer analysis.
on May 16, 2020 by guest
http://jcm.asm.org/
mg/ml; QIAGEN, Hilden, Germany) for 1 h at 37°C. The volume was increased by the addition of 200l of TE, and the salt concentration was adjusted to 0.7 M by addition of 5 M NaCl. A 10% CTAB (cetyltrimethylammonium bromide) solution in 0.7 M NaCl was added to a final concentration of 1%, followed by incubation at 65°C for 20 min in order to release DNA from polysaccharide DNA complexes. DNA was then extracted once with phenol-chloroform-isoamyl alco-hol (25:24:1) and once with chloroform-isoamyl alcoalco-hol (24:1) prior to precipi-tation with 1 volume of isopropanol. After centrifugation at 17,900⫻gfor 30 min, DNA pellets were washed in 70% ethanol and resuspended in 50 to 100l of TE. The concentrations, purities, and sizes of the purified DNA preparations were determined by UV spectrophotometry (Lambda 40; Perkin-Elmer, Boston. MA) and 1% agarose gel electrophoresis.
DNA labeling.Total DNA from clinical isolates and blood cultures was labeled by a nonenzymatic chemical labeling method using the Label-It Cy3/Cy5 kits (Mirus, Madison, WI) or the ULYSIS Alexa Fluor 647 nucleic acid labeling kit (Molecular Probes, Eugene, OR). Prior to labeling, PCR products amplified from three selected recombinant plasmids (1l each; 30 ng/l) were added to each reaction to serve as internal positive controls. For labeling with the Label-It Cy3/Cy5 kit, 5g of high-molecular-weight DNA (⬎12 kb) was mixed with 7.5l of reagent in a total volume of 50l, followed by incubation for 2 h at 37°C according to the recommendations by the supplier. After the volume was ad-justed to 200l with H2O and 0.1 volumes of 5 M NaCl were added, unbound label was removed by precipitation with 2 volumes of ice-cold absolute ethanol for at least 30 min at⫺20°C. The labeled DNA was recovered by centrifugation at 17,900⫻gfor 30 min. The pellet was washed with 70% ethanol and resus-pended in 70l of TE. For labeling with the Ulysis Alexa Fluor 647 kit, 1g of DNA was denatured at 95°C for 5 min, cooled on ice, and mixed with 20l of labeling buffer and 5l of reagent, followed by incubation at 80°C for 15 min according to the instructions of the manufacturer. Unbound dye was removed by ethanol precipitation as described above. The relative labeling efficiency of a reaction was evaluated by calculating the approximate ratio of bases to dye molecules (acceptable labeling ratios for nucleic acid wereⱕ60). This ratio and the amount of recovered labeled DNA was determined by measuring the absor-bance of the nucleic acids at 260 nm, and the absorabsor-bance of the dye at its absorbance maximum using a Lambda 40 UV spectrophotometer (Perkin-Elmer) and plastic disposable cuvettes for the range from 220 to 1,600 nm (UVette; Eppendorf, Hamburg, Germany).
Microarray construction.We used cloned PCR products to generate probes for the DNA microarray. Altogether, 120 gene segments representing virulence genes, antibiotic-resistant determinants, and species-specific metabolic and structural genes fromS. aureus(40),E. coli(31), andP. aeruginosa(49) were represented on the microarray (see Table S1 in the supplemental material).
S. aureus,E. coli, andP. aeruginosagenes were selected from the literature and databases and compared by BLAST analysis to all other sequences available in the NCBI database. Primers were designed to amplify gene segments 200 to 800 bp in length devoid of apparent homology with genes of other bacterial species andHomo sapiens. Gene segments were amplified by using puReTaq Ready-To-Go PCR beads (Amersham Biosciences, Freiburg, Germany) and cloned into the pDrive cloning vector (QIAGEN) according to the recommendations of the suppliers and transformed into competentEscherichia coli(XL1-Blue) cells using the calcium chloride protocol (31).
For quality control purposes, all gene probes were partially sequenced and verified (with the BigDye kit 1.1 and a 377 DNA sequencer; Applied Biosystems, Foster City, CA). All sequences obtained were identical or nearly identical to those obtained from the database. For DNA probe production, 120 recombinant plasmids containingS. aureus,E. coli, andP. aeruginosagene segments were used for reamplification. Amplicons were purified and spotted in four replicates per slide (Memorec, Cologne, Germany) (Fig. 1). Prior to spotting, the DNA con-centrations were normalized to ensure the deposition of equal DNA amounts. To verify probe deposition and spot morphology, for each batch a randomly selected microarray slide was stained by using SYBR Green DNA dye (Molecular Probes).
Hybridization and scanning.All experiments described in the present study represent dual cohybridizations of two different target DNA samples labeled, respectively, with Cy3, Cy5, or Alexa 647 (Fig. 1). After removal of unbound label, Cy3- and Cy5/Alexa 647-labeled DNAs were pooled and mixed with 10g of salmon sperm DNA and 50g of poly(A) DNA. The mixture was frozen in liquid nitrogen and lyophilized in the dark. Prior to hybridization the target DNA was reconstituted in 33l of H2O and 55 l of 2⫻hybridization solution (Memorec, Cologne, Germany), chemically denatured with 11l of denaturation buffer D1 (Mirus), and neutralized with 11l of buffer N1 (Mirus) according to the instructions of the supplier. Hybridization was automatically performed with a TECAN hybridization station (HS400; TECAN, Salzburg, Austria). The arrays
were prewashed at 60°C for 1 min with 0.2% sodium dodecyl sulfate and 4⫻SSC (1⫻SSC is 0.15 M NaCl plus 0.015 M sodium citrate) and prehybridized in 120
l of denatured prehybridization buffer (Memorec) for 30 min at 60°C with mild agitation. After injection of 110l of labeled DNA, hybridization was performed at 60°C for 18 h with mild agitation. The arrays were washed at 50°C in a primary wash buffer (Memorec) for five cycles of 1-min wash time and 30-s soak time and in a secondary wash buffer (Memorec) for five cycles of 20-s wash time and 30-s soak time, and finally dried at 30°C with N2(270 kPa) for 3 min. Hybridized arrays were scanned with a Scan Array 5000 laser scanner (Perkin-Elmer). Laser light of wavelengths at 532 and 635 nm were used to excite Cy3 dye and Cy5/ Alexa 647 dye, respectively. Fluorescent images were analyzed by using ImaGene software (BioDiscovery, El Segundo, CA). Spots were found and segmented in order to select areas of recognizable signals for analysis. The fluorescence in-tensity of each spot was measured, signal-to-local-background ratios were calcu-lated by ImaGene, and spot morphology and deviation from the expected spot position were considered using the default ImaGene settings. The data were imported into Microsoft Access and automatically processed. Spots with a signal-to-noise ratio of 1.2 and with at least 600 relative fluorescence units over the local background in all three replicas were considered positive. Cutoff values for these parameters were empirically determined in pilot experiments and used to tag spots either as positive or negative.
RESULTS
Specificity. In order to allow the simultaneous and rapid
identification ofS. aureus,E. coli, andP. aeruginosagrown in blood culture specimens from septicemic patients, a microar-ray comprising a set of 40 S. aureus, 31 E. coli, and 49 P. aeruginosagene probes 200 to 800 bp in length was developed (Table S1 in the supplemental material).
The specificity of the DNA-chip was validated first with 45 well-characterized clinical isolates and reference strains of the three target species, as well as other related bacteria and, second, with 13 blood cultures from patients with sepsis by following the workflow outlined in Fig. 1. Positive blood cul-tures were processed as they were encountered in the routine laboratory. Hybridization results were compared to conven-tional identification results obtained by routine diagnostics.
In all assays, three PCR-amplified DNA segments, which had been added to each DNA preparation as a positive inter-nal control, hybridized with the corresponding probes, indicat-ing that the labelindicat-ing and hybridization had performed effi-ciently.
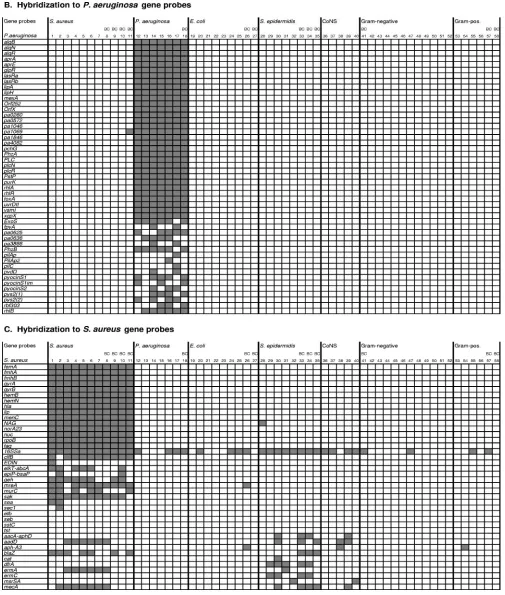
Hybridization experiments with S. aureus, E. coli, and P. aeruginosa target DNAs revealed specific hybridization with the species-specific gene probes (Fig. 2). There was no cross-hybridization between the three species, with the exception of theS. aureus16S rRNA gene probe (16SSa, Fig. 2C), which also hybridized withE. coliandP. aeruginosatarget DNA.
Identification of E. coli, P. aeruginosa, andS. aureus refer-ence strains, clinical isolates, and blood cultures by microarray analysis corresponded by 100% with the conventional identi-fication results (Fig. 2).
Detection and discrimination of E. coli. All DNA samples
from nineE. colistrains hybridized always with sevenE. coli
gene probes [envZ, fes(1) and fes(2), nfrB, yacH, yagX, and
ycdS] (Fig. 2A, columns 19 to 27); in the following discussion we will refer to these genes as core genes. With 14E. coligene probes, variable hybridization was observed, including the an-tibiotic resistance gene probesblaTEM-106,sul,strB, andaacC2.
Such a variable hybridization profile is expected for antibiotic resistance genes since acquired resistance to antimicrobials is isolate specific. For 11E. colivirulence gene probes (eae,eltB,
escR, escT, escU, espB, hlyA, hlyB, SLTII, toxA-LTPA, and
on May 16, 2020 by guest
http://jcm.asm.org/
VT2vaB) no hybridization signals were detected with any of the testedE. coliisolates and blood cultures. Since these virulence genes are known to be specific for particularE. colipathotypes (3), it was not surprising that they were not present in the tested strains. The eae, esc, andesp genes, for example, are encoded on a chromosomal pathogenicity island, which is typ-ical for enteropathogenic E. coliexhibiting the unique viru-lence mechanism known as attaching and effacing (13). The alpha-hemolysin (hly) operon is encoded on a large plasmid of enterohemorrhagicE. colistrains (32).
Detection and discrimination ofP. aeruginosa. DNA
sam-ples obtained fromP. aeruginosauniformly hybridized with 32 of 49P. aeruginosaspecific gene segments, including themexA
gene probe (core genes). Variable hybridization was observed with 17 probes, allowing for the discrimination of individualP. aeruginosaisolates (Fig. 2B, columns 12 to 18).
Detection and discrimination of S. aureus. Hybridization
experiments performed with 11 S. aureus target DNAs re-vealed signals in all assays with 16S. aureus gene segments (core genes) (Fig. 2C, columns 1 to 11). Variable hybridization was observed with 14S. aureusgene probes including the six antibiotic resistance gene segments aadD, aacA-aphD, blaZ,
dfrA,ermA, andmecAand the virulence genessak,sea,sec1, and EDIN. The gene probes geh, mreA, clfB, and elkT-abcA
hybridized with 8 (geh), 10 (mreAandclfB), and 6 (elkT-abcA)
target DNAs. However, PCR amplification of the four genes was positive for all 11 S. aureus target DNAs (results not shown), suggesting that the four genes were present in all of the strains investigated and that these gene probes did not allow reliable detection of the four genes inS. aureus.
No hybridization was observed with 10 probes, including the toxin genes seb, tst, and etb. In contrast to the community-acquired, multidrug-susceptible methicillin-resistantS. aureus
(MRSA) strain MW2 that hybridized tomecAandblaZonly, all six clinical MRSA strains showed the same multiresistant hybridization pattern, and their DNA hybridized to theermA
(erythromycin resistance), mecA (oxacillin resistance), and
aadD (tobramycin resistance) genes. As for the majority of multiresistant MRSA strains, theermAandaadDgenes were shown to be located upstream and downstream, respectively, of the mecA gene in themec chromosomal region (7, 26). Hy-bridization to the core gene probes permitted the identification ofS. aureus, while hybridization to antibiotic resistance gene probes allowed for the discrimination of strains.
Discrimination ofE. coli,P. aeruginosa, andS. aureusfrom
related bacterial species. Cohybridization experiments
per-formed with related bacterial species confirmed the high spec-ificity of the DNA-chip (Fig. 2). ForS. epidermidisand all other CoNS, cross-hybridization was observed only with theS. aureus
16S rRNA gene probe (16SSa, Fig. 2C) and several common
FIG. 2. DNA microarray analyses of 42 clinical isolates, 3 reference strains, and 13 blood cultures. Each column shows the results of an individual hybridization with target DNA prepared from:S. aureusATCC 29213 (column 1), MW2 (column 2), clinical isolates (columns 3 to 7), and positive blood cultures (columns 8 to 11);P. aeruginosaATCC 27853 (column 12), clinical isolates (columns 13 to 17), and positive blood cultures (column 18);E. coliATCC 25922 (column 19), clinical isolates (columns 20 to 25), and positive blood cultures (columns 26 and 27);S. epidermidisclinical isolates (columns 28 to 32) and blood cultures (columns 33 to 35); and clinical isolates of CoNSS. auricularis(column 36),S. capitis(column 37),S. haemolyticus(column 38),S. hominis(column 39), andS. warneri(column 40). Other gram-negative species included a Proteus mirabilispositive blood culture (column 41), clinical isolates ofProteus mirabilis(columns 42 and 43),Serratia marcescens(columns 44 and 45),Klebsiella pneumonia(columns 46 to 48),Stenotrophomonas maltophilia(column 49),Acinetobacter baumannii(column 50), Enterobacter cloacae(column 51), andEnterobacter aerogenes(column 52). Other gram-positive species included clinical isolates ofMicrococcusspp. (column 53),Enterococcusspp. (column 54),Enterococcus faecalis(column 55), andStreptococcus pneumoniae(column 56) and two positive blood cultures ofS. pneumoniae(columns 57 and 58). (A) Hybridization of DNA prepared from bacterial isolates, reference strains, and blood cultures withE. coligene probes; (B) hybridization withP. aeruginosagene probes; (C) hybridization withS. aureusgene probes. Gray boxes represent gene probes that hybridized with the respective target DNA; white boxes represent gene probes that showed no hybridization with the respective target DNA. Experiments performed with positive blood cultures are indicated (BC).
on May 16, 2020 by guest
http://jcm.asm.org/
staphylococcal antibiotic resistance determinants (aadD, aacA-aphD,aph-A3,blaZ,cat,dfrA,ermA,ermC,mdrSA, andmecA) (Fig. 2C, columns 28 to 36). There was no cross-hybridization with other metabolic or virulence genes ofS. aureus.
[image:5.585.41.546.69.664.2]TheMicrococcusspp. isolate showed no hybridization with the DNA-chip (column 53). Streptococci (columns 56 to 58) and enterococci (columns 54 and 55) showed hybridization with the staphylococcal 16S RNA gene probe and once with
FIG. 2—Continued.
on May 16, 2020 by guest
http://jcm.asm.org/
the staphylococcal aph-A3 aminoglycoside resistance gene probe (Enterococcusspp.) (Fig. 2C). Of twelve strains of seven gram-negative species (columns 41 to 52), two hybridized with theS. aureus16S rRNA gene probe (Klebsiella pneumoniaeand
Proteus mirabilis, Fig. 2C, columns 41 and 47), and one clinical isolate ofProteus mirabilishybridized with theE. coliresistance genesblaTEM-106(-lactam resistance),sul(sulfonamide
resis-tance), andstrB(streptomycin resistance) (Fig. 2A, column 42).
Serratia,Stenotrophomonas,Acinetobacter, andEnterobacter spe-cies showed no cross-hybridization with any gene probe.
Sensitivity. Although the majority ofP. aeruginosa probes
allowed unambiguous identification, some probes showed vari-able hybridization patterns when microarray hybridization was performed with different target DNA samples prepared from the same isolate (Table 1). Successful hybridization with strong fluorescent signals depends on efficiency of DNA labeling (ra-tio of bases per one dye molecule) and amount of labeled DNA. For the different target DNA preparations of four clin-ical isolates, variable hybridization was observed with 14 gene probes (uvrDII,vsmI,pa1069,rhlR,rhlA,rhlB,1046, pyocinS,
pyocinS1im,plcR,plcN,PHZb,rbf303, andpIIAp2). For exam-ple, for three different DNA preparations of isolate C4242, hybridization toPseudomonasgene probes varied from 31 to 43 probes, respectively, depending on the labeling efficiency and amount of DNA (Table 1). The lowest number of signals was detected with 382 ng of target DNA, which, however, showed a high base-to-dye ratio (BDR) of 75. Overall, our results suggest that various amounts of DNA and BDRs influ-enced the hybridization results of few gene probes. However, irrespective of the varying quality and quantity of the labeled target DNA, 35 of the 49P. aeruginosagene probes showed robust hybridization results in all performed experiments.
Detection and characterization of pathogens in blood
cul-tures.Although DNA prepared from blood cultures comprises
a mixture of human and bacterial DNA, the resulting hybrid-ization signals obtained with DNA from 1 ml of positive blood culture allowed a clear and unambiguous characterization of theS. aureus,E. coli, andP. aeruginosapresent in 7 of 13 tested
blood specimens (Fig. 2). In accordance with the VITEK2 characterization, positive BACTEC cultures were identified by microarray hybridization as multiresistant MRSA (Fig. 2C, column 8), penicillin-resistantS. aureus (columns 9 and 11), and multisusceptibleS. aureus(column 10), E. coli(Fig. 2A, columns 26 and 27), andP. aeruginosa(Fig. 2B, column 18) and discriminated from oxacillin-resistantStaphylococcus epidermi-dis (columns 33 to 35), Proteus mirabilis (column 43), and
Streptococcus pneumoniae(columns 57 and 58).
Correlation between susceptibility testing and microarray
hybridization of selected antibiotic resistance genes. (i)S.
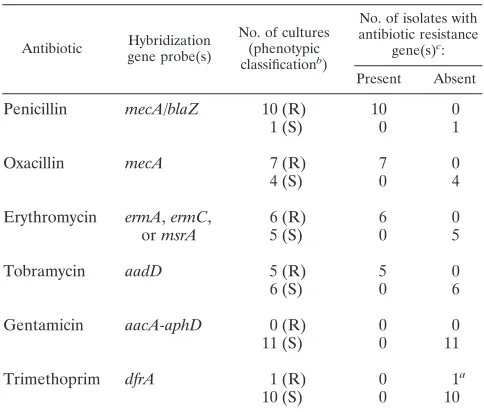
au-reus.For 11Staphylococcus aureusstrains and blood cultures, we compared susceptibility results determined by the VITEK2 system, Etest strips, and disk diffusion tests with the results of the microarray hybridization assay for the simultaneous detec-tion of antibiotic resistance genes (Table 2). The presence or absence of resistance genes as indicated by microarray hybrid-ization was confirmed by PCR with gene-specific primers (re-sults not shown).
For theS. aureus strains there was a 100% correlation be-tween phenotypic resistance to penicillin and hybridization to the mecAand/orblaZ gene (both genes confer resistance to penicillin) (see Table 2). Phenotypic resistance to oxacillin correlated 100% with the hybridization of themecAgene be-tween resistance to erythromycin and hybridization to the erythromycin resistance genesermA,ermC, ormsrSAand be-tween resistance to tobramycin and hybridization to theaadD
[image:6.585.300.542.88.294.2]gene. Furthermore, they all showed 100% correlation between phenotypic susceptibility to gentamicin and no hybridization to the resistance genesaacA-aphD. Notably, thedfrAgene of the trimethoprim-resistant strain MW2 (MIC of 1g/ml) was not detected by microarray hybridization, whereas PCR amplifica-tion revealed the presence of thedfrAgene.
TABLE 1. Microarray hybridization signals obtained with different target DNA preparations ofP. aeruginosaisolates
Isolate DNA amt (ng)a BDRb No. (%) of hybridized
gene probesc
C4242 130* 22 38 (88)
382* 75 31 (72)
1,350† 48 43 (100)
C3853 510* 29 36 (88)
⬎2,400† 30 41 (100)
C3045 550* 90 34 (89)
2,950† 41 38 (100)
C3755 1,180† 139 41 (95)
⬎1,600† 40 43 (100)
a
*, Labeled with Alexa 647; †, labeled with Cy3 or Cy5.
b
Base-to-dye ratio; the number of nucleotides per dye molecule.
c
That is, the number of signals obtained withP. aeruginosacapture probes (total of 49) after hybridization with different DNA preparations. The percent-age of specific hybridizations is compared to the highest number of signals obtained for each isolate (100%).
TABLE 2. Correlation between phenotypic and genotypic antibiotic resistance for 11S. aureusisolates and blood cultures
Antibiotic Hybridization gene probe(s)
No. of cultures (phenotypic classificationb)
No. of isolates with antibiotic resistance
gene(s)c:
Present Absent
Penicillin mecA/blaZ 10 (R) 10 0
1 (S) 0 1
Oxacillin mecA 7 (R) 7 0
4 (S) 0 4
Erythromycin ermA,ermC, 6 (R) 6 0
ormsrA 5 (S) 0 5
Tobramycin aadD 5 (R) 5 0
6 (S) 0 6
Gentamicin aacA-aphD 0 (R) 0 0
11 (S) 0 11
Trimethoprim dfrA 1 (R) 0 1a
10 (S) 0 10
a
ThedfrAgene was detected by PCR.
b
R, resistant; S, susceptible.
c
Genotypic antibiotic resistance was determined by microarray hybridization.
on May 16, 2020 by guest
http://jcm.asm.org/
[image:6.585.42.284.90.227.2](ii)E. coliand other gram-negative bacteria.The prototype microarray harbored only fourE. coliand one P. aeruginosa
resistance gene probes which do not yet allow a comprehensive prediction of antibiotic resistance. Nevertheless, hybridization with theE. coliresistance gene probeblaTEM-106was observed
in oneP. mirabilisand fourE. colistrains and correlated with phenotypic ampicillin resistance for all five strains (Table 3).
OneE. coliblood culture showed also resistance to tobra-mycin and gentamicin. This phenotypic resistance correlated with the hybridization of theaacC2gene probe for aminogly-coside resistance and theS. aureus aph-A3probe for tobramy-cin-kanamycin resistance (Table 3). For oneP. mirabilisand fourE. colistrains, phenotypic resistance to streptomycin cor-related with hybridization to thestrBprobe (Table 3).
All P. aeruginosa strains hybridized with the mexA gene probe (Fig. 2) and showed phenotypic resistance to tetracycline, trimethoprim-sulfamehoxazole, penicillin (ampicillin and me-zlocillin), and cephalosporin (cefazolin, cefixime, and cefu-roxime). ThemexA-mexB-oprMoperon is a determinant for a three-component efflux system responsible for intrinsic and acquired multiresistance in P. aeruginosa (-lactams, fluoro-quinolones, trimethoprim, sulfonamides, chloramphenicol, and others) (27).
DISCUSSION
We have established a novel, highly specific DNA microar-ray for the identification and characterization ofS. aureus,E. coli, andP. aeruginosa in blood cultures. Our DNA-chip al-lowed simultaneous species identification and detection of im-portant virulence and antibiotic resistance genes in a single assay. It was successful in identifying all 27E. coli,P. aerugi-nosa, andS. aureusstrains and in discriminating them from 21 closely related gram-positive and gram-negative bacterial strains which are also known to be causative agents of bac-teremia (30). Our prototype microarray demonstrates the feasibility of identifying and characterizing pathogens in 1
ml of positive blood culture without prior amplification of the target DNA.
The microarray consisted of 120 gene segments 200 to 800 bp in length amplified from recombinant plasmids. One im-portant feature of this format is that the panel of probes can be continually extended to include sequences for additional spe-cies, variant isolates, or antibiotic resistance determinants as they are characterized and available. The accuracy, range, and discriminatory power of the gene-segment-based microarray can be refined by adding or removing gene probes to the panel without significantly increasing the complexity or costs. In this pilot study, three important species causing bacteremia were selected to provide a proof of principle. The range of organ-isms that can be identified is easily expanded by increasing the number of gene probes in the array. For example, the addition of a few probes specific forS. epidermidisand other CoNS will allow for the species identification of CoNS. Furthermore, due to a specific hybridization pattern for each species, it will also allow the identification of mixed blood cultures with more than one pathogen.
A second important feature of this microarray format based on PCR products of extended length (100 to 3,000 bp) is that one probe per gene is usually sufficient to produce strong signals and high specificity (35). For long probes, minor point mutations are likely to only slightly reduce duplex formation, which does not lead to the loss of hybridization signals. In contrast, short oligonucleotide microarrays sometimes lack specificity and require multiple short oligonucleotides per one gene. Volokhov et al. (38) constructed a microarray for the analysis of erythromycin determinants; to increase confidence in the microarray analysis, each analyzed gene was represented by seven oligoprobes. However, for routine diagnostics this leads to an undesired increase in complexity.
A limitation of the long probes as used here is the reduced sensitivity to single or minor point mutations, such as those responsible for resistance conferred by extended-spectrum
-lactamases (ESBLs, e.g., TEM-, SHV-, and OXA-type ESBLs) (5). An extension of the probe panel for example, by OXA,
SHV, orCMYresistance genes, will allow the detection of the different-lactamase types (19) but not discrimination within one type.
As shown forP. aeruginosaprobes, the intensity of the hy-bridization signal largely relies on quality and quantity of the labeled target DNA. Under suboptimal conditions not all spe-cies-specific probes produced strong signals after hybridization with specific DNA. PCR analyses of some S. aureus genes revealed that the genesclfB, elkT-abcA, geh, andmreAwere present in all testedS. aureusstrains, whereas not all strains produced hybridization signals with the corresponding probes. Besides the quality and quantity of the target DNA, two cir-cumstances could explain the discrepancy. First, extensive se-quence variations between a specific probe and the corre-sponding gene present in a certain strain may produce false negatives (i.e., the gene is present but there is no hybridiza-tion). Second, it is impossible to predict the exact hybridization behavior of immobilized DNA probes. Therefore, it is impor-tant to judge the collection of probes experimentally using the prototype chip and to select only probes that produce robust results.
The use of a single protocol for all bacterial species,
com-TABLE 3. Correlation between ampicillin-penicillin resistance, gentamicin-tobramycin resistance, and streptomycin resistance
and hybridization with the resistance gene probes blaTEM-106,aacC2,aph-A3, andstrB
Species Resistance phenotypea
Hybridization with:
blaTEM-106b aacC2b aph-A3c strBb
E. coliATCC 25922 Susceptible – – – –
E. coliC4821 AMP STR ⫹ – – ⫹
E. coliF3437 AMP ⫹ – – –
E. coliC3941 AMP STR ⫹ – – ⫹
E. coliF1806d AMP GEN TOB
STR ⫹ ⫹ ⫹ ⫹
E. coliC4547 AMP (I) – – – –
E. coliC4230 AMP – – – –
E. coliC3940 Susceptible – – – –
E. coliF1642d STR – – – ⫹
P. mirabilisC4024 AMP STR ⫹ – – ⫹
P. mirabilisC4403 Susceptible – – – –
P. mirabilisF1738d Susceptible – – – –
a
AMP, ampicillin; GEN, gentamicin; STR, streptomycin; TOB, tobramycin; I, intermediate.
b
E. coligene probes.
c
S. aureusgene probes.
d
Positive blood culture.
on May 16, 2020 by guest
http://jcm.asm.org/
[image:7.585.43.282.105.246.2]prising all steps of DNA preparation and DNA-chip hybrid-ization, is essential for testing blood cultures where the bacte-rial diagnosis is usually uncertain. In regard to the processing time we decided to use a chemical labeling procedure using high-molecular-weight DNA (⬎12 kb). DNA fragmentation by sonication or enzymatic cleavage (restriction enzymes or DNase A) prior to the labeling reaction is difficult to control and increases the processing time and the chance of losing DNA by an additional required precipitation step. However, large target DNA molecules may hybridize poorly to immobi-lized probes due to spatial and steric constraints. Vora et al. (40) reported that the sensitivity is significantly increased by using fragmented DNA (40). A DNA preparation protocol using sonication for simultaneous cell disruption and target DNA fragmentation may be the method of choice to increase the sensitivity of the microarray, in particular toward low-copy-number and/or plasmid-encoded genes which may be under-represented in the target DNA.
Since the focus of the present study was to provide a proof of principle and to test the informative value of the selected capture probes, the protocols were not yet optimized for time. However, trials with commercial DNA kits showed that DNA extraction can be performed within 1 h. Hybridizations were performed according to standard protocols overnight. Prelim-inary experiments showed that this time may be shortened to 4 h, allowing the results to be obtained within 8 h after a blood culture becomes positive.
Previous studies have shown that the detection of antibiotic resistance genes by molecular techniques has good predictive power for the phenotypic resistance of clinicalS. aureus iso-lates (23, 36, 38). With our gene-segment-based microarray there was an excellent correlation between genotypic detection of antibiotic resistance determinants and phenotypic detection using conventional susceptibility testing. The detection of the resistance genesmecA, blaZ, ermA, ermC,msrSA, aadD, and
aacA-aphDby microarray hybridization allowed for the reli-able prediction of oxacillin, penicillin, erythromycin, tobramy-cin, and gentamicin resistance in a single assay.
By microarray hybridization it was possible to discriminate multidrug-resistant MRSA, resistant to methicillin-penicillin and other antibiotics, and MRSA resistant to methicillin-pen-icillin only, which is frequently encountered as community-acquired MRSA. Simultaneous comprehensive resistance geno-typing for oxacillin, macrolide, and aminoglycoside resistance genes (e.g.,mecA, aadD,aacA-aphD,ermA,ermB,ermC, and
msrSA) by microarray hybridization allows the rapid identifi-cation of multiresistant MRSA or macrolide- or aminoglyco-side-susceptible MRSA and, in consequence, permits other therapeutic options and may reduce reliance on vancomycin (26, 28).
Recently, a 23S rRNA gene oligonucleotide microarray (1) and a macroarray (96-well format) of DNA probes directed against rRNA (22) were shown to provide good discrimination of fungi and gram-positive and gram-negative bacteria causing bacteremia. However, since these approaches are solely based on rRNA they provide no information on antibiotic resistance determinants or virulence genes of the identified strains. An-other molecular approach applying the commercial Hyplex BloodScreen multiplex PCR-enzyme-linked immunosorbent assay system to positive blood cultures was shown to be well
suited for the direct and specific identification of the most common pathogenic bacteria and the direct detection of the
mecAgene ofS. aureus(41). However, due to the microtiter plate format of this assay, the number of gene probes is limited and themecAprobe was the only antibiotic-resistant determinant.
On the other hand, there are numerous studies on the de-tection of virulence genes and/or antibiotic resistance genes based on multiplex PCR (15, 21, 23, 33, 36, 37) or assays combining multiplex PCR and microarray detection systems (8, 38, 39). However, all assays using multiplex PCR have been limited by the number of genes that can be amplified in one reaction. By combining multiplex PCR and microarrays, the microarray detection system is superior to gel electrophoretic analysis, but in this way the great potential of microarrays allowing the analysis of hundreds of genes in parallel is aban-doned. Most of these systems can therefore be applied to preidentified pathogens only.
In contrast, the microarray presented here opens the possi-bility of identifying pathogens in blood cultures with concom-itant further characterization in terms of antibiotic resistance and virulence without preidentification. After extension and further automation, the DNA-chip has the potential to provide a clinical tool for microbiological diagnostics, as well as for epidemiological studies. It would allow clinicians to administer appropriate antibiotic chemotherapy in a timely fashion, im-prove the outcome of septic patients, and reduce the spread of antibiotic resistance genes.
ACKNOWLEDGMENTS
We thank the clinical laboratory staff for providing clinical isolates and blood cultures and Ludwig Eichinger for the use of the scanning facilities.
REFERENCES
1.Anthony, R. M., T. J. Brown, and G. L. French.2000. Rapid diagnosis of bacteremia by universal amplification of 23S ribosomal DNA followed by hybridization to an oligonucleotide array. J. Clin. Microbiol.38:781–788. 2.Barenfanger, J., C. Drake, and G. Kacich.1999. Clinical and financial
ben-efits of rapid bacterial identification and antimicrobial susceptibility testing. J. Clin. Microbiol.37:1415–1418.
3.Bekal, S., R. Brousseau, L. Masson, G. Prefontaine, J. Fairbrother, and J. Harel.2003. Rapid identification ofEscherichia colipathotypes by virulence gene detection with DNA microarrays. J. Clin. Microbiol.41:2113–2125. 4.Bourbeau, P. P., and J. K. Pohlman.2001. Three days of incubation may be
sufficient for routine blood cultures with BacT/Alert FAN blood culture bottles. J. Clin. Microbiol.39:2079–2082.
5.Bradford, P. A.2001. Extended-spectrum beta-lactamases in the 21st cen-tury: characterization, epidemiology, and detection of this important resis-tance threat. Clin. Microbiol. Rev.14:933–951.
6.Call, D. R., M. K. Bakko, M. J. Krug, and M. C. Roberts.2003. Identifying antimicrobial resistance genes with DNA microarrays. Antimicrob. Agents Chemother.47:3290–3295.
7.Chambers, H. F.1997. Methicillin resistance in staphylococci: molecular and biochemical basis and clinical implications. Clin. Microbiol. Rev.10:781–791. 8.Chizhikov, V., A. Rasooly, K. Chumakov, and D. D. Levy.2001. Microarray analysis of microbial virulence factors. Appl. Environ. Microbiol.67:3258– 3263.
9.Clinical and Laboratory Standards Institute.2006. Performance standards for antimicrobial susceptibility testing. Sixteenth informational supplement M100-S16. CLSI, Wayne, Pa.
10.Decousser, J. W., P. Pina, F. Picot, C. Delalande, B. Pangon, P. Courvalin, P. Allouch, et al.2003. Frequency of isolation and antimicrobial susceptibility of bacterial pathogens isolated from patients with bloodstream infections: a French prospective national survey. J. Antimicrob. Chemother.51:1213– 1222.
11.Dobrindt, U., F. Agerer, K. Michaelis, A. Janka, C. Buchrieser, M. Samuelson, C. Svanborg, G. Gottschalk, H. Karch, and J. Hacker.2003. Analysis of genome plasticity in pathogenic and commensalEscherichia coliisolates by use of DNA arrays. J. Bacteriol.185:1831–1840.
on May 16, 2020 by guest
http://jcm.asm.org/
12.Doern, G. V., R. Vautour, M. Gaudet, and B. Levy.1994. Clinical impact of rapid in vitro susceptibility testing and bacterial identification. J. Clin. Mi-crobiol.32:1757–1762.
13.Elliott, S. J., L. A. Wainwright, T. K. McDaniel, K. G. Jarvis, Y. K. Deng, L. C. Lai, B. P. McNamara, M. S. Donnenberg, and J. B. Kaper.1998. The complete sequence of the locus of enterocyte effacement (LEE) from en-teropathogenicEscherichia coliE2348/69. Mol. Microbiol.28:1–4. 14.Grimm, V., S. Ezaki, M. Susa, C. Knabbe, R. D. Schmid, and T. T.
Bach-mann.2004. Use of DNA microarrays for rapid genotyping of TEM beta-lactamases that confer resistance. J. Clin. Microbiol.42:3766–3774. 15.Huletsky, A., R. Giroux, V. Rossbach, M. Gagnon, M. Vaillancourt, M.
Bernier, F. Gagnon, K. Truchon, M. Bastien, F. J. Picard, A. van Belkum, M. Ouellette, P. H. Roy, and M. G. Bergeron.2004. New real-time PCR assay for rapid detection of methicillin-resistantStaphylococcus aureusdirectly from specimens containing a mixture of staphylococci. J. Clin. Microbiol.42:1875– 1884.
16.Jansen, G. J., M. Mooibroek, J. Idema, H. J. Harmsen, G. W. Welling, and J. E. Degener.2000. Rapid identification of bacteria in blood cultures by using fluorescently labeled oligonucleotide probes. J. Clin. Microbiol.38:
814–817.
17.Kempf, V. A., K. Trebesius, and I. B. Autenrieth.2000. Fluorescent In situ hybridization allows rapid identification of microorganisms in blood cultures. J. Clin. Microbiol.38:830–838.
18.Koneman, E. W., S. D. Allen, W. M. Janda, P. C. Schreckenberger, and W. C. Winn, Jr.1997. Color atlas and textbook of diagnostic microbiology, 5th ed. Lippincott, Philadelphia, Pa.
19.Lee, Y., C. S. Lee, Y. J. Kim, S. Chun, S. Park, Y. S. Kim, and B. D. Han.
2002. Development of DNA chip for the simultaneous detection of various beta-lactam antibiotic-resistant genes. Mol. Cells14:192–197.
20.Lyytikainen, O., J. Lumio, H. Sarkkinen, E. Kolho, A. Kostiala, and P. Ruutu.2002. Nosocomial bloodstream infections in Finnish hospitals during 1999–2000. Clin. Infect. Dis.35:e14–e19.
21.Maes, N., J. Magdalena, S. Rottiers, Y. De Gheldre, and M. J. Struelens.
2002. Evaluation of a triplex PCR assay to discriminateStaphylococcus au-reusfrom coagulase-negative staphylococci and determine methicillin resis-tance from blood cultures. J. Clin. Microbiol.40:1514–1517.
22.Marlowe, E. M., J. J. Hogan, J. F. Hindler, I. Andruszkiewicz, P. Gordon, and D. A. Bruckner.2003. Application of an rRNA probe matrix for rapid identification of bacteria and fungi from routine blood cultures. J. Clin. Microbiol.41:5127–5133.
23.Martineau, F., F. J. Picard, N. Lansac, C. Menard, P. H. Roy, M. Ouellette, and M. G. Bergeron.2000. Correlation between the resistance genotype determined by multiplex PCR assays and the antibiotic susceptibility patterns ofStaphylococcus aureusandStaphylococcus epidermidis. Antimicrob. Agents Chemother.44:231–238.
24.Oliveira, K., S. M. Brecher, A. Durbin, D. S. Shapiro, D. R. Schwartz, P. C. De Girolami, J. Dakos, G. W. Procop, D. Wilson, C. S. Hanna, G. Haase, H. Peltroche-Llacsahuanga, K. C. Chapin, M. C. Musgnug, M. H. Levi, C. Shoemaker, and H. Stender.2003. Direct identification ofStaphylococcus aureusfrom positive blood culture bottles. J. Clin. Microbiol.41:889–891. 25.Perreten, V., L. Vorlet-Fawer, P. Slickers, R. Ehricht, P. Kuhnert, and J.
Frey.2005. Microarray-based detection of 90 antibiotic resistance genes of gram-positive bacteria. J. Clin. Microbiol.43:2291–2302.
26.Polyzou, A., A. Slavakis, S. Pournaras, A. N. Maniatis, D. Sofianou, and A. Tsakris.2001. Predominance of a methicillin-resistantStaphylococcus aureus
clone susceptible to erythromycin and several other non-beta-lactam anti-biotics in a Greek hospital. J. Antimicrob. Chemother.48:231–234. 27.Poole, K.2004. Efflux-mediated multiresistance in gram-negative bacteria.
Clin. Microbiol. Infect.10:12–26.
28.Pournaras, S., A. Slavakis, A. Polyzou, D. Sofianou, A. N. Maniatis, and A. Tsakris.2001. Nosocomial spread of an unusual methicillin-resistant Staph-ylococcus aureusclone that is sensitive to all non-beta-lactam antibiotics, including tobramycin. J. Clin. Microbiol.39:779–781.
29.Reacher, M. H., A. Shah, D. M. Livermore, M. C. Wale, C. Graham, A. P. Johnson, H. Heine, M. A. Monnickendam, K. F. Barker, D. James, and R. C. George.2000. Bacteraemia and antibiotic resistance of its pathogens re-ported in England and Wales between 1990 and 1998: trend analysis. Bmj.
320:213–216.
30.Reimer, L. G., M. L. Wilson, and M. P. Weinstein.1997. Update on detection of bacteremia and fungemia. Clin. Microbiol. Rev.10:444–465.
31.Sambrook, J., and D. W. Russell.2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, New York, N.Y. 32.Schmidt, H., L. Beutin, and H. Karch. 1995. Molecular analysis of the
plasmid-encoded hemolysin ofEscherichia coliO157:H7 strain EDL 933. Infect. Immun.63:1055–1061.
33.Schmitz, F. J., M. Steiert, B. Hofmann, J. Verhoef, U. Hadding, H. P. Heinz, and K. Kohrer.1998. Development of a multiplex-PCR for direct detection of the genes for enterotoxin B and C, and toxic shock syndrome toxin-1 in
Staphylococcus aureusisolates. J. Med. Microbiol.47:335–340.
34.Shrestha, N. K., M. J. Tuohy, G. S. Hall, C. M. Isada, and G. W. Procop.
2002. Rapid identification ofStaphylococcus aureusand themecAgene from BacT/ALERT blood culture bottles by using the LightCycler system. J. Clin. Microbiol.40:2659–2661.
35.Stears, R. L., T. Martinsky, and M. Schena.2003. Trends in microarray analysis. Nat. Med.9:140–145.
36.Strommenger, B., C. Kettlitz, G. Werner, and W. Witte.2003. Multiplex PCR assay for simultaneous detection of nine clinically relevant antibiotic resis-tance genes inStaphylococcus aureus. J. Clin. Microbiol.41:4089–4094. 37.Vannuffel, P., J. Gigi, H. Ezzedine, B. Vandercam, M. Delmee, G. Wauters,
and J. L. Gala.1995. Specific detection of methicillin-resistant Staphylococ-cusspecies by multiplex PCR. J. Clin. Microbiol.33:2864–2867.
38.Volokhov, D., V. Chizhikov, K. Chumakov, and A. Rasooly.2003. Microarray analysis of erythromycin resistance determinants. J. Appl. Microbiol.95:787– 798.
39.Volokhov, D., A. Rasooly, K. Chumakov, and V. Chizhikov.2002. Identifi-cation ofListeriaspecies by microarray-based assay. J. Clin. Microbiol.40:
4720–4728.
40.Vora, G. J., C. E. Meador, D. A. Stenger, and J. D. Andreadis.2004. Nucleic acid amplification strategies for DNA microarray-based pathogen detection. Appl. Environ. Microbiol.70:3047–3054.
41.Wellinghausen, N., B. Wirths, A. Essig, and L. Wassill.2004. Evaluation of the Hyplex BloodScreen Multiplex PCR-Enzyme-linked immunosorbent as-say system for direct identification of gram-positive cocci and gram-negative bacilli from positive blood cultures. J. Clin. Microbiol.42:3147–3152. 42.Wolfgang, M. C., B. R. Kulasekara, X. Liang, D. Boyd, K. Wu, Q. Yang, C. G.
Miyada, and S. Lory.2003. Conservation of genome content and virulence determinants among clinical and environmental isolates ofPseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA100:8484–8489.
43.Ye, R. W., T. Wang, L. Bedzyk, and K. M. Croker.2001. Applications of DNA microarrays in microbial systems. J. Microbiol. Methods47:257–272.