continued acceleration of linear growth. Other features include fatigue, hyperhydrosis, goitre, osteoarthritis, carpal tunnel syndrome, and other peripheral neuropathies.1–3,5An increased risk of large-bowel polyps
and cancer has also been reported in large series of patients with acromegaly; thus, screening by colonoscopy is needed.6–8 Moreover, central or obstructive sleep
apnoea and cardiomyopathy are important risk factors for morbidity and mortality; both improve after suppression of GH and insulin-like growth factor I (IGF-I).9,10
Menstrual disorders and decreased libido or potency are related to anterior-pituitary insufficiency but also to cosecretion of prolactin in 30–40% of patients1–3,5
Acromegaly and gigantism are generally clinically clear and can be readily confirmed by measurement of GH concentrations, which in over 90% of patients are more than 10 µg/L.5Since secretion of GH is pulsatile, single
measurements are not reliable. The oral glucose tolerance test is the simplest and most specific dynamic test to diagnose and assess the cure of acromegaly. In healthy people, the oral glucose tolerance test (75–100 g glucose) suppresses GH concentrations to less than 2 µg/L after 2 h, whereas in patients with acromegaly such suppression does not occur, and a paradoxical GH increase is commonly seen.1 Measurement of plasma
IGF-I concentrations in one sample was proposed to replace the oral glucose tolerance test5 because IGF-I
concentrations are stable and are a function of the integrated 24 h GH concentration.1Assays of circulating
IGF-I-binding protein 3 (IGF-BP3) and free IGF-I do not, however, have a higher diagnostic sensitivity than the measurement of total IGF-I.11,12The greater sensitivity of
the GH assay facilitates the distinction of acromegalic patients and healthy people, as is shown by use of the chemiluminescence GH assay.13This assay may be useful
to show the persistence of GH hypersecretion after surgery or during medical therapy.
The last step is to investigate whether an anterior pituitary tumour is present. The refinement of imaging techniques, including high-field magnetic resonance imaging (which produces high signal-to-noise ratios and high spatial resolutions), with high temporal resolution studies has improved the detection of small pituitary or ectopic tumours.
Management
There are five objectives of treatment in acromegaly: removal of the tumour with resolution of its mass effects, preservation of normal residual pituitary function, and Growth hormone (GH) and prolactin are polypeptide
hormones (191 and 199 aminoacids, respectively) produced by specific populations of pituitary cells. The two hormones have 16% structural homology. The clinical consequences of excess production, however, differ substantially: acromegaly from excess of GH is a severe systemic disease that shortens life-expectancy owing to respiratory, metabolic, and cardiovascular complications and malignant disorders;1,2
hyperprolactinaemia from excess of prolactin leads to infertility and gonadal and sexual dysfunction.3 In most
patients with prolactin or GH excess, the cause is a pituitary adenoma.4The availability of effective and
well-tolerated somatostatin and dopamine analogues has changed the therapeutic approach to these patients.
GH concentrations can be measured either in µg/L or in mU/L, according to the assay used. We use µg/L. The equivalent ratio of mU/L to µg/L was 2 (2 mU/L=1 µg/L) before January, 1997, and 3 thereafter.
GH excess
EpidemiologyThe prevalence of GH-secreting adenomas is 50–80 cases per million, and the incidence is three to four new cases per million per year.1,2 In less than 2% of patients,
excessive GH secretion may be the consequence of a hypothalamic or ectopic tumour (gangliocytoma, bronchial, or pancreatic) that produces GH-releasing hormone (GHRH); such tumours lead to somatotroph hyperplasia or a well-defined adenoma. The ectopic secretion of GH itself is even rarer.1
Diagnosis
The clinical features of acromegaly develop insidiously and progressively over many years. The average delay between onset of symptoms and diagnosis is about 6 years.5 In adults, acromegaly due to chronic GH
hypersecretion is characterised by local bone overgrowth. In children and adolescents, acromegaly leads to gigantism because the associated secondary hypogonadism delays epiphysial closure, which allows
Growth-hormone and prolactin excess
Annamaria Colao, Gaetano Lombardi
Lancet 1998;352:1455–61
Department of Molecular and Clinical Endocrinology and Oncology, “Federico II” University, via S Pansini 5, 80131 Naples, Italy (A Colao MD, G Lombardi MD).
Correspondence to:Dr Annamaria Colao (e-mail: [email protected])
Seminar
The treatment of acromegaly and hyperprolactinaemia has been improved by the availability of effective and well-tolerated slow-release somatostatin analogues and dopamine agonists with long-lasting activity, such as cabergoline. The use of these drugs has extended the possibility of treatment to patients who would have responded poorly to the previously available compounds, such as octreotide or bromocriptine, and to those who were intolerant to pharmacotherapy. Moreover, the improvement in the management of acromegaly has enabled the reversal, at least partly, of cardiomyopathy and sleep apnoea, two important risk factors for morbidity and mortality in these patients.
prevention of recurrences; restoration of normal basal and stimulated GH secretion; relief from symptoms directly caused by GH excess; prevention of disabling long-term consequences (progressive disfigurement, bone expansion, osteoarthritis, and cardiomyopathy) and risk factors for vascular damage (hypertension, insulin resistance, diabetes mellitus, and lipid abnormalities); and reversal of the poor long-term outcome. Variables that remain unknown are the degree of GH suppression needed to achieve treatment success (especially for the last two objectives), whether the adverse long-term outlook can be reversed by any treatment, and what is the best treatment for suppression of GH, IGF-I, or both.
The currently available treatment options for acromegaly include surgery, irradiation, and pharmacological suppression of GH concentrations by dopamine agonists or somatostatin analogues. The clinical assessment of treatment efficacy for these approaches, alone or combined, is difficult because moderate improvement may occur even with partial decreases in GH concentrations, whereas some residual disease effects may persist once GH concentrations have been lowered. There is, however, no consensus on the criteria defining the cure of acromegaly. The most widely accepted criteria include mean serum GH concentrations of less than 2·5 µg/L (average of at least three to five samples), GH concentrations after suppression with the oral glucose tolerance test of less than 2·0 µg/L, and normal plasma IGF-I concentrations. One preliminary retrospective study reported that normal survival was attained in patients with GH concentrations after treatment of less than 2·5 µg/L, but not in those with higher concentrations.14Another study confirmed that the
lower the serum GH concentrations after treatment, the lower the mortality.15 Cure should be followed by
restoration of physiological patterns of GH secretion, but there are still questions about whether this approach really offers any practical advantage over basal GH and IGF-I values in routine management of patients with acromegaly.16 Therefore, although strict definition of
biochemical cure may not be necessary, what is essential is to define the GH and IGF-I values that reliably achieve clinical remission and decrease or eliminate long-term morbidity and mortality.16 This idea has led to the
concept of safe values, which is a more realistic concept in clinical practice. After any treatment, basal GH concentrations should be less than 2·5 µg/L or 1·7 µg/L, depending on the GH assay (ie, equivalent to 5 mU/L) with normal IGF-I concentrations for age.16
Other pituitary-hormone deficiencies that may be caused by the tumour or its treatment must be treated in the same way as in other types of pituitary tumours.
Surgery
Trans-sphenoidal adenomectomy is the first treatment for GH-secreting tumours.1,3 Among 224 consecutive
patients, this operation achieved endocrine remission in 72% of microadenomas, 50% of macroadenomas, and only 17% of giant adenomas.17 IGF-I concentrations
within the normal range and GH concentrations of less than 3·0 µg/L,18 or 1·6 µg/L,19 were achieved in 59% and
42%, respectively, of unselected patients. In microadenomas, surgery was successful in 61% of patients.19 Surgery relieves the compression on adjacent
structures such as the optic chiasm and the ventricles. Complications, such as leakage of cerebrospinal fluid,
arachnoiditis, and temporary or permanent diabetes insipidus, are rare. Pituitary failure is, however, reported in about 20–30% of patients with macroadenomas.1,19
Treatment with octreotide, a long-acting somatostatin analogue, has been reported to induce tumour shrinkage; Fahlbush and colleagues20therefore suggested this drug as
a preoperative treatment to improve surgical outcome. A controlled study did not support this hypothesis,21
but treatment for 3–6 months before surgery has been reported to improve metabolic and haemodynamic variables and to decrease substantially the duration of hospital stays for patients with acromegaly.22 To refine
pituitary surgery, a one-nostril endoscopic endonasal trans-sphenoidal procedure has been proposed to decrease damage to the nose and sphenoid sinus and to improve the management of the pituitary fossa.23
Radiation therapy
This treatment should be considered for patients with contraindications to surgery or whose operation has failed. Tumour response to radiotherapy is slow and up to 5 years may elapse before a clinically significant decrease in GH is achieved.24,25Serum GH concentrations
fall exponentially after irradiation, reportedly reaching less than 5 µg/L in 40–80% of patients.24,26Decreases of
GH concentrations to less than 2·5 µg/L occur in fewer patients,24 and achievement of normal IGF-I
concentrations is rare.27 The risks of complications such
as optic-nerve damage, cranial-nerve palsy, impaired memory, lethargy, and local tissue necrosis have been decreased through improved, precise isocentric simulators and accurate dosing techniques. Radiation damage to the surrounding normal pituitary tissue, however, leads to hypopituitarism in most patients within 10 years.1,24,25
The risk of malignant disorders of the central nervous system developing after radiotherapy should be considered. Brada and colleagues28 reported
that 2·4% of patients developed a second tumour, which suggests an actuarial risk of 1·3% at 10 years and 1·9% at 20 years, but no increased incidence of second tumours has been reported.29 ␥-knife radiosurgery is currently
being investigated, but it seems not to improve the efficacy of conventional radiotherapy, and its long-term side-effects are unknown.
Pharmacotherapy
Dopamine agonists and somatostatin analogues are used alone or in combination in selected patients1,2 to treat
acromegaly.
In healthy people, dopamine agonists stimulate GH secretion by a mechanism mediated through the central nervous system.30 By contrast, in about 50% of patients
with acromegaly, dopamine agonists inhibit GH secretion, presumably through the stimulation of D2 receptors.30 Dopamine agonists are primarily effective in
GH-secreting tumours that also secrete prolactin or that show immunostaining for prolactin.1,2,31 All dopamine
agonists, except quinagolide, are ergoline-derived compounds and are given orally. The frequency of administration varies from every 8–12 h (bromocriptine) to once weekly (cabergoline). In a collection of 28 series including more than 500 patients with acromegaly, bromocriptine lowered GH concentrations to less than 10 µg/L in 50% of cases, but to less than 5 µg/L in only 10–20%.30 Symptoms improved in up to 70% of
10–15%.30,32 Side-effects, such as nausea, vomiting,
hypotension, nasal congestion, and depression, in most patients can be prevented if lower doses are used at first. Since high doses are needed for therapeutic efficacy, however, side-effects are commonly a limiting factor. The published evidence does not suggest that any other dopamine agonist is more effective than bromocriptine or has different therapeutic/toxic ratios. Variable results have been reported with two selective D2-receptor agonists: cabergoline and quinagolide.31–34
Somatostatin suppresses GH concentrations in healthy people and patients with acromegaly, but its potential application in treating GH excess is limited by its short half-life (1–3 min).35Octreotide is a long-acting synthetic
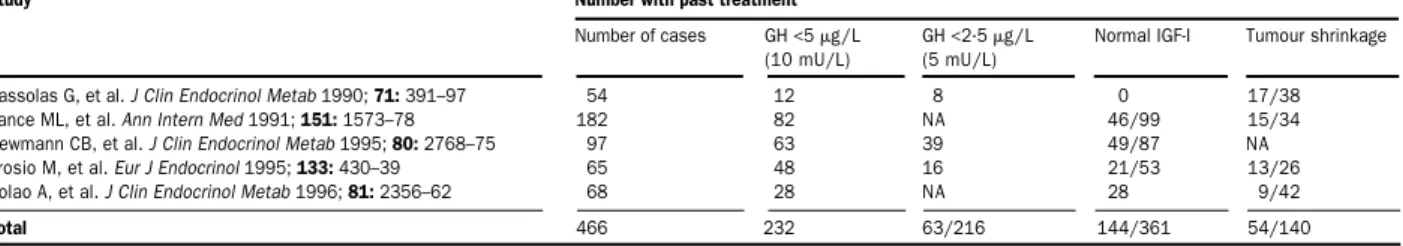
somatostatin analogue with a half-life of 80–100 min. In several studies, octreotide lowered GH in 94% and achieved normal IGF-I concentrations in about 70% of patients.1,2,36A combined analysis of 466 patients treated
worldwide showed that octreotide administration lowered GH concentrations to less than 2·5 µg/L in 29·2% of patients, achieved normal IGF-I concentrations in 39·9%, and achieved tumour shrinkage (>20% decrease in size) in 38·6% (table 1). Soon after administration of the first doses of octreotide, clinical signs and symptoms, especially headache, hyperhydrosis, and joint pain, are relieved. Octreotide treatment improves obstructive and central sleep apnoea (decrease in the number of apnoeic episodes and degree of blood oxygen desaturation)9and
significantly decreases left-ventricular mass (without improving cardiac performance),10 prostate size,37 and
joint thickness.38Despite the wide range of positive effects
of this treatment, including the evidence that thachyphylaxis does not occur even after 10 years of treatment,36octreotide is still reserved for patients whose
surgery fails or for control of symptoms during radiotherapy. The main reasons for the limited use of octreotide are its cost, the need for multiple daily injections, and its side-effects, especially on the gastrointestinal tract. Gallbladder abnormalities (sediment, sludge, microlithiasis, and gallstones) occur in about 20% of patients and are associated with low morbidity. Generally, these effects disappear spontaneously or after treatment with ursodeoxicolic acid in the absence of signs of symptomatic cholelithiasis or cholecystitis.39In patients with symptomatic cholelithiasis,
withdrawal of octreotide treatment is advisable.
Depot preparations have been developed to avoid the need for three injections of octreotide per day. Such preparations were developed to allow injections only once or twice every month and to achieve stable serum concentrations of the drug, good clinical control of symptoms and signs, sustained GH and IGF-I suppression, and good compliance.
At present, two depot preparations of long-acting somatostatin analogues are available in several European
countries. The first is a slow-release form of the cyclic octapeptide, lanreotide. Studies have reported that slow-release lanreotide is effective in patients previously responsive to octreotide but is better tolerated and improves compliance.40,41 13 of 22 acromegalic patients
treated with slow-release lanreotide for 3 years had normal GH and IGF-I concentrations after treatment. Nine of these patients, however, had to increase the injection frequency to one every 10 days for optimum control.41 Loose stools, nausea, abdominal pain, or a
combination of symptoms were reported by 11 of the patients for the first 2 days after injection, and four patients developed new gallstones.40
The second depot preparation is the slow-release form of octreotide incorporated in microspheres of a biodegradable polymer, poly-(DL-lactide-co-glycolide glucose).42 Decreases in GH concentrations to below
5 µg/L were recorded in 86–100%, to below 2 µg/L in 39–75%, and to below 1 µg/L in 24–40% in different series of patients with acromegaly including a total number of 175 cases.43 Normal plasma IGF-I
concentrations were achieved in 65·3% after the last injection.42 Tumour shrinkage by more than 20% was
recorded in 71·8% of patients.43After up to 34 months of
therapy, there was no evidence of tachyphylaxis. This slow-release octreotide preparation was well tolerated; up to 50% of patients experienced mild to moderate side-effects but they were of short duration and generally subsided with continued drug administration.43 The rate
of gallbladder abnormalities was higher among patients who had previously undergone long-term treatment with subcutaneous octreotide than among those who had not.43 Conclusions
Somastatin analogues are effective for most patients with acromegaly. Patients who have undergone unsuccessful trans-sphenoidal surgery or who are awaiting the therapeutic effect of external irradiation are candidates for treatment with these compounds. Because GH-secreting adenomas in elderly patients are more sensitive to somastatin analogues than those in younger people, these drugs can be recommended as the primary treatment for older patients. What is still unclear is whether the achievement of safe GH concentrations with any treatment improves the poor outlook for patients with acromegaly in terms of mortality from cardiorespiratory or neoplastic diseases.1–3,14,15 Furthermore, there are at
least five different somatostatin-receptor subtypes expressed in normal and neoplastic somatotrophs,36
whereas octreotide and lanreotide act mainly on receptor types 2 and 5. Therefore, whether these are the best analogues for treatment of acromegaly is doubtful. Lastly, 15–20% of patients with acromegaly do not respond well to somatostatin analogues, which suggests that abnormal expression or regulation of the receptor subtypes may be
Study Number with past treatment
Number of cases GH <5 g/L GH <2·5 g/L Normal IGF-I Tumour shrinkage (10 mU/L) (5 mU/L)
Sassolas G, et al. J Clin Endocrinol Metab 1990;71:391–97 54 12 8 0 17/38 Vance ML, et al. Ann Intern Med 1991;151:1573–78 182 82 NA 46/99 15/34 Newmann CB, et al. J Clin Endocrinol Metab 1995;80:2768–75 97 63 39 49/87 NA Arosio M, et al. Eur J Endocrinol 1995;133:430–39 65 48 16 21/53 13/26 Colao A, et al. J Clin Endocrinol Metab 1996;81:2356–62 68 28 NA 28 9/42
Total 466 232 63/216 144/361 54/140
NA=not assessed.
present. The results of trials of somatotroph-specific cytotoxic agents (targeted by an agonist or antibody to the GH-releasing-hormone receptor) for selective elimination of residual tumour cells, and GH-receptor antagonists to block the peripheral effects of GH will soon be available.
Prolactin excess
EpidemiologyThe aetiology of pathological hyperprolactinaemia is diverse (panel). Any process interfering with dopamine synthesis, its transport to the pituitary gland, or its action at lactotroph dopamine receptors can lead to hyperprolactinaemia.3 Once drugs are excluded,
microprolactinomas (<10 mm) or macroprolactinomas (>10 mm) are the most common causes of hyperprolactinaemia. The prevalence of prolactinomas is unknown; in necropsy series, the rate varies between 23% and 27% for microadenomas.44,45Macroadenomas are less
common than microadenomas, and occur more often in men than in women. This finding is probably related to duration of disease and the gradual development of symptoms, which primarily involve sexual and gonadal function in both sexes. Natural-history studies suggest, however, that more than 90% of microadenomas remain small and only a small proportion become macroadenomas.46 Macroadenomas may, therefore, be
biologically different from microadenomas.
Diagnosis
Secretion of prolactin is pulsatile, and at least three measurements are needed for the diagnosis to be made correctly. In rare cases, patients have an increased amount of high-molecular-weight prolactin (150–170 kDa), but the clinical implications of hyperprolactinaemia due to this molecular form of prolactin are still uncertain. During the past 20 years, many pharmacological tests have been used to distinguish tumoral from non-tumoral hyperprolactinaemia, with controversial results.3 These tests are no longer used,
because of the poor reliability in differential diagnosis. Generally, prolactin concentrations are correlated with tumour size.3Neuroradiological studies are mandatory to
complete the diagnostic protocol.
Management
The objectives of the treatment of hyperprolactinaemia are: suppression of excessive hormone secretion and its clinical consequences (particularly infertility, sexual dysfunction, and osteoporosis); tumour removal and relief of any disturbance in vision and cranial-nerve function; preservation of the residual pituitary function; and, if possible, prevention of disease recurrence or progression. Before medical treatment became available, therapy consisted of surgical removal with or without pituitary irradiation. Since the early 1970s, surgery and radiotherapy have been progressively replaced by dopamine agonists. There is controversy over the primary therapy (medical or surgical) of tumour-related hyperprolactinaemia, and over the treatment of women who wish to become pregnant.
Surgery
For microprolactinomas, trans-sphenoidal microsurgical resection restores normal prolactin concentrations and
normal menses and eliminates galactorrhoea in 85–90% of patients.3,47 Success rates are higher in patients who
have prolactin concentrations of less than 200 mg/L and who have had amenorrhoea for less than 5 years.3 The
risks of complications and hypopituitarism are small. Recurrence rates vary substantially between different series.3,47–50 In one report,47 71% of patients with
microprolactinoma were initially cured by means of surgery with a subsequent recurrence rate of 17% and a long-term cure rate of 59%. In another series,48the
long-term cure rate was 74% (initial cure in 90%, recurrence rate 16%). For macroprolactinomas, trans-sphenoidal surgery was less successful; 32% of patients were cured initially, with a 19% recurrence rate and a long-term cure rate of only 26%.48Long-term recurrence rates of 33·3%
and 12·0% were reported in macroprolactinomas.49,50
Despite these disappointing results, some centres still recommend surgery to debulk the tumour to protect vital structures such as the optic chiasm.3 Rapid tumour
shrinkage is, however, achieved with dopamine-agonist treatment in most patients.
Radiotherapy
Radiotherapy is applied in large invasive macroprolactinomas when medical treatment fails to suppress prolactin concentrations or induce tumour shrinkage. Radiotherapy gradually decreases prolactin concentrations, generally reaching normal values within 10 years.51 Radiotherapy prevents further growth of the
tumour but is less effective in promoting a prompt
Aetiology of hyperprolactinaemia Hypothalamic disorders
Tumours—craniopharyngioma, germinoma, third-ventricle tumour, cyst, glioma, hamartoma, metastasis
Infiltrative diseases—sarcoidosis, tuberculosis, Langerhans-cell histiocytosis Pseudotumour cerebri Cranial irradiation Pituitary disorders Microprolactinoma or macroprolactinoma Acromegaly Cushing’s disease Pituitary-stalk section Empty-sella syndrome
Pseudoprolactinomas—non-functioning adenoma, meningioma, intrasellar germinoma, metastasis that may produce functional stalk section
Infiltrative diseases—giant-cell granuloma, sarcoidosis
Drugs
Neuroleptics—perphenazine, fluphenazine, thorazine, promazine, trifluoperazine, haloperidol, chlorpromazine, dopamine-receptor blockers (such as metoclopramide, sulpiride, domperidone, cimetidine)
Antidepressants—amoxapine, imipramine, amitriptyline Antihypertensives—␣-methyldopa, reserpine
Oestrogens Opioids
Phenylalkylamine class N-type calcium-channel blockers—verapamil
Primary hypothyroidism Chronic renal failure Cirrhosis
Neurogenic
Chest wall or spinal-cord lesions, breast stimulation
Stress physical or psychological Idiopathic
decrease in prolactin concentrations.52 Radiobiological
damage rarely occurs as long as a linear accelerator is used, therapy is delivered through three fields, and a fractionated dose of less than 2 Gy daily is delivered to a total dose of 45 Gy.53 Owing to the high success rate of
pharmacotherapy, however, radiotherapy is rarely applied to prolactinomas.
Pharmacotherapy
For more than 20 years dopamine agonists, especially bromocriptine, but also pergolide and lisuride, have been the standard drugs for hyperprolactinaemia. They inhibit prolactin synthesis and secretion and decrease cellular DNA synthesis and tumour growth. Restoration of normal prolactin concentrations and tumour shrinkage after bromocriptine 2·5–5·0 mg daily are observed in about 80% of prolactinomas.3,47,53 Side-effects (nausea,
dizziness, and orthostatic hypotension) are limiting factors in continuing the treatment in 5–10% of patients.3,47,53 1·3% of patients are reported to have
developed psychoses.54 Complete or partial resistance to
bromocriptine treatment is seen in a few patients (about 5%). Such resistance is thought to be the consequence of abnormalities of the dopamine D2 receptor or post-receptor abnormalities.55 Quinagolide and cabergoline,
two selective D2 receptor agonists, are effective in some resistant patients.55,56
There is evidence that weekly administration of cabergoline is more effective and better tolerated than daily bromocriptine. In a randomised multicentre study of 459 women with hyperprolactinaemia, stable normal prolactin concentrations were achieved in 83% of patients treated with cabergoline compared with 59% of those treated with bromocriptine.57
Ovulatory cycles or pregnancies were recorded in 72% of women treated with cabergoline and in 52% of those treated with bromocriptine.57 A further advantage is that side-effects
were substantially less frequent, less severe, and shorter-lived (especially nausea and vomiting) with cabergoline-than with bromocriptine.57 Moreover, cabergoline
treatment at weekly low doses induced substantial tumour shrinkage.58–60
To overcome poor tolerability of the oral formulation of bromocriptine, a depot intramuscular injectable preparation of cabergoline was developed. This formulation (given at doses of 50–150 mg every 4 weeks) was well tolerated,61 and initial side-effects could be
prevented by administration of oral prednisone.62 This
preparation seems to be especially useful for patients who previously experienced severe side-effects from oral therapy.53 Striking tumour shrinkage and consequent
improvement of visual field were reported as early as 1–2 weeks from the first injection.61 To decrease
gastrointestinal side-effects, intravaginal administration of bromocriptine was also tested with partial efficacy.63
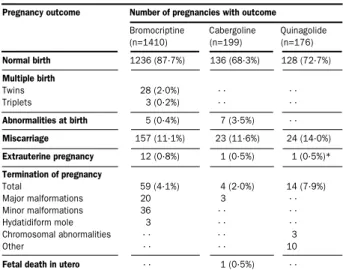
Bromocriptine, cabergoline, and quinagolide have not been associated with any adverse effect on fetal development or pregnancy outcome (table 2). Experience with bromocriptine is, however, wider than that with the other two drugs. Therefore, for women who want to retain fertility, this drug remains the treatment of choice in most centres, whereas cabergoline and quinagolide are acceptable second-line drugs in patients intolerant or resistant to bromocriptine. Current opinion is to stop treatment if patients with microprolactinomas become pregnant, whereas continued treatment is preferable in
those with macroprolactinomas to avoid the risk of tumour enlargement. In this case sellar magnetic resonance imaging is essential.
In women with microprolactinomas who do not want to retain fertility, oestrogen-replacement therapy is an option, since it does not increase tumour size.64 The
possibility of tumour enlargement in patients with oestrogen-sensitive tumours should, however, be monitored by serial magnetic resonance imaging.
Much information is available on the recovery of menses and fertility in hyperprolactinaemic women, but there are few data for male patients. Impairment of gonadal function in men seems to be a functional modification and can be reversed by long-term quinagolide, bromocriptine, or cabergoline.65,66
Restoration of normal sperm morphology and function occurred faster with cabergoline than with bromocriptine treatment.66 In patients with large tumours, irreversible
damage to the pituitary gonadotrophs can occur.
In both sexes osteoporosis can occur with hyperprolactinaemia; this disorder has been attributed to the concomitant hypogonadism rather than to hyperprolactinaemia per se, since the extent of bone loss is strictly related to the duration of hypogonadism.67, 68
Treatment with dopamine agonists restores gonadal function and increases vertebral bone-mineral density in most hyperprolactinaemic women.67 Few data are
available in men, but one study showed that 18 months of treatment with bromocriptine, quinagolide, or cabergoline did not completely eliminate osteopenia/osteoporosis in hyperprolactinaemic men, and a longer period of treatment may be necessary.69
Conclusions
Medical treatment with dopamine agonists is the most appropriate first-line treatment for macroprolactinomas except in poorly responsive or resistant patients. Dopamine agonists shrink tumours and can lead to tumour disappearance.31,58–60 Visual-field defects
substantially improve in most patients, in many cases without need for surgical tumour debulking. Theoretically, pharmacotherapy should be continued indefinitely, but the dose can be decreased over the years;
Pregnancy outcome Number of pregnancies with outcome
Bromocriptine Cabergoline Quinagolide (n=1410) (n=199) (n=176) Normal birth 1236 (87·7%) 136 (68·3%) 128 (72·7%) Multiple birth Twins 28 (2·0%) · · · · Triplets 3 (0·2%) · · · · Abnormalities at birth 5 (0·4%) 7 (3·5%) · · Miscarriage 157 (11·1%) 23 (11·6%) 24 (14·0%) Extrauterine pregnancy 12 (0·8%) 1 (0·5%) 1 (0·5%)* Termination of pregnancy Total 59 (4·1%) 4 (2·0%) 14 (7·9%) Major malformations 20 3 · · Minor malformations 36 · · · · Hydatidiform mole 3 · · · · Chromosomal abnormalities · · · · 3 Other · · · · 10
Fetal death in utero · · 1 (0·5%) · · Derived from Ferrari, et al. Hum Reprod 1995;10:1647–52; Turkalj, et al. JAMA 1982;247:1589–91; Webster. Drug Saf 1996;14:228–38. *This pregnancy was terminated.
Table 2: Pregnancy outcome in women treated with bromocriptine, cabergoline, or quinagolide
in 10–20% of patients drug discontinuation does not lead to recurrence of hyperprolactinaemia or rapid tumour regrowth.70In microprolactinomas, the treatment strategy
could be reconsidered on the basis of the high cure rate of trans-sphenoidal adenomectomy, which might be further improved by use of new endoscopic techniques. Surgery should also be recommended for patients who are severely intolerant to dopamine agonists.
We thank SWJ Lamberts for revising this paper.
References
1 Melmed S, Ho K, Klibanski A, Reichlin S, Thorner MO. Recent advances in pathogenesis, diagnosis and management of acromegaly.
J Clin Endocrinol Metab1995;80:3395–402.
2 Colao A, Merola B, Ferone D, Lombardi G. Extensive personal experience: acromegaly. J Clin Endocrinol Metab1997;82:2777–81. 3 Aron DC, Tyrrell B, Wilson CB. Pituitary tumors: current concepts in
diagnosis and management. World J Med1995;162:340–52. 4 Shimon I, Melmed S. Pituitary tumor pathogenesis. J Clin Endocrinol
Metab1997;82:1675–81.
5 Lamberts SWJ, van der Lely AJ, de Herder WW. Clinical and medical diagnosis of acromegaly. Metabolism1995;44(suppl 1): 15–17. 6 Delhougne B, Deneux C, Abs R, et al. The prevalence of colonic
polyps in acromegaly: a colonoscopic and pathological study in 103 patients.J Clin Endocrinol Metab1995;80:3223–26.
7 Jenkins PJ, Fairclough PD, Richards T, et al. Acromegaly, colonic polyps and carcinoma.Clin Endocrinol 1997;47:17–22.
8 Colao A, Balzano A, Ferone D, et al. Increased prevalence of colonic polyps and altered lymphocyte subset pattern in the colonic lamina propria in acromegaly. Clin Endocrinol 1997;47:23–28.
9 Barkan A. Acromegalic arthropathy and sleep apnea. J Endocrinol
1997;155:S41–44.
10 Lombardi G, Colao A, Marzullo P, et al. Is growth hormone bad for your heart? Cardiovascular impact of GH deficiency and of acromegaly.J Endocrinol1997;155:S33–36.
11 de Herder WW, van der Lely AJ, Janssen JAMJL, et al. IGFBP–3 is a poor parameter for assessment of clinical activity in acromegaly.
Clin Endocrinol 1995;43:501–15.
12 Stoffel-Wagner B, Springel W, Bidlingmeier F, Klingmuller D. A comparison of different methods for diagnosing acromegaly.
Clin Endocrinol 1997;46:531–37.
13 Chapman IM, Hartan ML, Straume M, Johnson ML, Veldhuis JD, Thorner MO. Enhanced sensitivity growth hormone (GH) chemiluminescence assay reveals lower postglucose nadir GH concentrations in men than in women. J Clin Endocrinol Metab 1994; 78:1312–19.
14 Bates A, vant’Hoff W, Jones J, Clayton R. An audit of outcome of treatment in acromegaly. QJM1993;86:293–99.
15 Rajasoorya C, Holdaway IM, Wrightson P, Scott DJ, Ibbertson AK. Determinants of clinical outcome and survival in acromegaly. Clin Endocrinol1994;41:95–102.
16 Clayton RN. New developments in the management of acromegaly: should we achieve absolute biochemical cure? J Endocrinol1997; 155:S23–29.
17 Fahlbush R, Honegger J, Buchfelder M. Acromegaly: the place of the neurosurgeon.Metabolism1996;45:65–66.
18 Yamada S, Aibat T, Takada K, et al. Retrospective analysis of long-term surgical results in acromegaly: preoperative and post-operative factors predicting outcome. Clin Endocrinol1996;45:291–98. 19 Sheaves R, Jenkins P, Blackburn P, et al. Outcome of transsphenoidal
surgery for acromegaly using strict criteria for surgical cure.
Clin Endocrinol1996;45:407–13.
20 Fahlbush R, Giovannelli M, Buchfelder M, Losa M, and participants of the Conference on Medical and Surgical Treatment of Pituitary Adenomas. Zurich, Oct 5, 1991: consensus statement in the medical and surgical treatment of pituitary adenomas: the role of long-acting somatostatin analogs. J Endocrinol Invest1993;16:449–60. 21 Ezzat S, Horvath E, Harris AG, Kovacs K. Morphological effects of
octreotide on growth hormone-producing pituitary adenomas. J Clin Endocrinol Metab1994;79:113–18.
22 Colao A, Ferone D, Cappabianca P, et al. Effect of octreotide pretreatment on surgical outcome in acromegaly. J Clin Endocrinol Metab1997;82:3308–14.
23 Jho HD, Carrau RL, Ko Y. Endoscopic pituitary surgery. In: Wilkins RH, Rengachary SS, eds. Neurosurgical operative atlas.Park Ridge, Ill: Am Assoc Neurol Surg1996;5:1–12.
24 Wass JAH. Evidence for the effectiveness of radiotherapy in the treatment of acromegaly. J Endocrinol1997;155:S57–58.
25 Eastman RC, Gorden P, Glatstein E, et al. Radiation therapy for acromegaly.Endocrinol Metab Clin North Am1992;21:693–712. 26 Brierle JD, Gosbodarowicz MK, Panzanellat J, Sutcliffe SB,
Simpson WJ, Tsang RW. The role of radiation therapy in clinically hormonally active pituitary adenomas. Radiat Oncol1996;41:55–53. 27 Barkan A, Halasz I, Dornfeld KJ, et al. Pituitary irradiation is
ineffective in normalizing plasma insulin-like growth factor I in patients with acromegaly. J Clin Endocrinol Metab1997;82:3187–91. 28 Brada M, Ford D, Aschley S, et al. Risk of second brain tumor after
conservative surgery and radiotherapy for pituitary adenoma. BMJ
1992;304:1343–46.
29 Jones A. Radiation oncogenesis in relation to the treatment of pituitary tumours.Clin Endocrinol1991;35:379–97.
30 Jaffe CA, Barkan AL. Acromegaly: recognition and treatment. Drugs
1994;47:425–45.
31 Colao A, Ferone D, Marzullo P, et al. Effect of different dopaminergic agents in the treatment of acromegaly. J Clin Endocrinol Metab1997; 82: 518–23.
32 Bevan JS, Webster J, Burke CW, Scanlon MF. Dopamine agonists and pituitary tumor shrinkage. Endocr Rev1992;13:221–35.
33 Abs R, Versholst J, Maiter D, et al. Cabergoline in the treatment of acromegaly: a study in 64 patients.J Clin Endocrinol Metab1998;83: 374–78.
34 Chiodini PG. Attanasio R, Cozzi R, et al. CV 205–502 in acromegaly.
Acta Endocrinol (Copenh) 1993;128: 389–93.
35 Hale R, Besser GM, Schally A, et al. Actions of growth hormone release inhibitory hormone in healthy men and in acromegaly. Lancet
1973; ii: 581–84.
36 Lamberts SWJ, van der Lely A-J, de Herder WW, Hofland LJ. Octreotide.N Engl J Med 1996;334:246–54.
37 Colao A, Marzullo P, Ferone D, et al. Prostatic hyperplasia: an unknown feature of acromegaly. J Clin Endocrinol Metab1998;83: 775–79.
38 Colao A, Marzullo P, Vallone G, et al. Reversibility of joint thickening in acromegalic patients: an ultrasonography study.J Clin Endocrinol Metab1998;83: 2121–25.
39 Redfern JS, Fortuner WJ II.Octreotide-associated biliary tract dysfunction and gallstones formation: pathophysiology and management.Am J Gastroenterol. 1995; 90:1042–52. 40 Giusti M, Gussoni G, Cuttica CM, Giordano G, and the Italian
Multicenter Slow Release Lanreotide Study Group. Effectiveness and tolerability of slow release lanreotide treatment in active acromegaly; six month report on an Italian multicenter study. J Clin Endocrinol Metab1996;81:2089–97.
41 Caron P, Morange-Ramos I, Cogne M, Jaquet P. Three year follow up of acromegalic patients treated with intramuscular slow-release lanreotide.J Clin Endocrinol Metab1997;82:18–22.
42 Lancranjan I, Bruns C, Grass P, et al. Sandostatin-LAR: a promising therapeutic tool in the management of acromegalic patients.
Metabolism1996;45:67–71.
43 Gillis JC, Noble S, Goa KL. Octreotide long-acting release (LAR): a review of its phamacological properties and therapeutic use in the management of acromegaly. Drugs1997;53:681–99.
44 Costello RT. Subclinical adenoma of the pituitary gland. Am J Pathol
1936;12:205–16.
45 Burrow GN, Wortzman G, Rewcastle NB, et al. Microadenomas of the pituitary gland and abnormal sellar tomograms in an unselected autopsy series. N Engl J Med1980;304:156–58.
46 Schlechte J, Dolan K, Sherman B, Chapler F, Luciano A. The natural history of untreated hyperprolactinemia: a prospective analysis. J Clin Endocrinol Metab1989;68:412–18.
47 Molitch ME, Thorner MO, Wilson C. Management of prolactinomas.
J Clin Endocrinol Metab1997;82:996–1000.
48 Molitch ME. Prolactinoma. In: Melmed S, ed. The pituitary. Boston: Blackwell Scientific, 1995: 443–77.
49 Ciccarelli E, Ghigo E, Miola C, Gandini G, Muller EE, Camanni F. Long-term follow-up of cured prolactinoma patients after successful adenomectomy.Clin Endocrinol1990;32:583–92.
50 Webster J, Page MD, Bevan JS, Richards SH, Douglas-Jones AG, Scanlon MF. Low recurrence rate after partial hypophysectomy for prolactinoma: the predictive value of dynamic prolactin function test.
Clin Endocrinol1992;36:35–44.
51 Grossman A, Cohen BC, Charlesworth M, et al. Treatment of prolactinomas with megavoltage radiotherapy. BMJ1984;228: 1105–09. 52 Sheline GE, Grossman A, Jones AE, et al. Radiation therapy for
prolactinomas. In: Black PMcL, Zervas NT, Ridgway EC, et al, eds. Secretory tumors of the pituitary gland: progress in endocrine research and therapy. New York: Raven Press, 1984: 93–108.
53 Cunnah D, Besser GM. Management of prolactinomas.
Clin Endocrinol1991;34:231–35.
54 Turner TH, Cookson JC, Wass JAH, Drury PL, Price PA, Besser GM. Psychotic reactions during treatment of pituitary tumours with dopamine agonist. BMJ1984;289:1101–03.
55 Brue T, Pellegrini I, Gunz G, et al. Effects of dopamine agonist CV 205–502 in human prolactinomas resistant to bromocriptine. J Clin Endocrinol Metab1992;74:577–84.
56 Colao A, Di Sarno A, Sarnacchiaro F, et al. Prolactinomas resistant to standard dopamine agonists respond to chronic cabergoline treatment.
J Clin Endocrinol Metab1997; 82: 876–83.
57 Webster J, Piscitelli G, Polli A, et al. A comparison of cabergoline and bromocriptine in the treatment of hyperprolactinemic amenorrhea.
N Engl J Med1994;331:904–09.
58 Biller BMK, Molitch ME, Vance ML, et al. Treatment of prolactin-secreting macroadenomas with the once-weekly dopamine agonist cabergoline.J Clin Endocrinol Metab1996;81:2338–43.
59 Ferrari C, Abs R, Bevan JS, et al. Treatment of macroprolactinoma with cabergoline: a study of 85 patients. Clin Endocrinol 1997;46:409–13. 60 Colao A, Di Sarno A, Landi ML, et al. Long-term and low-dose
treatment with cabergoline induces macroprolactinoma shrinkage.
J Clin Endocrinol Metab1997;82:3574–79.
61 Schettini G, Lombardi G, Merola B, et al. Rapid and long-lasting suppression on prolactin secretion and shrinkage of prolactinomas with improved tolerability after injection of long-acting repeatable form of bromocriptine (Parlodel LAR). Clin Endocrinol 1990;33:161–69. 62 Jenkins PJ, Jain A, Jones SL, Besser GM, Grossman AB. Oral
prednisolone supplement abolishes the acute adverse effects following initiation of depot bromocriptine therapy. Clin Endocrinol (Oxf)1996; 45:447–51.
63 Kletzky OA, Wermesch M. Effectiveness of vaginal bromocriptine in treating women with hyperprolactinemia. Fertil Steril1989;51: 269–72.
64 Corenblum B, Donovan L. The safety of physiological estrogen plus progestin replacement therapy and with oral contraceptive therapy in women with pathological hyperprolactinemia. Fertil Steril1993;59: 671–73.
65 Colao A, De Rosa M, Sarnacchiaro F, et al. Chronic treatment with CV 205–502 restores the gonadal function in hyperprolactinemic males.Eur J Endocrinol1996;135:548–52.
66 De Rosa M, Colao A, Di Sarno A, et al. Cabergoline treatment rapidly improves gonadal function in hyperprolactinemic males: a comparison with bromocriptine. Eur J Endocrinol1998;138:286–93.
67 Klibanski A, Neer RM, Beitins IZ, Ridgway C, Zarvas NT,
McArthur JW. Decreased bone density in amenorrheic women. N Engl J Med1980;303: 1511–14.
68 Greenspan SL, Oppenheim DS, Klibanski A. Importance of gonadal steroids to bone mass in men with hyperprolactinemic hypogonadism.
Ann Intern Med1989;110: 526–31.
69 Di Somma C, Colao A, Di Sarno A, et al. Bone markers and bone density responses to dopamine agonist therapy in hyperprolactinemic males.J Clin Endocrinol Metab1998;83: 807–13.
70 Johnston DG, Hall K, Kendall-Taylor P, Patrick D, Watson M, Cook DB. Effect of dopamine agonist withdrawal after long-term therapy in prolactinomas. Lancet1984; ii: 187–92.
Further reading
GH excess
Alexander L, Appleton D, Hall R, Ross WM, Wilkinson R. Epidemiology of acromegaly in the Newcastle region. Clin Endocrinol (Oxf) 1980; 12:71–79.
Andrews DW. Pituitary adenomas. Curr Opin Oncol 1997;9: 55–60.
Bengtsson B-A, Eden S, Ernest I, Oden A, Sjogren B. Epidemiology and long-term survival in acromegaly. Acta Med Scand 1988;223: 327–35.
Ezzat S, Forster MJ, Berchtold P, Redelmeier DA, Boerlin V, Harris A. Acromegaly. clinical and biochemical features in 500 patients. Medicine 1994;73:233–40.
Lamberts SWJ, Krenning EP, Reubi J-C. The role of somatostatin and its analogs in the diagnosis and treatment of tumors. Endocr Rev 1991; 12:450–82.
Melmed S, Dowling RH, Frohman LA, et al. Consensus statement: benefits versus risk of medical therapy for acromegaly. Am J Med 1994;97:468–73.
Nabarro JDN. Acromegaly. Clin Endocrinol (Oxf) 1980;26: 81–512.
Saccá L, Cittadini A, Fazio F. Growth hormone and the heart. Endocr Rev 1994;15:555–73.
Wass JAH. Acromegaly treatment after 100 years. BMJ 1993;307: 1505–06.
Prolactin excess
Berezin M, Shimon I, Hadani M. Prolactinoma in 53 men: clinical characteristics and modes of treatment. J Endocrinol Invest 1995; 18:436–41.
Jackson JA, Kleerekoper M, Parfitt M. Symptomatic osteoporosis in a man with hyperprolactinemic hypogonadism. Ann Intern Med 1986; 105:543–45.
Klibanki A, Greenspan SL. Increase in bone mass after treament of hyperprolactinemic amenorrhea. N Engl J Med 1986;315:542–46. Liuzzi A, Oppizzi G. Microprolactinomas: why requiem for surgery?
J Endocrinol Invest 1996;19:196–98.
Maor Y, Berezin M. Hyperprolactinemia in postmenopausal women. Fertil Steril 1997;67:693–96.
Rains CP, Bryson HM, Fitton A. Cabergoline: a review of its pharmacological properties and therapeutic potential in the treatment of hyperprolactinaemia and inhibition of lactation. Drugs 1995;49:255–79.
Serri O. Progress in the management of hyperprolactinemia. N Engl J Med 1994;331:942–44.
Soule SG, Farhi J, Conway GS, Jacobs HS, Powell M. The outcome of hypophysectomy for prolactinomas in the era of dopamine agonist therapy.Clin Endocrinol 1996;44:711–16.
Stewart PM, Maheshwaran S, Griffith J, et al. Pituitary imaging is essential for women with moderate hyperprolactinaemia. BMJ 1993; 306:507–08.
Webster J. A comparative review of the tolerability profile of DA agonists in the treament of hyperprolactinemia and inhibition of lactation.Drug Saf 1996;14:228–38.