mammary tumor progression in vivo,
demonstrating a critical interaction in the
tumor/stroma microenvironment
Puneeth Iyengar, … , Paolo Bonaldo, Philipp E. Scherer
J Clin Invest.
2005;
115(5)
:1163-1176.
https://doi.org/10.1172/JCI23424
.
The interactions of transformed cells with the surrounding stromal cells are of importance for
tumor progression and metastasis. The relevance of adipocyte-derived factors to breast
cancer cell survival and growth is well established. However, it remains unknown which
specific adipocyte-derived factors are most critical in this process. Collagen VI is abundantly
expressed in adipocytes. Collagen
–/–mice in the background of the mouse mammary tumor
virus/polyoma virus middle T oncogene (MMTV-PyMT) mammary cancer model
demonstrate dramatically reduced rates of early hyperplasia and primary tumor growth.
Collagen VI promotes its growth-stimulatory and pro-survival effects in part by signaling
through the NG2/chondroitin sulfate proteoglycan receptor expressed on the surface of
malignant ductal epithelial cells to sequentially activate Akt and
b
-catenin and stabilize
cyclin D1. Levels of the carboxyterminal domain of collagen VI
a
3, a proteolytic product of
the full-length molecule, are dramatically upregulated in murine and human breast cancer
lesions. The same fragment exerts potent growth-stimulatory effects on MCF-7 cells in vitro.
Therefore, adipocytes play a vital role in defining the ECM environment for normal and
tumor-derived ductal epithelial cells and contribute significantly to tumor growth at early
stages through secretion and processing of collagen VI.
Article
Oncology
Find the latest version:
Adipocyte-derived collagen VI affects
early mammary tumor progression in vivo,
demonstrating a critical interaction in the
tumor/stroma microenvironment
Puneeth Iyengar,1 Virginia Espina,2 Terence W. Williams,3 Ying Lin,1 David Berry,4,5
Linda A. Jelicks,6 Hyangkyu Lee,3 Karla Temple,7 Reed Graves,8 Jeffrey Pollard,9 Neeru Chopra,10
Robert G. Russell,11 Ram Sasisekharan,12 Bruce J. Trock,13 Marc Lippman,14 Valerie S. Calvert,15
Emanuel F. Petricoin III,15 Lance Liotta,2 Ekaterina Dadachova,16 Richard G. Pestell,11
Michael P. Lisanti,3 Paolo Bonaldo,17 and Philipp E. Scherer1
1Departments of Cell Biology and Medicine, Albert Einstein Cancer Center, Albert Einstein College of Medicine, New York, New York, USA. 2Laboratory of Pathology, National Cancer Institute, Bethesda, Maryland, USA. 3Department of Molecular Pharmacology, Albert Einstein Cancer Center,
Albert Einstein College of Medicine, New York, New York, USA. 4Harvard Medical School, Boston, Massachusetts, USA. 5Harvard–Massachusetts Institute of Technology Division of Health Sciences and Technology, Cambridge, Massachusetts, USA. 6Department of Physiology and Biophysics, Albert Einstein Cancer Center, Albert Einstein College of Medicine, New York, New York, USA.
7Committee on Human Nutrition and Nutritional Biology, University of Chicago, Chicago, Illinois, USA. 8Department of Biochemistry,
State University of New York at Buffalo, School of Medicine and Biomedical Sciences, Buffalo, New York, USA. 9Department of Developmental
and Molecular Biology and 10Department of Pathology, Albert Einstein Cancer Center, Albert Einstein College of Medicine, New York, New York, USA. 11Lombardi Comprehensive Cancer Center, Georgetown University Medical School, Georgetown, Washington, DC, USA. 12Center for Biological Engineering,
Massachusetts Institute of Technology, Cambridge, Massachusetts, USA. 13Department of Urology, Johns Hopkins University, Baltimore, Maryland, USA. 14Department of Internal Medicine, University of Michigan, Ann Arbor, Michigan, USA. 15FDA–National Cancer Institute Clinical Proteomics Program,
Office of Cellular and Gene Therapy, Center for Biologics Evaluation and Research, FDA, Bethesda, Maryland, USA. 16Department of Nuclear Medicine,
Albert Einstein Cancer Center, Albert Einstein College of Medicine, New York, New York, USA. 17Department of Histology,
Microbiology and Medical Biotechnologies, University of Padova, Padova, Italy.
The interactions of transformed cells with the surrounding stromal cells are of importance for tumor
progres-sion and metastasis. The relevance of adipocyte-derived factors to breast cancer cell survival and growth is well
established. However, it remains unknown which specific adipocyte-derived factors are most critical in this
process. Collagen VI is abundantly expressed in adipocytes. Collagen
–/–mice in the background of the mouse
mammary tumor virus/polyoma virus middle T oncogene (MMTV-PyMT) mammary cancer model demonstrate
dramatically reduced rates of early hyperplasia and primary tumor growth. Collagen VI promotes its
growth-stimulatory and pro-survival effects in part by signaling through the NG2/chondroitin sulfate proteoglycan
receptor expressed on the surface of malignant ductal epithelial cells to sequentially activate Akt and
β
-catenin
and stabilize cyclin D1. Levels of the carboxyterminal domain of collagen VI
α
3, a proteolytic product of the
full-length molecule, are dramatically upregulated in murine and human breast cancer lesions. The same fragment
exerts potent growth-stimulatory effects on MCF-7 cells in vitro. Therefore, adipocytes play a vital role in
defin-ing the ECM environment for normal and tumor-derived ductal epithelial cells and contribute significantly to
tumor growth at early stages through secretion and processing of collagen VI.
Introduction
The interactions between malignant ductal epithelial cells and the surrounding stromal cells play a critical role in mammary tumor progression (1–3). Myofibroblasts, macrophages, fibroblasts, and adipocytes have been demonstrated to interact with breast can-cer cells (4, 5). The adipocyte is one of the predominant stromal cell types in the microenvironment of mammary tissue as well as in bone marrow, an area frequently fostering metastases dur-ing breast cancer progression. A supportive role of adipocytes for
tumor growth has previously been demonstrated by coinjection of tumor cells with adipocytes (6), and many more studies have elaborated on the vital role of tumor-stromal interactions for the development and progression of cancer (7, 8).
The adipocyte is a potent source of signaling molecules, several of which are uniquely produced in this cell type (reviewed in ref. 9). We have previously shown that type VI collagen is upregulated during murine mammary tumor progression (4). Type VI collagen, while expressed by a number of other cell types, is abundantly pro-duced and secreted by adipocytes. Our earlier studies have shown that adipose tissue represents the single most abundant source of collagen VI systemically (10). Collagen VI is composed of 3 chains,
α1, α2, and α3, which associate to form higher-order complexes (11). The 3 chains of collagen VI form intracellular heterotrimers that subsequently form higher-order complexes of tetramers of trimers before being secreted. Collagen VI contributes essential
Nonstandard abbreviations used: CSPG, chondroitin sulfate proteoglycan; GDI-3, GDP dissociation inhibitor 3; GSK, glycogen synthase kinase; MIN, mammary intraepithelial neoplasia; MMTV, mouse mammary tumor virus; PyMT, polyoma virus middle T oncogene; TDLU, terminal duct lobular unit.
Conflict of interest: The authors have declared that no conflict of interest exists.
functions to the local ECM environment by providing structural support for cells and enrichment of growth factors, cytokines, and other ligands on cell surfaces and, in fact, can itself assume impor-tant signaling effects (12).
Increased stromal expression of collagen VI has been correlated with various aspects of tumorigenesis and malignant progres-sion. Specifically, the α3 subunit of collagen VI is upregulated in the stroma surrounding colonic tumors compared with that surrounding normal tissue (13). Exposure of fibroblasts to colla-gen VI promotes proliferation and upregulation of cyclin D1 (14). Collagen VI can also increase cell migration and invasion in cells expressing the NG2/chondroitin sulfate proteoglycan 1 (NG2/ CSPG) receptor (15). Furthermore, the protein inhibits apoptosis in a variety of cell types (16). TGF-β–expressing melanoma cells induce collagen VI expression in mammary stroma, facilitating tumor progression and invasiveness (17). Most recently, increased collagen VI expression in the ECM has been associated with the promotion of chemoresistance in ovarian cancer cells (18).
Here, we explored the bi-directional interactions between adipocytes and malignant ductal epithelial cells via the ECM. Using mice lacking the gene encoding the α1 chain of collagen VI, which effectively gives rise to a functional null phenotype for holo-collagen VI (19), we demonstrate that collagen VI promotes the development of hyperplastic foci and primary tumor growth, as judged by a markedly reduced rate of early hyperplasia and pri-mary tumor growth in the collagen VI–/– mice in the background of
a breast cancer–prone mouse strain (MMTV-PyMT) (20). Collagen VI activates the pro-survival and proliferation pathways involving Akt, β-catenin, and cyclin D1 to achieve this effect. Use of laser-cap-ture microdissection and reverse-phase protein arrays verifies this mechanism in vivo and illustrates the important role of adipocyte-derived collagen VI in the process. Immunohistochemistry, protein arrays, in vivo imaging, and signaling studies with recombinant protein argue for an important role for a collagen VIα3 carboxy-terminal proteolytic fragment during early tumor development. Transplantation studies verify the important role of adipocytes and collagen VI in regulation of early tumor development. Com-bined, our observations provide critical evidence for the important involvement of adipocyte-derived factors at early, but not at late, stages of mammary tumor progression and offer unique signaling mechanisms to account for these effects.
Results
The absence of collagen VI delays the onset of early hyperplasia. Northern blot analysis has previously shown that expression of collagen VI is highly enriched in adipose tissue and strongly induced during dif-ferentiation (10). Immunohistochemistry and immunofluorescence showed a strong positive signal for collagen VI protein on the sur-face of murine mammary adipocytes. Given the upregulation of collagen VI during tumor progression (4), we wanted to determine whether this upregulation is a secondary result of tumor growth or whether it has direct functional importance for tumor progres-sion. To test a possible in vivo role of collagen VI, we opted to take advantage of MMTV-PyMT transgenic mice. In addressing the rel-evance of collagen VI, we crossed MMTV-PyMT mice with collagen VI–/– mice (19). Collagen VI–/– mice carry a genetic deletion in the α1 locus, resulting in failure to secrete the α2 and α3 subunits, and are functional knockouts for the entire collagen VI molecule. The initial characterization of these collagen VIα1–/– animals revealed
no striking phenotypic changes except mild histological signs of
myopathy in skeletal muscle of both homo- and heterozygous mutant animals (19). This myopathy resembled pathologically Bethlem myopathy, a human inherited syndrome correlated with collagen VI genes (21–23). Subsequent characterization has revealed mitochondrial dysfunction and increased levels of apoptosis in myocytes of these mice (24).
The mammary epithelium of PyMT transgenic mice in the WT background versus the collagen VI–/– background was compared
at 3–4 and 5–6 weeks. Whole mounts were generated from 1 set of inguinal mammary glands (the fourth), and H&E staining was performed on the corresponding contralateral glands. Early dys-plastic foci representing mammary hyperplasia could be seen as early as 3–4 weeks in the PyMT+ColVI+/+ mice (Figure 1A). These
foci occupied approximately 5 times the area of the dysplastic foci in the PyMT+ColVI–/– mice. The PyMT+ColVI+/– mice exhibited
lev-els of early hyperplasia closer to those of the PyMT+ColVI+/+ mice
than to those of the PyMT+ColVI–/– mice. The differential levels of
early hyperplasia were maintained through the 5- to 6-week time period, when a similar 4- to 5-fold difference in area occupied by early dysplastic foci or mammary intraepithelial neoplasias (MINs) was observable (Figure 1B). Importantly, there was less than a 10% difference in the number of foci counted at weeks 3 and 6 in PyMT+ColVI+/+ mice compared with PyMT+ColVI–/– mice (Figure
1C). Hence, the actual number of transformed cells leading to foci was comparable, excluding nonspecific effects on the PyMT anti-gen in a collaanti-gen VI–/– background. There was, however, a dramatic
difference with respect to the ability of the foci to proliferate and progress through development in the collagen VI–/– background.
The whole mounts and H&E-stained slides were subsequently examined according to the recommendations from the Annapolis meeting (25). At approximately 4 weeks, mammary glands from the knockout mice showed few small MINs involving the ducts and the terminal duct lobular units (TDLUs) (Figure 1D). These lesions were low grade and characterized by layers of atypical hyper-chromatic epithelial cells with little cytoplasm. Some of the ducts showed surrounding fibrosis and inflammatory cells both in the wall and surrounding the involved ducts. The lesions seen in the PyMT+ColVI–/– mice were also present in the PyMT+ColVI+/+ and
PyMT+ColVI+/– mice, though the lesions were more extensive in
the latter 2, in that they involved more ducts and TDLUs. In some of the more extensive lesions, MIN-involved ducts were filled with cells such that no lumen was apparent. The cells were of medium-grade atypia with 4–7 mitotic figures per high-powered field. These lesions were multifocal. Histologically, the MIN lesions were seen as simple (semicystic bulges along the ducts containing 1 to sev-eral layers of atypical cells), solid (larger lesions containing dense masses of atypical cells in sheets), cystic (containing central fluid-filled spaces, lined by multilayered epithelium that was frequently papillary), and mixed solid and cystic.
Collagen VI promotes primary tumor growth in vivo. The effects observed at early stages of tumor development are rather dra-matic. To determine the long-term consequences of the lack of collagen VI for tumor growth, another cohort of PyMT+ColVI+/+,
PyMT+ColVI+/–, and PyMT+ColVI–/– mice was generated, and
pri-mary tumor formation was monitored over 10 months. Both male and female PyMT+ColVI+/+ mice exhibited quicker and
more widespread induction of primary tumor development than PyMT+ColVI–/– mice. In both males and females, PyMT+ColVI+/–
mice had palpable tumors as early as 1 month, while PyMT+ColVI–/–
mice did not produce palpable tumors until 3 months. This differ-ence in tumor size remained evident throughout the bulk of the observation period (10 months) (Figure 1E). The PyMT+ColVI+/+
mice exhibited tumors that were 4- to 6-fold larger, based on the 3 largest tumors in a given mouse, than those found in PyMT+ColVI–/–
mice. For instance, at month 5, tumors in the collagen VI+/+ females
were 2,488 ± 237 mm3 on average, whereas those in the knockout
background averaged 445 ± 27 mm3. Tumors in the heterozygote
collagen VI background were between the 2 averages but closer to WT levels (Figure 1E, left graph). Only by the latest stages (10 months), the tumors in the female PyMT+ColVI–/– mice caught up
in size and number with those in the PyMT+ColVI+/+ mice. Results
from the male transgenics displayed similar differences in pat-terns of tumor development (Figure 1E, right graph). Important-ly, PyMT+ColVI+/+ and PyMT+ColVI–/– mice were injected with an
equal number (105) of metastatic tumor cells (Met1 cells) derived
from an isogenic PyMT+ mouse. The number of lung metastases
was determined at several time points after injection. No difference in the number of lung metastases was observed at any stage (see Supplemental Figure 1; supplemental material available online with this article; doi:10.1172/JCI200523424DS1). Therefore, the homing and growth of metastatic lesions are unaffected by the absence of collagen VI.
[image:4.585.114.465.80.406.2]As an additional control, we examined murine mammary ductal architecture to determine whether the absence of collagen VI influ-ences normal ductal development of the mammary tree. To assess this possibility, we examined whole mounts of knockout mam-mary glands at various stages of development. No striking differ-ences can be observed between WT and null mice. This is further corroborated by a quantitative analysis, since there was statistically no significant difference in the amount of terminal branching in comparable regions of the mammary glands (data not shown). In addition, there were no functional differences in mammary tissue
Figure 1
Lack of collagen VI leads to a reduction in tumor growth.(A)Whole-mount analysis of early hyperplasia in 6-week-old collagen VI+/+ or collagen
VI–/– MMTV-PyMT transgenic mice. Bottom panel: whole mount from a WT mouse. (B) Collagen VI reduction in hyperplastic foci size. Quantitation
of hyperplastic areas at 3 and 6 weeks. n = 5 mice per group. *P < 0.05 vs. WT. (C) Number of hyperplastic foci does not significantly depend on collagen VI presence or absence in the MMTV-PyMT mouse. Quantitation of the number of foci formed in the mammary glands of 3-week-old MMTV-PyMT mice. n = 5 mice per group. (D)A representative H&E stain of a mammary section taken from PyMT+ColVI–/– and PyMT+ColVI+/+
mice at 6 weeks of age. (E)Quantitation of tumor sizes of female and male PyMT+ mice at different ages. n = 10 for each group. The 3 largest
between the genotypes, since collagen VI–/– mice are fully fertile,
have normal litter sizes, lactate, and have pup survival rates com-parable to those of WT.
Another important analysis involved identifying the local sources and relative contributions of collagen VI in mammary microenvi-ronments. Our previous paperhad suggested that adipocytes were the primary local producers of collagen VI as determined by in situ hybridization (4). To further illustrate the prime importance of adipocyte-specific collagen VI expression within mammary glands, we conducted several experiments. RT-PCR of human adipocytes and cultured MCF-7 cells suggested that the adipocytes expressed collagen VI but not the cell surface receptor NG2/CSPG whereas the MCF-7 cells expressed the latter gene but were not express-ing measurable quantities of collagen VI message (Figure 1F). To take this experiment one step further, we took murine adipose tis-sue and sections of early- and late-stage tumors and performed quantitative real-time PCR for collagen VI expression. Adipocytes were specifically isolated from the mammary adipose tissue using
collagenase I treatment of the specimen. Multiple early- and late-stage tumor tissues from 3 mice each were taken, pooled, and snap-frozen. RNA isolation, reverse transcription, and quantita-tive real-time PCR were performed. The data highlighted the dif-ferential levels of collagen VIα3 message in the local mammary microenvironments, illustrating that adipocytes represented the major source of collagen VI at all stages of tumor growth (Figure 1G). As the tumors became larger, they expressed 4 times as much collagen VI as early tumors but still significantly lagged behind the adipocytes normalized for β-actin message.
Collagen VI–secreting adipocytes are more permissive of breast tumor growth in vivo and have a greater impact on the progression of early tumor development. Given the dramatic reduction of tumor growth in collagen VI–/– mice in the background of an MMTV-PyMT
transgene, we wanted to assay the ability of collagen VI secreted from adipocytes to support mammary tumor growth indepen-dently. Athymic nude mice were injected with estrogen receptor– negative SUM-159PT breast cancer cells (26) mixed with purified adipocytes isolated from collagen VI+/+, collagen VI+/–, and collagen
VI–/– mice or 3T3-L1 adipocytes. For these experiments, primary
adipocytes were prepared by collagenase digestion, and all other stromal cell types were removed by centrifugation. This procedure yielded a highly purified preparation of adipocytes. There was no difference in the level of structural integrity between WT and null adipocytes. Upon coinjection, tumors were allowed to grow for a period of 4 weeks. At the end of the experiment, tumor size was measured, and the tumor mass was excised and stained for the presence of the human ErbB2 antigen to confirm the presence of the human SUM-159PT cells. Primary collagen VI+/+-derived
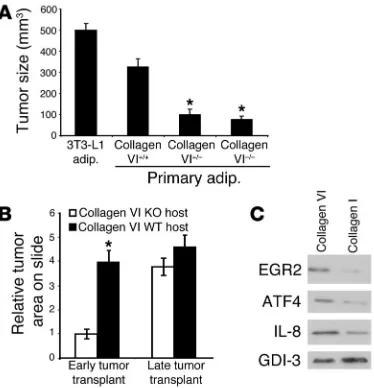
adipocytes and 3T3-L1 adipocytes best supported tumor growth and promoted the development of foci up to 300 mm3 and 500
mm3, respectively (Figure 2A). Adipocytes from either collagen
VI+/– mice or collagen VI–/– mice supported foci only up to 100
mm3. In line with our previously published results (4), 3T3-L1
adipocytes promoted SUM-159PT foci formation in vivo very well. Asterisks highlight the results that were statistically signifi-cant and differed from the growth of tumor foci provided WT, collagen VI–expressing adipocytes (Figure 2A). Similar studies were performed with estrogen receptor–positive MCF-7 cells, cor-roborating the data obtained with the estrogen receptor–negative SUM cells. Adipocytes derived from WT mice and the 3T3-L1 cell line accommodated development of MCF-7 foci to an average size of approximately 289 mm3 and 533 mm3, respectively. The
adipocytes taken from mice heterozygous and null for collagen VI had reduced capacity to promote MCF-7 growth, with foci reach-ing an average volume of 115 mm3.
An additional study was performed to assess the capacity of collagen VI–secreting cells in influencing early versus late tumor progression. These experiments suggested that the lack of adipose tissue (and collagen VI) has no impact on the tumor progression (size) and metastatic potential of late-stage malignant lesions (Supplemental Figure 2). Figure 1 pointed to the role of collagen VI in early neoplastic processes. With the experiment described below, we were able to specifically study the effect of collagen VI on early- and late-stage tumor progression. Small sections of tumors were isolated from early- and late-stage lesions in PyMT+ColVI+/+ mice
as outlined in Methods. The tumor pieces of equal size were then transplanted into the partially cleared fat pads of collagen VI+/+
and collagen VI–/– mice. After 3 weeks the mice were sacrificed and
[image:5.585.72.259.82.276.2]the transplanted lesions examined with whole-mount and H&E
Figure 2
Adipocytes from collagen VI+/– and collagen VI–/– mice are less potent
stimulators of tumor growth. Collagen VI also has greater impact on early than on late tumor progression. (A) SUM159-PT cells (1 × 105)
were coinjected into 8-week-old nude mice with the same number of isolated primary mammary adipocytes from collagen VI+/+, collagen
VI+/–, collagen VI–/– mice, or 3T3-L1 adipocytes. Four weeks after
injec-tion, the sizes of the resulting foci were measured. n = 4 mice in each cohort. Results are shown as mean ± SEM. (B) MMTV-PyMT trans-genic mice were sacrificed, and large, late tumor sections (from lesions larger than 1,500 mm3) and some small transformed mammary tissue
for early tumor samples (lesions smaller than 300 mm3) were isolated.
Tumor pieces were transplanted into partially cleared mammary glands of collagen VI+/+ and collagen VI–/– mice (n = 4). After 3 weeks, whole
analysis (Figure 2B). These studies indicated that early tumor cells display significantly enhanced growth in a WT host compared with a host lacking collagen VI. Collagen VI has, however, less of an impact on the tumor growth rate upon transplantation of late tumor cells. Combined, these results underlined the importance of adipocyte-derived collagen VI for early tumor growth.
Collagen VI induces genes associated with breast cancer progression. In order to obtain a global perspective of transcriptional changes induced by type VI collagen in mammary ductal epithelial cells, DNA microarray experiments were performed. To identify changes specifically brought about by collagen VI, we compared the col-lagen VI–induced transcriptional profile of MCF-7 cells with that of cells treated with collagen I. Collagen I treatment served in this case as a suitable base line, as it represents another abundant colla-gen that the epithelial cells are exposed to, prepared under compa-rable conditions. The cells were treated for 6 hours, and total RNA was isolated, reverse transcribed, and used to probe a microarray of approximately 9,500 human genes. Collagen VI induced the upregulation of a host of genes that either promote tumor devel-opment or have been positively correlated with breast cancer in some other context. These genes
included human transcription factor ETR101, growth factor PC4 homolog, protein tyrosine kinase tyk2, CDC28 protein kinase 1, and human metallothionein-1f and -1e (Supplemental Table 1). Notably, CDC28 protein kinase 1 promotes cellular proliferation via p27 Kip1 degradation, and metallothionein-1f/1e is corre-lated with poor patient prognosis in breast cancer. In line with the mRNA data, collagen VI mediated the upregulation of metallothio-nein also at the protein level as judged by immunohistochemistry in metastatic breast cancer cells treated with exogenous collagen VI protein (data not shown). Since metallothioneins are critically involved in cisplatin resistance, the observed upregulation of metallo-thionein by collagen VI provided, for the first time to our knowledge, a mechanistic explanation of why cultivation of cisplatin-sensitive cells in the presence of collagen VI protein promotes cisplatin resis-tance (18). In addition, collagen VI also specifically upregulated ATF3 and ATF4, transcription factors that are positive modulators of tumor development and invasion. Several other genes that promote angiogenesis, tumor growth, and/ or tumor invasion were also found to be upregulated (Supplemental Table 1). These genes included VEGF, calpain 4, IL-8, and
angio-poietin-2. Figure 2C provides representative data for a subset of proteins, confirming by Western blot analysis that the observed changes at the mRNA level translated effectively into changes at the protein level. Proteins tested include EGR2, ATF4, and IL-8. Compared with collagen I, collagen VI treatment led to a much greater induction of these proteins, at a magnitude similar to that of the transcriptional induction seen in the DNA microarrays.
Collagen VI activates Tcf/Lef signaling downstream of glycogen synthase kinase-3β phosphorylation. Given the strong evidence for a direct involvement of collagen VI in early tumor growth, we wanted to gain a better understanding of the underlying molecular mech-anism that leads to such a strong pro-proliferative and pro-sur-vival/anti-apoptotic response in breast cancer cells. The Akt-gly-cogen synthase kinase-3β–β-catenin–Tcf/Lef pathway is relevant to tumorigenesis, both in colonic adenocarcinomas and in breast tumors (27). The activation of Tcf/Lef, the downstream effector in this signaling cascade, induces the transcription of various pro-oncogenes, including cyclin D1, NF-κB, cFOS, and ATF3 (28). Events that lead to the accumulation of β-catenin in the cytoplasm increase Tcf/Lef activity. We had previously demonstrated that
Figure 3
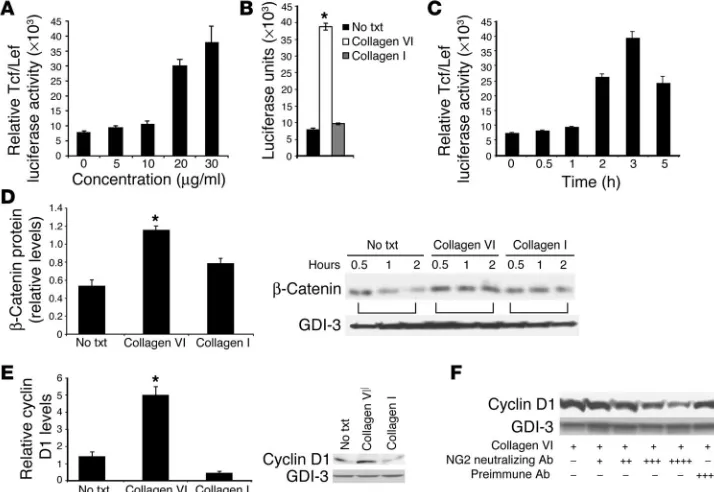
[image:6.585.183.540.325.571.2]collagen VI caused phosphorylation of GSK3β (4). Protein kinase A and Akt are known upstream kinases that target GSK3β for phosphorylation. The TOPFLASH reporter luciferase assay system was used here to determine whether collagen VI–induced GSK3β
phosphorylation led to downstream Tcf/Lef transcriptional acti-vation in MCF-7 cells. Initial experiments revealed that collagen VI works to maximally induce Tcf/Lef luciferase activity to levels 6- to 8-fold above base line at approximately 3 hours in a time- and dose-dependent fashion (Figure 3, A and C). This was a specific effect, since exposure to collagen VI, but not to collagen I, caused a 6- to 8-fold induction of Tcf/Lef activity (Figure 3B). To corrobo-rate this and subsequent data in another cell line, SUM cells were used. The capacity of collagen VI to signal for increased Tcf/Lef activity and β-catenin and cyclin D1 stabilization was preserved in the latter cell type as well.
Collagen VI prevents degradation of endogenous β-catenin. Increased cel-lular β-catenin levels were a likely result of the observed increase in GSK3β phosphorylation and Tcf/Lef activity in mammary cancer
cells induced by collagen VI. To test this, β-catenin levels were monitored over the course of sev-eral hours after a pulse-chase reac-tion. Degradation of 35S-labeled β-catenin was evident in untreated cells, with decreased intracellular levels apparent as early as 0.5 to 1 hour followed by a further progressive decrease apparent by 2 hours. The pres-ence of collagen VI dramatically reduced the rate of degrada-tion and maintained levels of
β-catenin over the course of the experiment (Figure 3D). In con-trast, the signal for GDP dis-sociation inhibitor 3 (GDI-3) is relatively long-lived and, con-sistent with our previously pub-lished observations, does not significantly turn over within a 2-hour chase period (29). Data from several independent experi-ments were analyzed and quanti-tated. A marginal stabilization of
β-catenin was also evident at early time points upon treatment with collagen I, but increased degradation compared with col-lagen VI–stabilized β-catenin was evident after 2 hours. Col-lagen VI therefore specifically enhanced intracellular β-catenin activity by decreasing its rate of degradation.
[image:7.585.42.409.81.337.2]Collagen VI increases cyclin D1 protein stability through NG2/ CSPG. Since cyclin D1 is a known target of Tcf/Lef and is affected by GSK3β and β-catenin activ-ity, we tested whether collagen VI could regulate cyclin D1 levels in breast cancer cells. MCF-7 cells were treated with vehicle, collagen VI, or collagen I for 3 hours, and protein extracts were generated. Collagen VI indeed maintained and substantially increased cyclin D1 levels, whereas collagen I showed no significant change compared with the vehi-cle condition (Figure 3E). A possible candidate to mediate these effects is the collagen VI receptor NG2/CSPG, a molecule that has previously been implicated as a growth mediator in a number of cancers (reviewed in ref. 30). The role of NG2/CSPG in the context of breast cancer has not, however, been examined. A previously described neutralizing antibody preparation against NG2/CSPG was used to determine whether the collagen VI effects on cyclin D1 expression are indeed exerted through this receptor (31–37). MCF-7 cells pretreated with the anti-NG2/CSPG antibody were exposed to collagen VI protein, and cyclin D1 levels were moni-tored. Increasing concentrations of the anti-NG2/CSPG antibody in the presence of collagen VI led to progressively lower levels of intracellular cyclin D1 (Figure 3F). Antibody treatment in the
Figure 4
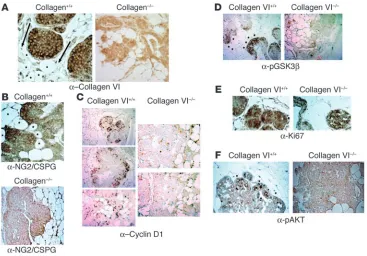
Immunohistochemical analysis of pathologically matched tumors from collagen+/+ and collagen–/– mice in
the MMTV-PyMT background. (A) Absence of collagen VIα3 signal in collagen–/– mice. Staining for
col-lagen VIα3 (with carboxyterminal polyclonal antibody) on mammary sections from pathologically matched hyperplasias developed in collagen+/+ and collagen–/– mice. Arrows point in the direction of increased
levels of staining. Asterisks denote local mammary adipocytes. Magnification, ×25. (B) NG2/CSPG is expressed at the highest levels in malignant regions adjacent to adipocytes in WT mice. Note the positive staining in WT backgrounds on the leading edges of the tumor mass. Magnification, ×25. (C) Cyclin D1 is expressed at the highest levels in malignant regions adjacent to adipocytes. Magnification, ×10. (D) GSK3β is phosphorylated at the highest levels in malignant regions adjacent to adipocytes. Magnification,
absence of added collagen VI had no effect on cyclin D1 stabil-ity. Pretreatment with a nonspecific control antibody preparation before collagen VI exposure did not impair the collagen VI–medi-ated stabilization of cyclin D1. This suggested that collagen VI mediates its effects on intracellular cyclin D1 levels at least in part through binding to and activation of the cell surface receptor NG2/CSPG. Equal protein loading for all the Western blots and labeling experiments was verified by probing for GDI-3.
Immunohistochemical evidence for effects of adipocyte-enriched collagen VI on GSK3β and cyclin D1. To lend further support to our in vitro observations, we examined sections of hyperplasia and early pri-mary lesions by immunohistochemistry. Staining for the carboxy terminus of collagen VIα3 in PyMT+ColVI+/+ mice was localized
mainly to the periphery of the tumor masses, appearing in a ring pattern around the region of early hyperplasia and around the periphery of adipocytes (Figure 4A). A gradient in colla-gen signal was observed with increased staining of MIN cell peripheries closer to adipocytes (Figure 4A; arrows point in the direction of increased staining). Staining on the interior of regions of early hyperplasia showed weaker collagen VI expression. Asterisks denote the local adipocytes in the mam-mary glands in the various sections. No collagen VI staining was observed in PyMT+ColVI–/– mice. Immunohistochemical
staining for NG2/CSPG demonstrated localization to cell sur-faces throughout the tumor masses in PyMT+ColVI+/+ mice
(Figure 4B). The staining was notably more pronounced on the surfaces of tumor cells closer to the adipocyte-rich regions. In PyMT+ColVI–/– sections that were stained with NG2/CSPG,
a much lower signal intensity was detected (Figure 4B, bot-tom panel). When staining for cyclin D1, we observed a ring of malignant cells on the outside of the MIN regions that exhib-ited positive staining in PyMT+ColVI+/+ mice, similar to the
pattern that was recently reported by Pollard and colleagues (38). This ring of cyclin D1 staining was reduced, however, in pathologically matched PyMT+ColVI–/– mice (Figure 4C).
The type of cyclin D1 staining seen in Figure 4 represented what would be seen with MINs or at the stage of early tumor development (39). The staining pattern for phospho-GSK3β
(pGSK3β) was similar to that for cyclin D1 (Figure 4D). A gra-dient was again seen with increased staining intensities closer to adipocytes on the leading edges of the hyperplastic lesions. Staining was present in matched PyMT+ColVI–/– tumors,
though the levels were substantially reduced.
To verify that cells in the interiors of the tumor masses were still proliferating, immunohistochemistry was performed using the polyclonal antibody against the proliferation marker Ki67. Early lesions from collagen VI+/+ and collagen VI–/– mice
were removed, sectioned, and stained for Ki67. As is apparent from both panels in Figure 4E, the brown diaminobenzidine stain indicative of cells positively expressing Ki67 was present throughout the tumor masses in the presence or absence of collagen VI. While not all the cells were proliferating, a sig-nificant proportion of the cells continued to do so both in the presence and in the absence of collagen VI.
Along with differences in cyclin D1 and pGSK3β expres-sion levels in the presence and absence of collagen VI, it is also apparent that the phosphorylation of Akt at ser473 also varied in the tumors derived from the 2 mouse models. Based on the increased cyclin D1 and pGSK3β expression in mammary tumors from WT mice, it would be expected that
ser473 phosphorylation of Akt would also be more prevalent in the collagen VI+/+ background compared with the null mice.
Early tumor sections from the 2 mouse lines ColVI+/+MMTV+ and
ColVI–/–MMTV+ were probed with the anti-pAkt ser473 antibody.
Figure 4F further underlines that the absence of collagen VI pro-tein strictly curtailed signaling through the GSK–Akt–cyclin D1 pathway. The presence of collagen VI allowed for increased pAkt expression (arrowheads) in cells on the periphery of the malignan-cy closest to the interacting adipomalignan-cytes (asterisks).
[image:8.585.286.542.259.560.2]Collagen VI activates similar pathways in vivo and in vitro. In light of the effects of collagen VI on the generation of dysplastic foci and growth of primary tumors, we wanted to better understand which signaling pathways critically contribute to this pheno-type in vivo. Laser-capture microdissection was used to isolate
Figure 5
Breast tumor cell protein expression and activation differences between col-lagen VI+/+ and collagen VI–/– mice in the background of the MMTV-PyMT
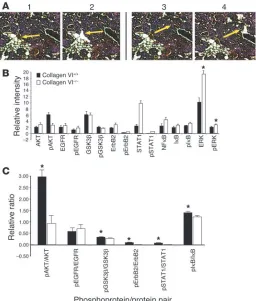
transgene. (A) Laser-capture microdissection was used to isolate hyper-plastic cells in a 100-μM radius from adipocytes. A representative example is shown in the top panels, before (panel 1) and after (panel 2) removal of cells. Similarly, cells not associated at all with adipocytes (> 500 μM from the nearest adipocyte) were isolated from pathologically matched sections from mice carrying the MMTV-PyMT transgene in either a collagen VI+/+ or
a collagen VI–/– background (panels 3 and 4). Arrows indicate the targeted
specific cells prior to reverse-phase protein array analysis. Speci-mens of stage- and size-matched early malignancies isolated from PyMT+ColVI+/+, and PyMT+ColVI–/– mice were analyzed. Cells were
chosen from clusters located at the periphery of the hyperplasias in close juxtaposition to adipocytes and from clusters more distal to adipocytes (Figure 5A). Proteomic reverse-phase array analy-sis revealed highly significant differences that demonstrated the
variable activation states of a number of relevant signaling modules in the presence and absence of collagen VI and adipocytes in general (Figure 5, B and C). Most notably, while total Akt levels were comparable in cells from PyMT+ColVI+/+ and PyMT+ColVI–/–
mice, a much larger portion of the protein was phosphorylated and activated in PyMT+ColVI+/+ mice
(Figure 5C). Similarly, greater per-centages of GSK3β, IκB, and ErbB2 were phosphorylated in MIN cells of PyMT+ColVI+/+ mice compared with
pathologically matched hyperplasias in PyMT+ColVI–/– mice. While not
investigated in our in vitro analy-ses, both STAT1 and NF-κB (p52) exhibited increased expression and phosphorylation, and STAT1 also exhibited increased activation, in PyMT+ColVI–/– hyperplastic cells.
Finally, though there were slightly increased levels of the mitogen-acti-vated protein kinase ERK in hyper-plasias from knockout animals, there were far greater levels of total ERK in these same cells. This results in an overall lowered activation state of ERK in these knockout mice (Figure 5B). Perhaps most importantly, the differ-ences in the activation state of all of these signaling components between PyMT+ColVI+/+ and PyMT+ColVI–/–
cells were only observed near the periphery of the tumor in proxim-ity to adipocytes, whereas no signifi-cant differences in signaling could be observed in cells isolated from the center (not shown). Several impor-tant conclusions can be made from this study. Firstly, without local adipocytes, very few differences could be observed in the signaling pathways of the neoplastic cells. Secondly, the presence or absence of collagen VI had a profound influence on signaling and posttranslational modifications in the malignant breast cancer cells close to the adipocytes. Perhaps most signifi-cantly, these in vivo results corrobo-rated our previously summarized in vitro signaling findings.
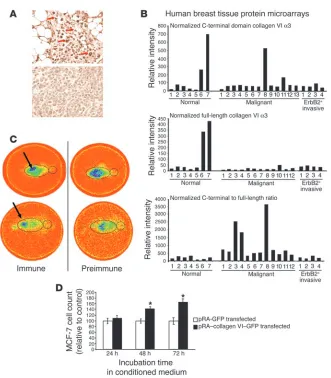
[image:9.585.48.379.83.461.2]Human breast tumor specimens display a collagen VI staining pattern similar to that observed in mice. We have previously reported increased staining for collagen VI during progression of tumorigenesis in the mouse (4). To test whether these observations are relevant to human disease, we probed human tumor and normal human mammary tissue sections with 2 different antibody preparations: a polyclonal antibody raised against the carboxyterminal domain of the collagen
Figure 6
VIα3 subunit and a commercially available mAb against collagen VI that recognizes another, not defined epitope on one of the other
α chains. Normal mammary glands exhibited relatively low levels of collagen VI staining with both antibodies, with a predominant signal detectable around adipocytes (data not shown). Late-stage carcinomas, however, showed a very intense staining pattern for collagen VI around the tumor masses when the antibody directed against the carboxyterminal domain of the collagen VIα3 subunit was used (Figure 6A). Immunostaining was particularly prominent in tumor cells near adipocytes, and less pronounced in cells located more distantly from adipocytes. The 2 panels of Figure 6A were taken from the same slide and represent human tumor cells clos-er to (top panel) and furthclos-er away from (bottom panel) the local mammary human adipocytes. Interestingly, in a direct comparison of staining patterns, the mAb recognizing the full-length collagen complex was associated with diffuse staining around adipocytes, whereas the polyclonal antibody against the carboxyterminal domain was associated with enhanced staining around tumor cells (not shown). This strongly suggested that the α3 carboxyterminal domain may have been cleaved from the polymeric remainder of the collagen VI. Furthermore, this small soluble domain may in turn be the only portion of collagen VI that is directly interacting with tumor cells. This points to the α3 carboxyterminal domain as a factor that is potentially relevant to cancer progression.
Collagen VI regulation in the human mammary carcinoma. The poten-tial importance of collagen VI protein may be as an early diagnostic marker of mammary hyperplasia or as a growth-promoting factor that can be inhibited to reduce tumor progression. To that end, we undertook experiments to look at collagen VI protein expression in normal and tumor cells from human cancer patients at various stages of the disease by using reverse-phase protein microarrays. Both the polyclonal antibody against the collagen VIα3 domain and the mAb against the collagenous domain of the full-length protein were used. Interestingly, the relative levels of the collagen VIα3 carboxyterminal domain (Figure 6B, top panel) versus the full-length collagen VI protein (Figure 6B, middle panel) were strongly biased toward the collagen VIα3 domain in tumor, but not in healthy tissue, as judged by the ratio of the signal intensi-ties obtained with the 2 different antibodies (Figure 6B, bottom panel). This suggested that the ratio of the collagen VIα3 carboxy terminus to full-length collagen VI may serve as a useful diagnostic marker, even in human specimens. Interestingly, this observation did not hold up for ErB2-positive tumors.
In vivo imaging of tumors with antibodies recognizing the carboxytermi-nal domain of collagen VIα3 shows that the recombinant carboxyterminal fragment of the collagen VIα3 protein has the functional capacity to signal and promote tumor cell proliferation. In order to gain a systemic over-view of the relative enrichment of the carboxyterminal domain of collagen VIα3, we isolated polyclonal IgGs from immune and non-immune preparations and radiolabeled them with 188-rhenium. Preparations were injected either i.p. (Figure 6C, top) or i.v. in the tail vein (Figure 6C, bottom) into an established murine breast can-cer model, a transgenic mouse expressing the polyoma middle T antigen under the control of the MMTV promoter (MMTV-PyMT) (20). These mice were subsequently imaged on a gamma camera. Whereas the preimmune IgGs rapidly accumulated in the kidneys, the immune IgGs specifically accumulated on the tumor mass; this suggests that the carboxyterminal domain of collagen VIα3 is highly enriched on the surface of tumors cells and may serve as a potent biomarker for breast tumor cells. Finally, to demonstrate
that this carboxyterminal fragment was not merely a byproduct of tumor progression but an active pro-mitogenic player during the process, we transiently transfected HEK-293T cells with a cDNA construct encoding a signal sequence fused to the C3 to C5 domains of collagen VIα3. This protein was efficiently secreted from the producer HEK-293T cells (not shown). MCF-7 cells were plated in 6-well dishes and exposed to conditioned (serum-free) supernatant harvested from cells expressing this C3–C5 collagen VIα3 fragment, or to conditioned medium from cells transfected with a plasmid lacking this cDNA insert. The fragment exerted potent growth-stimulatory properties on MCF-7 cells even in this isolated in vitro setting (Figure 6D).
Discussion
The ECM affects breast ductal epithelial cells at multiple levels, including by helping to remodel ductal architecture, influencing cell morphology, modulating proliferation through direct action on ductal epithelial cells, and acting as a reservoir for circulating growth factors. Under normal conditions, the basement mem-brane forms an effective barrier between the ductal epithelium and the surrounding stromal cells, but upon neoplastic transforma-tion, the ductal cells break through the basement membrane and are subsequently in direct contact with a new extracellular milieu. Adipocytes, the most abundant cell type in the stroma, are highly active endocrine cells that not only secrete a host of soluble factors but also contribute very significantly to the unique makeup of the ECM. Our previous work characterized the effects that the entire complement of soluble secretory products from adipocytes exerts on survival and growth of ductal epithelial cells (4). Here, we have focused on the role of an abundant, adipocyte-enriched ECM com-ponent, type VI collagen, and, for the first time to our knowledge, we implicate collagen VI both genetically and biochemically in the pathophysiology of breast cancer.
The upregulation of collagen VI observed during murine tumor progression suggested that collagen VI has the potential to influ-ence breast cancer progression in vivo. Our genetic evidinflu-ence, derived from the analysis of PyMT+ColVI+/+, PyMT+ColVI+/–, and
PyMT+ColVI–/– mice, strongly supports the involvement of
colla-gen VI in the early developmental progression of dysplastic foci and the promotion of primary tumor growth. The absence of col-lagen VI did not have an effect on the number of dysplastic foci observed as measured by MINs and TDLUs. No other systemic effects were observed. We have demonstrated here that the effi-ciency of early hyperplastic and primary tumor growth was dra-matically reduced in the absence of collagen VI.
Reconstitution studies were used to further examine whether collagen VI is a necessary driving force for tumor progression. 3T3-L1 adipocytes (an established adipocyte cell line) and isolated primary adipocytes that secrete collagen VI both supported for-mation and growth of tumor foci in coinjection experiments with SUM-159PT cells. Those adipocytes lacking collagen VI, isolated from collagen VI–/– mice, were unable to sustain tumor growth to
the same extent. Our studies also demonstrated that early mam-mary lesions transplanted into cleared mammam-mary fat pads of null mice did not progress pathologically. Though these implants had functional NG2 receptor, they did not develop into larger primary tumors without the collagen VI. These studies further demon-strate that collagen VI is a critical factor secreted by adipocytes, promoting dysplasia and early tumor growth in vivo.
An important issue is how collagen VI manages to exert these pro-mitogenic/pro-survival effects on breast cancer cells. Consis-tent with the role of collagen as a soluble paracrine factor, released by adipocytes and acting on adjacent tumor cells, we show by immunohistochemistry that murine mammary hyperplasias express NG2/CSPG, a previously characterized collagen VI recep-tor. However, a role for additional receptors for collagen VI, such as integrin α2β1, cannot be excluded, particularly in light of the
fact that our previously published microarray studies that exam-ined transcriptional changes induced by the total complement of secretory proteins in cancer cells showed that integrin α2β1 is
sig-nificantly upregulated upon exposure to adipocyte-conditioned medium (4). Nevertheless, the reduction of collagen VI–mediated signaling in breast cancer cells (decreased cyclin D1 stabilization) using neutralizing antibodies against NG2 suggests that NG2 is a major player in the context of mammary tumor growth and col-lagen VI. The NG2/CSPG receptor has not previously been demon-strated to promote the phosphorylation of GSK3β or activate the downstream Wnt pathway to regulate cyclin D1. In MCF-7 cells, we found that collagen VI, deviating from several other ECM pro-teins, uniquely promotes Tcf/Lef activity and inhibits β-catenin degradation. Therefore, collagen VI derived from adipocytes can induce a pro-mitogenic signaling cascade downstream of the NG2/CSPG receptor, using relatively well-established pathways in breast cancer cells to stabilize β-catenin, thereby elevating Tcf/Lef activity, which in turn upregulates cyclin D1. The effect of collagen VI on breast cancer cells is not limited to MCF-7 cell lines, as our in vivo data suggest.
Immunohistochemistry of dysplastic foci and primary tumors revealed staining patterns that reflected the in vitrofindings. Col-lagen VI was found in a gradient from adipocytes to breast can-cer cells. The NG2/CSPG receptor displayed a similar expression gradient. Furthermore, relative expressions of pGSK3β and cyclin D1 in the presence of collagen VI, compared with the absence of collagen VI, resembled those found in MCF-7 cells under similar conditions. These results argue that the promotion of dysplasia and tumor growth by collagen VI may occur by the same mecha-nism as illustrated by our in vitro studies. At later stages, the PyMT model generates malignant tumors that lead to metastases. Colla-gen VI did not have an effect on the number of metastases found. Collagen VI activity through NG2/CSPG, therefore, is likely to be involved in early tumor growth. Subsequently, upon reaching pri-mary tumor stages and metastatic potential, the tumor cells lose their dependence on collagen VI.
Proteomic data derived from PyMT+ColVI+/+, PyMT+ColVI+/–,
and PyMT+ColVI–/– mice supported our in vitro data. The most
striking difference was seen in the activation of Akt in malig-nant cells close to adipocytes expressing collagen VI. There was a sharp gradient with respect to the activation state of the pro-survival factors in the hyperplastic cells adjacent to collagen VI–null adipocytes. The increased phosphorylation of Akt sug-gests that Akt may be the dominant kinase responsible for the phosphorylation of GSK3β in vivo.
Detailed proteomic analysis using immunohistochemistry pro-vides additional insight into the role of collagen VI. The presence of collagen VI leads to upregulated cyclin D1 expression. Hyperplas-tic cells near the periphery of MINs, which are closer to adipocytes, express higher levels of cyclin D1 than those closer to the middle of MINs. Correspondingly, cells on the periphery of the tumor have been found to be more proliferative than those at the core, further emphasizing the importance of adipocyte factors including colla-gen VI in tumor proliferation. Also, in agreement with the in vitro signaling data, there was increased staining for pGSK3β on the leading edge of tumor cells in the PyMT+ColVI+/+ mice, whereas
matched tumors from collagen VI–/– mice stained very poorly for
the pGSK3β molecule. DNA microarray data from MCF-7 cells treated with soluble collagen VI additionally demonstrated gene induction patterns consistent with the activation of a number of genes associated with breast cancer growth and progression.
Immunohistochemistry of human mammary carcinoma tis-sue reveals strong collagen VI staining around the tumor masses, focused primarily around the adipocytes and producing a gradient similar to that seen in murine sections. Normal human mammary tissue exhibits low-level collagen VI staining. These results dem-onstrate collagen VI protein presence in human breast tumors. While these results do not define collagen VI as a critical factor in the development and proliferation of human tumors, the strong upregulation of collagen VI expression in human malignant lesions combined with the highly provocative mouse data suggests that collagen VI may be relevant to human breast cancer as well.
Several independent lines of evidence point toward the potential importance of the collagen VIα3 carboxyterminal domains in mod-ulating the protein’s activity. Published observations described that a proteolytic cleavage event that releases this fragment can indeed occur upon secretion of the holo-collagen VI complex (40). The resulting cleavage product is soluble and may be the fragment that has a high affinity for the surrounding breast cancer cells. Consistent with such a conclusion is the observation that only antibodies to this carboxyterminal domain display a highly signifi-cant increase in staining intensity in immunohistochemical slides during tumor progression. Antibodies recognizing other domains on collagen VI display the expected staining pattern primarily surrounding adipocytes with little signal detected on the tumor cells directly. It is probable that, like other collagens, type VI may be cleaved either at the adipocyte cell surface or by the influx of proteases in and around tumor cells in the local mammary envi-ronments during early pathological changes.
tumor extracts that the carboxyterminal domain and the full-length collagen VI protein are detected at different relative levels, depending on whether normal or transformed human breast tis-sue is examined. The increased C-terminal fragment/full-length collagen VI ratio may therefore be a potent prognostic indicator. Future studies on a much larger number of specimens will have to address the general usefulness of this ratio with respect to predic-tion of future disease outcome and whether neutralizapredic-tion of the carboxyterminal domain of collagen VIα3 fragment through the use of specific antibodies may serve as a therapeutic modality for early-stage breast cancer therapy in humans.
In summary, we propose a model that involves adipocyte-derived collagen VI, particularly a carboxyterminal fragment of collagen VIα3 that is either produced at higher levels or has an increased half-life during tumor progression, presumably as a result of the local paracrine interactions between the tumor cells and the sur-rounding adipocytes. This fragment accumulates on tumor cells, where, through activation of the NG2 receptor, it triggers the acti-vation of specific signaling pathways that involve β-catenin, lead-ing to a further pro-mitogenic response for the cancer cells.
Methods
Proteins and reagents. DMEM was prepared at the Albert Einstein Cancer Center media preparation facility. Human type VI collagen (99.9% homoge-neity) was obtained from Research Diagnostics Inc. Human collagen I was obtained from Collaborative Biomedical Products. Collagen I and collagen
VI were used at a concentration of 30 μg/ml unless otherwise stated. The
mAb to β-catenin was obtained from Transduction Laboratories. A rabbit
polyclonal anti–collagen VI antibody specific to the C-terminal of the α3
subunit was generated using a bacterially produced GST-fusion protein
raised against the last 70 amino acids of mouse collagen VIα3. A
monoclo-nal anti–collagen VI antibody was obtained from Chemicon Internatiomonoclo-nal Inc. The antibody against NG2 was a kind gift from Joel M. Levine (State University of New York at Stony Brook, Stony Brook, New York, USA). The anti–GDI-3 antibody was a gift from Perry Bickel (Washington University, St. Louis, Missouri, USA).
Laboratory animals. Mice were housed in groups of 2–5 in filter-top cages and given free access to water and standard laboratory chow diet. The colonies were maintained in a pathogen-free Association for Assessment and Accredi-tation of Laboratory Animal Care–accredited facility at the Albert Einstein College of Medicine under controlled environment settings (22–25°C, 40– 50% humidity, 12-hour/12-hour light/dark cycle). All mouse strains (MMTV-PyMT, collagen VI–null mutant, and aP2-DTA) were maintained in a pure FVB background. All animal protocols were approved by the Albert Einstein Animal Committee of the Albert Einstein College of Medicine.
Cell culture. 3T3-L1 cells were propagated and differentiated as previ-ously described (41). NIH-3T3 cells were grown and propagated in DMEM containing 10% donor calf serum and antibiotics. SUM-159PT cells were grown in 5% charcoal-stripped FCS, 10% Ham’s F12 nutrient medium, sodium bicarbonate, 10 mM HEPES, and 25 mM glucose (26). The SUM-159PT medium was adjusted to pH 7.1. Met1 and dB7 cells were a generous gift of Robert Cardiff (University of California, Davis, California, USA) and were grown in 10% FCS-DMEM. MCF-7 cells were used for in vitro signal-ing experimentation and were grown in 10% FCS-DMEM as well.
In vivo tumorigenesis assay. The reconstituted in vivo tumor model used 3T3-L1 adipocytes or primary adipocytes (obtained by collagenase treat-ment of adipose tissue followed by centrifugation and removal of stromal cells) in combination with fluorescently tagged SUM-159PT cells. Mice received injections of 3 × 105 adipocytes and 1 × 104 SUM-159PT cells. Cells
were implanted by s.c. injection in the flank of male 6- to 8-week-old, nude
mice obtained from The Jackson Laboratory. Mice were killed 4 weeks after injection. After excision, tumor size (area and wet weight) was recorded.
Immunoblotting. Separation of proteins by SDS-PAGE fluorography and immunoblotting were performed as previously described (42). For Western blots used to examine phosphorylated protein products, TNET lysis buffer (1% Triton X-100, 150 mM NaCl, 5 mM EDTA pH 8.0, 50 mM Tris pH 8.0) was supplemented with 50 mM NaF, 30 mM sodium pyrophosphate, and
100 μM sodium orthovanadate.
Luciferase assays. MCF-7 or SUM-159PT cells were seeded in 12-well plates sufficient to reach 50–60% confluence the next day. Two days before the luciferase readout, cells were transiently transfected with the TOPFLASH luciferase promoter construct (which contains 4 Tcf/Lef–binding elements) as well as a thymidine kinase–driven renilla luciferase construct as a transfection control vector (Promega Corp.) (39). SuperFect transfection reagent (QIAGEN
Corp.) was used, and, for 12-well plates, 1.5 μg of DNA per well.
Approxi-mately 30 hours after the transfection, collagen I or VI at the indicated levels was added to the cells for 3 hours. Subsequently, cells were processed using a dual-luciferase assay system for luciferase activity from both the TOPFLASH constructs and the renilla constructs. All results were standardized to the rela-tive transfection efficiencies as observed from the renilla values.
Metabolic labeling for β-catenin. MCF-7 cells, seeded in 6-well plates, were treated in methionine/cysteine–deficient DMEM for 1 hour. Samples
were pulsed with 500 μCi/ml [35S]-methionine/cysteine for 20 minutes
and chased in the presence of cycloheximide supplemented with either vehicle or 30 μg/ml collagen I or VI for various time periods. After pulsing, the cells were washed with DMEM twice and lysed in a TNET buffer
(con-taining 1 μM phenylmethanesulfonyl fluoride and 60 mM
octylgluco-side). The lysates were cleared with Sepharose 4CL beads and subjected to
β-catenin immunoprecipitation and anti–GDI-3 immunoprecipitation.
The immunoprecipitates were subjected to electrophoresis on an SDS-PAGE gel and treated in 1 M sodium salicylate for 30 minutes. Subse-quently, the gel was dried and exposed to film. We have previously shown that GDI-3 has an extended half-life and that its signal remains stable for at least 2 hours (29).
Microarray analysis. MCF-7 cells at 60% confluence were treated with type VI or type I collagen for 7 hours. Total RNA was isolated by treatment with TRIzol. Human microarrays (9,596 spots) from the Albert Einstein College of Medicine were obtained and used for experimentation. Reverse transcription with the concomitant incorporation of Cy3- and Cy5-labeled
nucleotides was performed on 100 μg of total RNA. The reverse-transcribed
RNA was incubated on the microarrays and subsequently analyzed using Scanalyze software (Digital Michelangelo Project, Stanford University) according to the standardized protocol of the Albert Einstein College of Medicine (as described in ref. 4). Each independent experiment incorpo-rated data from 3 separate chips and 3 separate RNA isolations as well as a reversal of the incorporated labeled nucleotides.
Tissue recovery for protein microarray. Whole mammary tissue samples were
recovered from MMTV-PyMT+ mice that were in either a collagen VI WT
or a collagen VI knockout background. Mammary glands were removed and placed in cryomolds, covered with OCT compound, and immediately frozen in isopentane and stored at –80°C.
Sectioning and staining. The OCT-embeddedtissue blocks were cut with a
Tissue-Tek 2000 cryostat (Sakura Products) into 8-μm sections and placed
clear-ing agent; Surgipath Medical Industries Inc.). Protease inhibitors (Roche Diagnostics Corp.) were added to both the 70% ethanol and the water solu-tions. The tissue section was air-dried before microdissection.
Murine immunohistochemistry. Mammary tumor tissues were excised from
MMTV-PyMT transgenic mice euthanized with CO2, and the tissues were
fixed in 10% neutral-buffered formalin at room temperature overnight. The
tissue was embedded in paraffin, and 6- to 8-μm sections were made. The
sections represented breast tumors from various stages of development, from adenomas to late-stage carcinomas. An avidin-biotin-peroxidase
staining system was used along with 3,3′-diaminobenzidine
tetrahydro-chloride as the chromogen. Sections were blocked to prevent nonspecific binding with 10% horse serum, and the polyclonal collagen VI antibody was added to the sections and incubated in a humidified chamber at 4°C overnight. Sections were washed twice in PBS between each incubation.
Endogenous peroxidase was quenched with 3% H2O2 in methanol for 30
minutes at room temperature. A biotin-labeled rabbit secondary anti-body was applied for 30 minutes at room temperature, followed by incuba-tion with a preformed avidin-biotinylated enzyme complex for 30 minutes. Chromogen was subsequently added, the cells were counterstained with H&E when necessary to improve contrast of the images, and the slides were finally mounted and examined by light microscopy. Polyclonal antibody to Ki67 was obtained from Santa Cruz Biotechnology Inc., pAkt mAb to serine residue 473 from Cell Signaling Technology Inc., and pGSK3b anti-body to serine residue 9 from Cell Signaling Technology Inc., and collagen VI antibodies were generated in the laboratory.
Magnetic resonance imaging. All images were obtained using a 9.4-tesla hor-izontal imaging system. Mice were anesthetized with isoflurane (1.5% in oxygen) delivered via a nose cone and were positioned in a 25-mm birdcage 1H coil. Body temperature was maintained with a homeothermic system. To quantitatively assess whole-body fat and water, each mouse was sub-jected to a 16-scan pulse-acquire sequence, and spectra, including the water and fat peaks, were obtained and integrated. For imaging, several data sets of 9 slices of 1-mm thickness spanning the whole body (the gap between slices was 1 mm) were obtained. Imaging was conducted using a routine spin-echo pulse sequence (18-millisecond echo time, 600-millisecond rep-etition time, and averages of 2 signals per scan).
In vivo imaging of the carboxyterminal domain of collagen VIα3. Preimmune and collagen-specific polyclonal antibodies were radiolabeled directly with 188-rhenium (188Re) via reduction of antibody disulfide bonds with dithiothreitol
(43). An amount of 2–4 mCi 188ReO4– in saline was reduced by incubation
with 20 mg sodium gluconate and 20 μl 20 mg/ml SnC12 in 0.1 M HC1 at
37°C for 60 minutes. Reduced 188Re(V)-gluconate was combined with reduced
and purified antibodies and kept at 37°C for 60 minutes. Radioactivity not bound to the antibody was removed by centrifugal purification on Centricon-30 microconcentrators (Millipore Corp.). Tumor-bearing mice were injected i.p. or i.v. with 0.5 mCi 188Re-Ab (100 μg). At 1.5 hours after injection, the
animals were anesthetized with isoflurane and scintigraphically imaged on a LEM+ZLC DIGITRAC gamma camera (Siemens AG) equipped with a pinhole collimator and ICON image-processing software (ICON Corp.).
Whole-mount analysis. The fourth and fifth mammary glands of female mice were excised. The glands were maintained in a 75% acetic acid/meth-anol fixative for 4 hours. Subsequently, the sections were rehydrated in increasing ratios of water to ethanol for 20 minutes and put in carmine red solution overnight. The samples were then treated with xylene, and photo-graphs were taken. The area of early hyperplasia per gland was determined using NIH ImageJ software.
Patients and surgically resected frozen tissues. The study set consisted of 17 pri-mary human breast cancer samples and 7 healthy control samples taken from patients undergoing breast reduction surgery. Both human and animal stud-ies were approved by the Institutional Review Boards of the National Cancer
Institute and Lombardi Comprehensive Cancer Center, Georgetown Uni-versity. All carcinomas were infiltrating ductal carcinomas. Frozen sections were prepared for laser-capture microdissection on plain glass microscope slides. Microdissection was performed with a PixCell II system (Arcturus Engineering Inc.). Microdissected areas represented tumor from the breast cancer cases or normal-appearing mammary ducts adjacent to the tumor from the same patient, or mammary ductal epithelium from the unaffected patients undergoing reduction mammoplasty. Protein was extracted from
the microdissected cells using 10 μl of extraction buffer per 1,000
micro-dissected cells (1:1 dilution of 4% β-mercaptoethanol in 2×
Tris-glycine-SDS buffer [Invitrogen Corp.] with liquid tissue protein extraction reagent [T-PER; Pierce Chemical Co.]) for 1 hour at 70°C. After protein extraction, samples were boiled for 5 minutes, and serial dilutions representing 1:1, 1:3, and 1:9 concentrations and a negative control were prepared in extrac-tion buffer. Three nanoliters of the sample and control lysates were arrayed onto glass-backed nitrocellulose slides (FAST slides; Schleicher & Schuell BioScience) using a GMS 417 pin-and-ring-style arrayer (Affymetrix Inc.). Arrays were treated with mild ReBlot solution (Chemicon International Inc.)
for 15 minutes, washed twice for 5 minutes each in 1× PBS without calcium
or magnesium, and blocked for a minimum of 30 minutes at room tempera-ture with I-Block protein block solution (Applied Biosystems). Arrays were probed with antibodies to total and cleaved collagen VI using an automated slide stainer (DakoCytomation) with a catalyzed signal amplification system (DakoCytomation). A rabbit polyclonal anti–collagen VI antibody specific to
the C-terminal of the α3 subunit was generated using a bacterially produced
GST-fusion protein raised against the last 70 amino acids of mouse collagen
VIα3. A monoclonal anti–collagen VI antibody was obtained from Chemicon
International Inc. Polyclonal primary antibody was used at a final dilution of 1:1,000. Monoclonal primary antibody was used at a final dilution of 1:250. One slide was stained for total protein with SYPRO Ruby Protein Blot stain (Invitrogen Corp.) according to the manufacturer’s instructions. The slide was imaged with an Alpha Innotech Corp. imager equipped with a SYPRO Red/Texas red filter. Exposure time was 20 seconds. Quantification of the relative pixel density of each array spot was performed with ImageQuant software version 5.2 (Molecular Dynamics).
Arrays were allowed to air dry, and images were scanned on an Epson 1640SU flatbed scanner (Epson Corp.). Quantitation of the relative pixel density of each array spot was performed with ImageQuant software ver-sion 5.2 (Molecular Dynamics). Each intensity value was normalized to its total protein relative intensity value. The ratio of the spot intensity of the
cleaved carboxyterminal domain of collagen VIα3 to that of the full-length
collagen VI transcript was calculated.
Laser-capture microdissection and protein extraction for mouse samples. Microdis-section was carried out using a PixCell II and AutoPix Laser Capture Micro-dissection System (Arcturus Engineering Inc.). Four areas were microdis-sected from the WT-phenotype frozen tissue sections, representing 34,000 cells. Ten areas were microdissected from the knockout-phenotype tissue sections, representing 26,830 cells. Within each tissue section, only mam-mary tumor cells were microdissected. The microdissected samples were frozen at –80°C until molecular analysis. Protein was extracted from the
microdissected cells using 10 μl of extraction buffer per 1,000
microdis-sected cells (1:1 dilution of 4% β-mercaptoethanol in 2× Tris-glycine-SDS
buffer [Invitrogen Corp.] with liquid tissue protein extraction reagent [T-PER; Pierce Chemical Co.]) for 1 hour at 70°C.