organic papers
Acta Cryst.(2005). E61, o1999–o2000 doi:10.1107/S1600536805016909 Wanget al. C
10H14O4
o1999
Acta Crystallographica Section E Structure Reports Online
ISSN 1600-5368
3-(2-Methoxyphenoxy)propane-1,2-diol
Yongli Wang, Ming Li,* Lijun Liu, Lina Zhou and Jingkang Wang
School of Chemical Engineering and
Technology, Tianjin University, Tianjin 300072, People’s Republic of China
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study
T= 293 K
Mean(C–C) = 0.003 A˚
Rfactor = 0.048
wRfactor = 0.148
Data-to-parameter ratio = 17.7
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2005 International Union of Crystallography Printed in Great Britain – all rights reserved
The title compound, C10H14O4, is used to cure coughs and
clear up phlegm and it is also used as an intermediate in the synthesis of other medicinal products. The entire molecule, except for atoms C10 and O4, is essentially planar (to within 0.001 A˚ ).
Comment
The title compound, (I), also known as guaiphenesin, is an ingredient usually found in cold preparations. In the year 1530, it was first extracted from guaiacum and used to treat rheu-matism. Over 20 years ago, it was synthesized, named guai-phenesin, and pressed into tablets (Starlanyl, 2001). To the best of our knowledge [using Chemical Abstracts and the Cambridge Structural Database (Allen, 2002)], the single-crystal structure has not been reported previously.
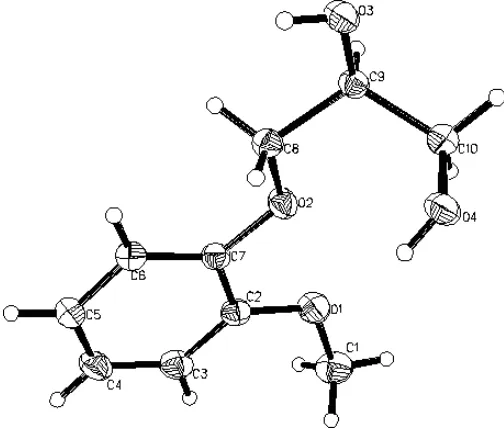
The molecular structure of (I) is shown in Fig. 1. It can be seen that the entire molecule, except for atoms C10 and O4
[image:1.610.271.380.357.443.2] [image:1.610.205.457.495.710.2]Received 14 April 2005 Accepted 26 May 2005 Online 10 June 2005
Figure 1
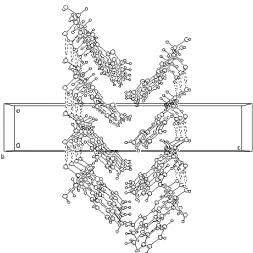
and the H atoms, is essentially planar (to within 0.001 A˚ ). The crystal packing projected on to theacface is shown in Fig. 2. There are two kinds of intermolecular hydrogen bonds between molecules (Table 1). The hydrogen bond O3— H3 O4 is approximately parallel to theacface, while O4— H4 O3 is approximately perpendicular to theacface.
Experimental
The title compound was provided by Tianjin Zhongxin Pharmaceu-tical Co. Ltd. A saturated solution of guaiphenesin in ethanol was prepared at 338–343 K and then a small quantity of seeds was added when the temperature fell to 308 K. After a long time, a large quantity of white crystals was obtained by recrystallization. The product was characterized by NMR, IR and elemental analyses, and its purity was 99%. The melting point determined by DSC (differ-ential scanning calorimetry) is 355.4 K. Colorless block-shaped single crystals suitable for X-ray diffraction were obtained by adding a small quantity of seeds to a room-temperature solution of the above product and placing it in a refrigerator for 3 d.
Crystal data
C10H14O4 Mr= 198.21
Orthorhombic,P212121 a= 4.9836 (10) A˚ b= 7.6562 (15) A˚ c= 25.698 (5) A˚ V= 980.5 (3) A˚3 Z= 4
Dx= 1.343 Mg m 3 MoKradiation Cell parameters from 9593
reflections = 3.1–27.5
= 0.10 mm1 T= 293 (2) K Block, colorless
0.380.200.09 mm
Data collection
Rigaku R-AXIS RAPID IP area-detector diffractometer !scans
Absorption correction: multi-scan (ABSCOR; Higashi, 1995) Tmin= 0.962,Tmax= 0.991 9631 measured reflections
2248 independent reflections 1862 reflections withI> 2(I) Rint= 0.092
max= 27.5
h=6!6 k=9!9 l=33!32
Refinement
Refinement onF2 R[F2> 2(F2)] = 0.048 wR(F2) = 0.148 S= 1.00 2248 reflections 127 parameters
H-atom parameters constrained
w= 1/[2(F
o2) + (0.0903P)2 + 0.0695P]
whereP= (Fo2+ 2Fc2)/3 (/)max= 0.001
max= 0.31 e A˚3 min=0.42 e A˚3
Table 1
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
O3—H3B O4i 0.82 1.96 2.744 (2) 161 O4—H4C O3ii
0.82 1.99 2.729 (2) 149
Symmetry codes: (i)xþ1;y1 2;zþ
1
2; (ii)xþ1;y;z.
H atoms were placed in calculated positions and constrained to ride on their parent atoms, with C—H = 0.93–0.98 A˚ andUiso(H) =
1.2Ueq(C). In the absence of significant anomalous dispersion effects,
Friedel equivalents were merged prior to the final refinements, and the absolute configuration was assigned to correspond with the known chiral centers of the precursor molecule, which remained unchanged during the synthesis of the title compound.
Data collection:RAPID-AUTO (Rigaku, 2001); cell refinement:
RAPID-AUTO; data reduction:RAPID-AUTO; program(s) used to solve structure: SHELXS97(Sheldrick, 1997); program(s) used to refine structure:SHELXL97(Sheldrick, 1997); molecular graphics:
ORTEPII (Johnson, 1976); software used to prepare material for publication:SHELXL97.
The authors gratefully acknowledge support from the SRCICT of Tianjin University and the materials afforded by Tianjin Zhongxin Pharmaceutical Co. Ltd.
References
Allen, F. H. (2002).Acta Cryst.B58, 380–388.
Higashi, T. (1995).ABSCOR. Rigaku Corporation, Tokyo, Japan.
Johnson, C. K. (1976).ORTEPII. Report ORNL-5138. Oak Ridge National Laboratory, Tennessee, USA.
Rigaku (2001).RAPID-AUTO. Rigaku Corporation, Tokyo, Japan. Sheldrick, G. M. (1997). SHELXS97 and SHELXL97. University of
Go¨ttingen, Germany.
[image:2.610.44.297.67.320.2]Starlanyl, D. J. (2001).Fibromyalgia and Chronic Myofascial Pain: A Survival Manual, 2nd ed., p. 201. Oakland: New Harbinger.
Figure 2
supporting information
sup-1
Acta Cryst. (2005). E61, o1999–o2000
supporting information
Acta Cryst. (2005). E61, o1999–o2000 [https://doi.org/10.1107/S1600536805016909]
3-(2-Methoxyphenoxy)propane-1,2-diol
Yongli Wang, Ming Li, Lijun Liu, Lina Zhou and Jingkang Wang
3-(2-Methoxyphenoxy)propane-1,2-diol
Crystal data
C10H14O4 Mr = 198.21
Orthorhombic, P212121
a = 4.9836 (10) Å
b = 7.6562 (15) Å
c = 25.698 (5) Å
V = 980.5 (3) Å3
Z = 4
F(000) = 424
Dx = 1.343 Mg m−3
Melting point: 355.4 K
Mo Kα radiation, λ = 0.71073 Å
Cell parameters from 9631 reflections
θ = 3.1–27.5°
µ = 0.10 mm−1
T = 293 K
Needle, colorless 0.38 × 0.20 × 0.09 mm
Data collection
Rigaku R-axis Rapid IP area-detector diffractometer
Radiation source: rotating anode Graphite monochromator Oscillation scans
Absorption correction: multi-scan (ABSCOR; Higashi, 1995) Tmin = 0.962, Tmax = 0.991
9593 measured reflections 2248 independent reflections 1862 reflections with I > 2σ(I) Rint = 0.092
θmax = 27.5°, θmin = 3.1°
h = −6→6
k = −9→9
l = −33→32
Refinement
Refinement on F2
Least-squares matrix: full R[F2 > 2σ(F2)] = 0.048 wR(F2) = 0.148
S = 1.00
2248 reflections 127 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained w = 1/[σ2(F
o2) + (0.0903P)2 + 0.0695P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max = 0.001
Δρmax = 0.31 e Å−3
Δρmin = −0.42 e Å−3
Special details
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is
not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those
based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
C1 1.2993 (5) 0.5757 (4) 0.02231 (11) 0.0575 (7)
H1A 1.2735 0.6945 0.0115 0.086*
H1B 1.3004 0.5009 −0.0077 0.086*
H1C 1.4674 0.5655 0.0403 0.086*
C2 1.0968 (4) 0.3583 (3) 0.07477 (8) 0.0361 (4)
C3 1.2737 (5) 0.2306 (3) 0.05803 (8) 0.0450 (5)
H3A 1.3976 0.2562 0.0321 0.054*
C4 1.2676 (5) 0.0650 (3) 0.07965 (10) 0.0497 (6)
H4A 1.3853 −0.0203 0.0677 0.060*
C5 1.0883 (5) 0.0258 (3) 0.11871 (11) 0.0483 (6)
H5A 1.0859 −0.0855 0.1332 0.058*
C6 0.9106 (5) 0.1529 (3) 0.13648 (9) 0.0409 (5)
H6A 0.7896 0.1264 0.1629 0.049*
C7 0.9138 (4) 0.3183 (3) 0.11489 (7) 0.0339 (4)
C8 0.5792 (4) 0.4210 (3) 0.17351 (8) 0.0348 (4)
H8A 0.6862 0.3980 0.2043 0.042*
H8B 0.4656 0.3204 0.1670 0.042*
C9 0.4095 (4) 0.5827 (3) 0.18138 (7) 0.0319 (4)
H9A 0.2902 0.5954 0.1513 0.038*
C10 0.5733 (4) 0.7480 (3) 0.18666 (9) 0.0395 (5)
H10A 0.6559 0.7752 0.1535 0.047*
H10B 0.4554 0.8441 0.1958 0.047*
O1 1.0869 (3) 0.5255 (2) 0.05609 (6) 0.0469 (4)
O2 0.7492 (3) 0.45292 (19) 0.12978 (6) 0.0394 (4)
O3 0.2484 (3) 0.5507 (2) 0.22650 (6) 0.0406 (4)
H3B 0.2657 0.4485 0.2355 0.061*
O4 0.7766 (3) 0.7314 (2) 0.22526 (7) 0.0499 (4)
H4C 0.8962 0.6676 0.2144 0.075*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
C1 0.0508 (13) 0.0691 (19) 0.0527 (14) −0.0041 (13) 0.0085 (11) 0.0197 (13)
C2 0.0337 (10) 0.0410 (11) 0.0336 (9) −0.0013 (9) −0.0008 (9) −0.0026 (8)
C3 0.0392 (11) 0.0528 (14) 0.0432 (12) 0.0021 (11) 0.0050 (10) −0.0095 (10)
C4 0.0380 (11) 0.0470 (14) 0.0643 (15) 0.0119 (11) 0.0007 (12) −0.0128 (11)
C5 0.0398 (11) 0.0368 (12) 0.0684 (14) 0.0045 (10) −0.0046 (12) 0.0005 (10)
C6 0.0340 (9) 0.0383 (12) 0.0505 (12) −0.0002 (10) 0.0023 (10) −0.0011 (9)
supporting information
sup-3
Acta Cryst. (2005). E61, o1999–o2000
C8 0.0313 (9) 0.0362 (11) 0.0370 (10) 0.0012 (9) 0.0029 (8) −0.0020 (8)
C9 0.0295 (9) 0.0328 (10) 0.0335 (9) 0.0028 (8) −0.0010 (8) 0.0016 (8)
C10 0.0348 (9) 0.0357 (11) 0.0480 (11) 0.0010 (10) −0.0002 (10) 0.0009 (9)
O1 0.0460 (8) 0.0486 (10) 0.0462 (8) 0.0021 (8) 0.0115 (8) 0.0087 (7)
O2 0.0408 (7) 0.0353 (8) 0.0420 (8) 0.0051 (7) 0.0102 (7) 0.0022 (6)
O3 0.0369 (7) 0.0400 (9) 0.0450 (8) 0.0080 (7) 0.0089 (7) 0.0098 (6)
O4 0.0340 (7) 0.0476 (10) 0.0682 (10) 0.0102 (7) −0.0105 (7) −0.0186 (8)
Geometric parameters (Å, º)
C1—O1 1.422 (3) C6—H6A 0.9300
C1—H1A 0.9600 C7—O2 1.372 (2)
C1—H1B 0.9600 C8—O2 1.428 (2)
C1—H1C 0.9600 C8—C9 1.513 (3)
C2—O1 1.368 (3) C8—H8A 0.9700
C2—C3 1.385 (3) C8—H8B 0.9700
C2—C7 1.410 (3) C9—O3 1.431 (2)
C3—C4 1.384 (4) C9—C10 1.512 (3)
C3—H3A 0.9300 C9—H9A 0.9800
C4—C5 1.377 (4) C10—O4 1.424 (3)
C4—H4A 0.9300 C10—H10A 0.9700
C5—C6 1.393 (3) C10—H10B 0.9700
C5—H5A 0.9300 O3—H3B 0.8200
C6—C7 1.383 (3) O4—H4C 0.8200
O1—C1—H1A 109.5 C6—C7—C2 120.0 (2)
O1—C1—H1B 109.5 O2—C8—C9 107.24 (16)
H1A—C1—H1B 109.5 O2—C8—H8A 110.3
O1—C1—H1C 109.5 C9—C8—H8A 110.3
H1A—C1—H1C 109.5 O2—C8—H8B 110.3
H1B—C1—H1C 109.5 C9—C8—H8B 110.3
O1—C2—C3 125.1 (2) H8A—C8—H8B 108.5
O1—C2—C7 115.88 (18) O3—C9—C10 111.92 (16)
C3—C2—C7 119.0 (2) O3—C9—C8 106.34 (15)
C4—C3—C2 120.5 (2) C10—C9—C8 113.31 (16)
C4—C3—H3A 119.7 O3—C9—H9A 108.4
C2—C3—H3A 119.7 C10—C9—H9A 108.4
C5—C4—C3 120.4 (2) C8—C9—H9A 108.4
C5—C4—H4A 119.8 O4—C10—C9 111.82 (18)
C3—C4—H4A 119.8 O4—C10—H10A 109.3
C4—C5—C6 120.0 (2) C9—C10—H10A 109.3
C4—C5—H5A 120.0 O4—C10—H10B 109.3
C6—C5—H5A 120.0 C9—C10—H10B 109.3
C7—C6—C5 120.1 (2) H10A—C10—H10B 107.9
C7—C6—H6A 120.0 C2—O1—C1 116.11 (19)
C5—C6—H6A 120.0 C7—O2—C8 116.46 (16)
O2—C7—C6 124.70 (19) C9—O3—H3B 109.5
O1—C2—C3—C4 179.8 (2) C3—C2—C7—C6 −0.8 (3)
C7—C2—C3—C4 1.2 (3) O2—C8—C9—O3 −177.81 (14)
C2—C3—C4—C5 −1.0 (4) O2—C8—C9—C10 −54.4 (2)
C3—C4—C5—C6 0.4 (4) O3—C9—C10—O4 68.2 (2)
C4—C5—C6—C7 0.1 (4) C8—C9—C10—O4 −52.0 (2)
C5—C6—C7—O2 −179.5 (2) C3—C2—O1—C1 −9.1 (3)
C5—C6—C7—C2 0.1 (3) C7—C2—O1—C1 169.5 (2)
O1—C2—C7—O2 0.2 (3) C6—C7—O2—C8 5.2 (3)
C3—C2—C7—O2 178.88 (18) C2—C7—O2—C8 −174.42 (16)
O1—C2—C7—C6 −179.51 (19) C9—C8—O2—C7 −177.72 (16)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
O3—H3B···O4i 0.82 1.96 2.744 (2) 161
O4—H4C···O3ii 0.82 1.99 2.729 (2) 149