organic papers
Acta Cryst.(2006). E62, o255–o257 doi:10.1107/S1600536805039231 Kolevet al. C
18H19NO3
o255
Acta Crystallographica Section E Structure Reports
Online
ISSN 1600-5368
Codeinone
Tsonko Kolev,aRumyana Bakalska,bBoris Shivachevcand Rosica Petrovac*
aBulgarian Academy of Sciences, Institute of Organic Chemistry, Acad G. Bonchev Street Building 9, 1113 Sofia, Bulgaria,bUniversity of Plovdiv, Department of Organic Chemistry, 24 Tzar Asen Street, 4000 Plovdiv, Bulgaria, and c
Bulgarian Academy of Sciences, CL of Mineralogy and Crystallography, Acad G. Bonchev Street Building 107, 1113 Sofia, Bulgaria
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study T= 290 K
Mean(C–C) = 0.006 A˚ Rfactor = 0.060 wRfactor = 0.118
Data-to-parameter ratio = 10.2
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2006 International Union of Crystallography Printed in Great Britain – all rights reserved
The title compound, 4,5
-epoxy-3-methoxy-17-methyl-morphin-7-en-6-one, C18H19NO3, the molecular structure exhibits features typical for morphine derivatives, with a T-shaped configuration. The crystal packing is stabilized by weak intermolecular C—H O interactions.
Comment
The structure of codeinone, (I), was elucidated as part of our synthetic, spectroscopic and structural investigations of morphine alkaloids, which constitute a major class of pain-alleviating drugs. Codeinone has been found to possess anti-tumour potential, with high cytotoxic activity against human promyelocytic leukaemic cell lines (Hitosugiet al., 2003). The transformation of morphine derivatives into different meta-bolites is a matter of practical interest for detecting opiates in blood or urine. It is known that codeinone in the living cell is produced from the reaction of codeine with nicotinamide adenine dinucleotide phosphate (NADP+).
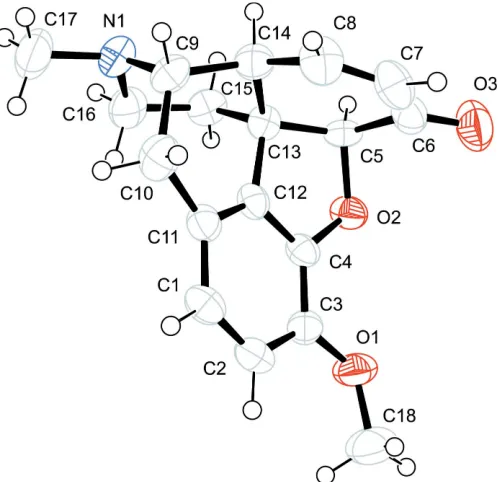
The overall configuration of (I) and the atom-numbering scheme are shown in Fig. 1. The absolute configuration of the chiral centres in the molecule is identical to those of the starting material, codeine. The main structural features of the molecule are very close to those of codeine (Canfield et al., 1987), heroin (Canfieldet al., 1979), morphine (Gylbert, 1973) and acetylcodeine (Sonaret al., 2005; Kolevet al., 2005).
The molecule of (I) exhibits the T-shaped configuration characteristic of classical morphine opiates.
The ring fusions and conformations are similar to those previously reported for morphine derivatives (Gelders & de Ranter, 1979; Petrickova et al., 2002; Moody et al., 1997). Aromatic ringAis planar, ringBis close to an envelope, rings CandDassume half-chair conformations and ringEassumes a chair form (Table 1). The oxidation of codeine to codeinone should mainly affect the shape and properties of ring D.
However, no major differences between the geometric para-meters of ringDin codeinone, codeine and even 6-O-codeine could be established.
The conformation about the single C—C bonds within the rings is staggered, as in codeine; exceptions such as the C5— C6 eclipsed bond in 6-O-codeine are not present. This difference between the codeinone and 6-acetylcodeine conformations, along with the absence or presence of a chiral centre at the C6 position, is associated with the different functional groups attached to atom C6.
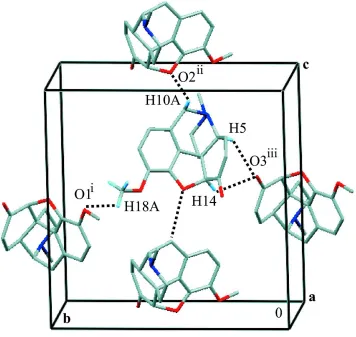
In the three-dimensional arrangement of the molecules of (I) (Fig. 2), no classical hydrogen bonds could be found. A subsequent examination of intermolecular contacts suggested that molecules of (I) are linked in the crystal structure through weak C—H O interactions (Desiraju, 1996; Steiner & Desiraju, 1998; Zhu et al., 2005). The interactions involving atoms O1 and O3 (Table 2) connect the molecules to form undulating ‘pseudo’-layers perpendicular to the c axis. The C10—H10A O2 interaction connects the layers along thec axis, extending the structure stabilization in all three direc-tions.
Experimental
Codeinone was prepared according to the method of Bakalskaet al. (2002). Crystals suitable for X-ray diffraction were obtained by slow evaporation of an ethanol solution at 277 K.
IR spectra were measured on a Bomem–Michelson 100 FT–IR spectrometer in the range 4000–400 cm1, with 2 cm1resolution and 150 scans. Solid-state IR spectra were recorded using the KBr pellet technique. Chloroform (Merck) solutions, at a concentration of 0.01M, were measured using 0.05 cm KBr pellets. The bands at 2840
1 s
in the IR spectrum of codeinone belongs to the(C O) mode of the conjugated C O group. The maxima at 1374 and 1388 cm1indicate s
[CH3(N)] and s
[CH3(O)], respectively. Typical for morphine compounds are peaks at about 1633, 1604 and 1506 cm1, which are assigned as(C C), 8a and 19a in-plane (A1) phenyl modes. The series of in-plane peaks of 1,2,3,4-o-tetrasubstituted benzene at about 1150 and 1050 cm1 is observed in the 1200–800 cm1 frequency region. Below 1000 cm1, an intense maximum at 940 cm1and a pair of maxima at about 936 and 804 cm1are present, but the exact assignment with conventional IR techniques is ambiguous. A detailed spectroscopic study, combined withab initioUHF calculations of (I), are in progress and will be published at a later date.
Spectroscopic analysis for codeinone: 1H NMR (Bruker 250, 250 MHz, CDCl3,, p.p.m.): 6.67 (d, 1H,J= 8.2 Hz, H-2), 6.62 (d, 1H,
J= 10.2 Hz, H-8), 6.59 (d, 1H,J= 8.2 Hz, H-1), 6.07 (dd, 1H,J= 10.2 and 2.9 Hz, H-7), 4.68 (s, 1H, H-5), 3.85 (s, 3H OCH3), 3.45–3.35 (m, 1H, H-9), 3.25–3.17 (m, 1H, H-14), 3.10 (d, 1H,J= 18.5 Hz, H-10), 2.61 (dm, 1H,J= 11.8Hz, H-16e), 2.45 (s, 3H, NCH3), 2.30 (dd, 1H,J= 18.5 and 5.5 Hz, H-10), 2.30 (td, 1H,J= 11.9 and 3.7 Hz, H-16a), 2.06 (td, 1H,J= 12.0 and 4.8 Hz, H-15a), 1.85 (dm, 1H,J= 12.5 Hz, H-15e); 13
C NMR (CDCl3,, p.p.m.): 119.7 (C-1), 114.7 (C-2), 142.3 (C-3), 146.2 4), 88.0 5), 194.1 6), 132.2 7), 149.1 8), 58.9 (C-9), 20.4 (C-10),126.1 (C-11), 129.0 (C-12), 43.1 (C-13), 41.4 (C-14), 33.9 (C-15), 46.7 (C-16), 42.9 (NMe), 56.7 (OMe).
Crystal data
C18H19NO3
Mr= 297.34
Orthorhombic,P212121
a= 7.2541 (10) A˚
b= 14.0943 (14) A˚
c= 14.3507 (15) A˚
V= 1467.2 (3) A˚3
Z= 4
MoKradiation Cell parameters from 22
reflections
= 16.3–17.7
= 0.09 mm1
[image:2.610.48.297.71.312.2]T= 290 (2) K Prism, colourless 0.180.160.15 mm
Figure 1
The molecular structure of (I), showing 50% probability displacement ellipsoids.
Figure 2
The molecular packing in (I). Intermolecular C—H O contacts are denoted by dotted lines. H atoms not participating in C—H O contacts have been omitted for clarity. [Symmetry codes: (i)x1
2,y+ 3 2,z+ 1;
(ii)x+3
2,y+ 1,z+ 1 2; (iii)x+
1 2,y+
[image:2.610.325.545.73.279.2]Data collection
Enraf–Nonius CAD-4 diffractometer Non–profiled!/2scans Absorption correction: none 7258 measured reflections 2032 independent reflections 987 reflections withI> 2(I)
Rint= 0.172
max= 28.0
h= 0!9
k=18!18
l=18!18 3 standard reflections
frequency: 120 min intensity decay: 2%
Refinement
Refinement onF2 R[F2> 2(F2)] = 0.060
wR(F2) = 0.118
S= 1.02 2032 reflections 200 parameters
H-atom parameters constrained
w= 1/[2
(Fo2) + (0.0344P)2] whereP= (Fo2+ 2Fc2)/3 (/)max< 0.001
max= 0.16 e A˚ 3
min=0.18 e A˚ 3
Extinction correction:SHELXL97
(Sheldrick, 1997)
Extinction coefficient: 0.0128 (18)
Table 1
Selected torsion angles ().
C16—N1—C9—C14 62.5 (5) O2—C4—C12—C13 4.2 (5) C13—C5—C6—C7 1.6 (5) O2—C5—C13—C14 144.8 (4) C6—C7—C8—C14 1.2 (8) C7—C8—C14—C13 26.9 (7)
C14—C9—C10—C11 36.2 (6) C10—C11—C12—C13 5.8 (7) C4—C12—C13—C5 21.0 (4) C11—C12—C13—C15 86.9 (5) C15—C13—C14—C9 64.1 (5) C13—C15—C16—N1 52.0 (5)
Table 2
Intermolecular C—H O contacts (A˚ ,).
D—H A D—H H A D A D—H A
C18—H18A O1i
0.96 2.75 3.285 (6) 116 C10—H10A O2ii
0.97 2.62 3.423 (5) 140 C14—H14 O3iii 0.98 2.66 3.389 (6) 131 C5—H5 O3iii
0.98 2.70 3.260 (5) 117
Symmetry codes: (i) x1 2;yþ
3
2;zþ1; (ii) xþ 3
2;yþ1;zþ 1 2; (iii)
xþ1 2;yþ
1 2;zþ1.
All H atoms were placed in idealized positions, with C—H = 0.93– 0.98 A˚ , and constrained to ride on their parent atoms, withUiso(H) =
1.2Ueq(Csp 3
and Caromatic), or 1.5Ueq(CMe). In the absence of signifi-cant anomalous dispersion effects, Friedel pairs were averaged.
Data collection: CAD-4 EXPRESS (Enraf–Nonius, 1994); cell refinement:CAD-4 EXPRESS; data reduction:XCAD4(Harms & Wocadlo, 1995); program(s) used to solve structure: SHELXS97 (Sheldrick, 1997); program(s) used to refine structure:SHELXL97 (Sheldrick, 1997); molecular graphics: ORTEP-3 for Windows (Farrugia, 1997) andMERCURY(Version 1.3; Bruno et al., 2002); software used to prepare material for publication:WinGX(Farrugia, 1999).
This work was supported by the Bulgarian National Fund of Scientific Research, Contract Nos. X-1213 and BYX 01.05.
References
Bakalska, R. I. & Chervenkova, V. B. (2002).Bulg. Chem. Ind.73, 108–111. Bruno, I. J., Cole, J. C., Edgington, P. R., Kessler, M., Macrae, C. F., McCabe, P.,
Pearson, J. & Taylor, R. (2002).Acta Cryst.B58, 389–397.
Canfield, D., Barrick, J. & Giessen, B. C. (1979).Acta Cryst.B35, 2806–2809. Canfield, D., Barrick, J. & Giessent, B. C. (1987).Acta Cryst.C43, 977–979. Desiraju, G. R. (1996).Acc. Chem. Res.29, 441–449.
Enraf–Nonius (1994). CAD-4 EXPRESS. Version 5.1/1.2. Enraf–Nonius, Delft, The Netherlands.
Farrugia, L. J. (1997).J. Appl. Cryst.30, 565. Farrugia, L. J. (1999).J. Appl. Cryst.32, 837–838.
Gelders, Y. G. & de Ranter, C. J. (1979).Acta Cryst.B35, 1111–1116. Gylbert, L. (1973).Acta Cryst.B29, 1630–1635.
Harms, K. & Wocadlo, S. (1995).XCAD4. University of Marburg, Germany. Hitosugi, N., Hatsukari, I., Ohno, R., Hashimoto, K, Mihara, S., Mizukami, S., Nakamura, S., Sakagami, H., Nagasaka, H., Matsumoto, I. & Kawase, M. (2003).Anesthesiology,98, 643–650.
Kolev, T., Bakalska, R., Shivachev, B. L. & Petrova, R. (2005).Acta Cryst.E61, o2582–o2584.
Moody, P. C. E., Shikotra, N., French, C. E., Bruce, N. C. & Scrutton, N. S. (1997).Acta Cryst.D53, 619–621.
Petrickova, H., Jegorov, A., Husak, M. & Cisarova, I. (2002).Acta Cryst.A58, C-126.
Sheldrick, G. M. (1997). SHELXL97 and SHELXS97. University of Go¨ttingen, Germany.
Sonar, V. N., Parkin, S. & Crooks, P. A. (2005).Acta Cryst.E61, o2579–o2581. Steiner, T. & Desiraju G. R. (1998).Chem. Commun.pp. 891–892.
Zhu, X.-Q., Wang, J.-S. & Cheng, J.-P. (2005).Tetrahedron Lett.46, 877–879.
organic papers
Acta Cryst.(2006). E62, o255–o257 Kolevet al. C
supporting information
Acta Cryst. (2006). E62, o255–o257 [doi:10.1107/S1600536805039231]
Codeinone
Tsonko Kolev, Rumyana Bakalska, Boris Shivachev and Rosica Petrova
S1. Comment
The structure of codeinone, (I), was elucidated as part of our synthetic, spectroscopic and structural investigations of
morphine alkaloids, which constitute a major class of pain-alleviating drugs. Codeinone has been found to possess
antitumour potential, with high cytotoxic activity against human promyelocytic leukaemic cell lines (Hitosugi et al.,
2003). The transformation of morphine derivatives into different metabolites is a matter of practical interest for detecting
opiates in blood or urine. It is known that codeinone in the living cell is produced from the reaction of codeine with
nicotinamide adenine dinucleotide phosphate (NADP+).
The overall configuration of (I) and the atom-numbering scheme are shown in Fig. 1. The absolute configuration of the
chiral centres in the molecule is identical to that of the starting material, codeine. The main structural features of the
molecule are very close to those of codeine (Canfield et al., 1987), heroin (Canfield et al., 1979), morphine (Gylbert,
1973) and acetylcodeine (Sonar et al., 2005; Kolev et al., 2005).
The molecule of (I) exhibits the T-shaped configuration characteristic of classical morphine opiates. The value of
86.62 (6)° for the dihedral angle between the mean planes of the A/B/C and D/E rings in (I) is comparable with that in
codeine (88.94°; Canfield et al., 1987) and differs somewhat with that observed in 6-O-codeine [80.56 (8)°; Sonar et al.,
2005; Kolev et al., 2005].
The ring fusions and conformations are similar to those previously reported for morphine derivatives (Gelders & de
Ranter, 1979; Petrickova et al., 2002; Moody et al., 1997). Aromatic ring A is planar, B is close to an envelope, C and D
assume half-chair conformations and E assumes a chair form (Table 1). The oxidation of codeine to codeinone should
mainly affect the shape and properties of ring D. However, no major differences between the geometric parameters of
ring D in codeinone, codeine and even 6-O-codeine could be established.
The conformation about the single C—C bonds within the rings is staggered, as in codeine; exceptions such as the C5—
C6 eclipsed bond in 6-O-codeine are not present. This difference between the codeinone and 6-acetylcodeine
conformations, along with the absence or presence of a chiral centre at the C6 position, is associated with the different
functional groups attached to atom C6.
In the three-dimensional arrangement of the molecules of (I) (Fig. 2), no classical hydrogen bonds could be found. A
subsequent examination of intermolecular contacts suggested that molecules of (I) are linked in the crystal structure
through weak C—H···O interactions (Desiraju, 1996; Steiner & Desiraju, 1998; Zhu et al., 2005). The interactions
involving atoms O1 and O3 (Table 2) connect the molecules to form undulating `pseudo′-layers perpendicular to the c
axis. The C10—H10A···O2 interaction connects the layers along the c axis, spreading the structure stabilization in all
supporting information
sup-2
Acta Cryst. (2006). E62, o255–o257
S2. Experimental
Codeinone was prepared according to the method of Bakalska et al. (2002). Crystals suitable for X-ray diffraction were
obtained by slow evaporation from ethanol at 277 K.
IR spectra were measured on a Bomem–Michelson 100 F T–IR spectrometer in the range 4000–400 cm−1, with 2 cm−1
resolution and 150 scans. Solid-state IR spectra were recorded using the KBr pellet technique. Chloroform (Merck)
solutions, at a concentration of 1.10−2M, were measured using 0.05 cm KBr pellets. The bands at 2840 and 2807 cm−1 of
codeinone are assigned to νs[CH
3(O)] and νs[CH3(N)] modes, respectively. The most intense peak at 1677 cm−1 in the IR
spectrum of codeinone belongs to the ν(C═O) mode of the conjugated C═O group. The maxima at 1374 and 1388 cm−1
indicate δs[CH3(N)] and δs[CH3(O)], respectively. Typical for morphine compounds are peaks at about 1633, 1604 and
1506 cm−1, which are assigned as ν(C ═ C), 8a and 19a in-plane (A
1) phenyl modes. The series of in-plane peaks of
1,2,3,4-o-tetrasubstituted benzene at about 1150 and 1050 cm−1 are observed in the 1200–800 cm−1 frequency region.
Below 1000 cm−1, an intense maximum at 940 cm−1 and a pair of maxima at about 936 and 804 cm−1 are present, but the
exact assignment with conventional IR techniques is ambiguous. A detailed spectroscopic study, combined with ab initio
UHF calculations of (I), are in progress and will be published at a later date.
Spectroscopic analysis for codeinone: 1H NMR (Bruker 250, 250 MHz, CDCl
3, δ, p.p.m.): 6.67 (d, 1H, J = 8.2 Hz, H-2),
6.62 (d, 1H, J = 10.2 Hz, H-8), 6.59 (d, 1H, J = 8.2 Hz, H-1), 6.07 (dd, 1H, J = 10.2 and 2.9 Hz, H-7), 4.68 (s, 1H, H-5),
3.85 (s, 3H OCH3), 3.45–3.35 (m, 1H, H-9), 3.25–3.17 (m, 1H, H-14), 3.10 (d, 1H, J = 18.5 Hz, H-10β), 2.61 (dm, 1H, J
= 11.8 Hz, H-16e), 2.45 (s, 3H, NCH3), 2.30 (dd, 1H, J = 18.5 and 5.5 Hz, H-10α), 2.30 (td, 1H, J = 11.9 and 3.7 Hz,
H-16a), 2.06 (td, 1H, J = 12.0 and 4.8 Hz, H-15a), 1.85 (dm, 1H, J = 12.5 Hz, H-15e); 13C NMR (CDCl3, δ, p.p.m.): 119.7
(C-1), 114.7 (C-2), 142.3 (C-3), 146.2 (C-4), 88.0 (C-5), 194.1 (C-6), 132.2 (C-7), 149.1 (C-8), 58.9 (C-9), 20.4
(C-10),126.1 (C-11), 129.0 (C-12), 43.1 (C-13), 41.4 (C-14), 33.9 (C-15), 46.7 (C-16), 42.9 (NMe), 56.7 (OMe).
S3. Refinement
All H atoms were placed in idealized positions, with C—H = 0.93–0.98 Å, and constrained to ride on their parent atoms,
Figure 1
supporting information
sup-4
[image:7.610.128.484.71.408.2]Acta Cryst. (2006). E62, o255–o257
Figure 2
The molecular packing in (I). Intermolecular C—H···O contacts are denoted by dotted lines. H atoms not participating in
C—H···O contacts have been omitted for clarity. [Symmetry codes: (i) x − 1/2, −y + 3/2, −z + 1; (ii) −x + 3/2, −y + 1, z +
1/2; (iii) x + 1/2, −y + 1/2, −z + 1.]
4,5α-epoxy-3-methoxy-17-methylmorphin-7-ene-6-one
Crystal data
C18H19NO3
Mr = 297.34
Orthorhombic, P212121
Hall symbol: P 2ac 2ab
a = 7.2541 (10) Å
b = 14.0943 (14) Å
c = 14.3507 (15) Å
V = 1467.2 (3) Å3
Z = 4
F(000) = 632
Dx = 1.346 Mg m−3
Melting point: not measured K Mo Kα radiation, λ = 0.71073 Å Cell parameters from 22 reflections
θ = 16.3–17.7°
µ = 0.09 mm−1
T = 290 K
Prismatic, colourless 0.18 × 0.16 × 0.15 mm
Data collection
Enraf–Nonius CAD-4 diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
Non–profiled ω/2θ scans
7258 measured reflections 2032 independent reflections 987 reflections with I > 2σ(I)
Rint = 0.172
h = 0→9
k = −18→18
l = −18→18
3 standard reflections every 120 min intensity decay: 2%
Refinement
Refinement on F2
Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.060
wR(F2) = 0.118
S = 1.02 2032 reflections 200 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained
w = 1/[σ2(F
o2) + (0.0344P)2]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001
Δρmax = 0.16 e Å−3
Δρmin = −0.18 e Å−3
Extinction correction: SHELXL97 (Sheldrick, 1997), Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4
Extinction coefficient: 0.0128 (18)
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
N1 1.2910 (6) 0.4138 (3) 0.7428 (2) 0.0481 (10) O1 0.5791 (5) 0.6486 (2) 0.5119 (2) 0.0575 (9) O2 0.7983 (4) 0.48246 (17) 0.49336 (18) 0.0429 (8) O3 0.6008 (6) 0.3076 (2) 0.4889 (2) 0.0700 (11) C1 0.7832 (7) 0.6168 (3) 0.7470 (3) 0.0467 (12)
H1 0.7759 0.6433 0.8062 0.056*
C2 0.6847 (8) 0.6569 (3) 0.6747 (3) 0.0468 (12)
H2 0.6181 0.7122 0.6858 0.056*
C3 0.6818 (7) 0.6173 (3) 0.5859 (3) 0.0409 (11) C4 0.7905 (6) 0.5387 (3) 0.5723 (3) 0.0367 (10) C5 0.8760 (6) 0.3921 (3) 0.5255 (3) 0.0382 (11)
H5 0.9501 0.3635 0.4758 0.046*
C6 0.7138 (7) 0.3272 (3) 0.5490 (3) 0.0474 (12) C7 0.6934 (8) 0.2932 (3) 0.6438 (3) 0.0580 (14)
H7 0.5832 0.2643 0.6609 0.070*
C8 0.8256 (8) 0.3018 (3) 0.7074 (3) 0.0564 (14)
H8 0.8029 0.2795 0.7673 0.068*
C9 1.0988 (7) 0.3944 (3) 0.7720 (3) 0.0436 (12)
H9 1.1039 0.3481 0.8228 0.052*
supporting information
sup-6
Acta Cryst. (2006). E62, o255–o257
H10A 0.8951 0.4574 0.8506 0.066*
H10B 1.0703 0.5214 0.8415 0.066*
C11 0.8938 (7) 0.5371 (3) 0.7325 (3) 0.0403 (11) C12 0.8970 (7) 0.5025 (3) 0.6428 (3) 0.0374 (11) C13 0.9998 (6) 0.4165 (3) 0.6086 (3) 0.0370 (11) C14 1.0065 (7) 0.3447 (3) 0.6874 (3) 0.0432 (12)
H14 1.0889 0.2934 0.6677 0.052*
C15 1.1965 (7) 0.4410 (3) 0.5809 (3) 0.0467 (12)
H15A 1.2568 0.3850 0.5562 0.056*
H15B 1.1949 0.4889 0.5324 0.056*
C16 1.3037 (7) 0.4777 (3) 0.6639 (3) 0.0529 (13)
H16A 1.4321 0.4854 0.6466 0.063*
H16B 1.2561 0.5394 0.6816 0.063*
C17 1.4038 (8) 0.4485 (4) 0.8207 (3) 0.0691 (16)
H17A 1.3950 0.4049 0.8720 0.104*
H17B 1.3601 0.5097 0.8399 0.104*
H17C 1.5300 0.4534 0.8012 0.104*
C18 0.4366 (8) 0.7148 (4) 0.5298 (4) 0.082 (2)
H18A 0.3755 0.7304 0.4725 0.122*
H18B 0.4884 0.7712 0.5566 0.122*
H18C 0.3494 0.6875 0.5724 0.122*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
Geometric parameters (Å, º)
N1—C16 1.450 (5) C9—C10 1.535 (6)
N1—C17 1.469 (6) C9—C14 1.553 (6)
N1—C9 1.482 (6) C9—H9 0.9800
O1—C3 1.371 (5) C10—C11 1.506 (6)
O1—C18 1.415 (6) C10—H10A 0.9700
O2—C4 1.384 (4) C10—H10B 0.9700
O2—C5 1.468 (4) C11—C12 1.377 (5)
O3—C6 1.221 (5) C12—C13 1.505 (6)
C1—C2 1.380 (6) C13—C14 1.519 (5)
C1—C11 1.396 (6) C13—C15 1.521 (6)
C1—H1 0.9300 C14—H14 0.9800
C2—C3 1.391 (5) C15—C16 1.513 (6)
C2—H2 0.9300 C15—H15A 0.9700
C3—C4 1.374 (5) C15—H15B 0.9700
C4—C12 1.370 (5) C16—H16A 0.9700
C5—C6 1.527 (6) C16—H16B 0.9700
C5—C13 1.532 (6) C17—H17A 0.9600
C5—H5 0.9800 C17—H17B 0.9600
C6—C7 1.450 (6) C17—H17C 0.9600
C7—C8 1.329 (7) C18—H18A 0.9600
C7—H7 0.9300 C18—H18B 0.9600
C8—C14 1.473 (6) C18—H18C 0.9600
C8—H8 0.9300
C16—N1—C17 110.6 (4) H10A—C10—H10B 107.5
C16—N1—C9 113.3 (4) C12—C11—C1 115.7 (4)
C17—N1—C9 111.7 (4) C12—C11—C10 118.0 (4)
C3—O1—C18 117.9 (4) C1—C11—C10 125.8 (4)
C4—O2—C5 104.8 (3) C4—C12—C11 123.3 (4)
C2—C1—C11 121.0 (4) C4—C12—C13 109.8 (4)
C2—C1—H1 119.5 C11—C12—C13 126.7 (4)
C11—C1—H1 119.5 C12—C13—C14 108.0 (3)
C1—C2—C3 122.1 (4) C12—C13—C15 111.6 (4)
C1—C2—H2 118.9 C14—C13—C15 108.4 (4)
C3—C2—H2 118.9 C12—C13—C5 98.3 (4)
O1—C3—C4 117.5 (4) C14—C13—C5 116.6 (4)
O1—C3—C2 126.1 (4) C15—C13—C5 113.4 (4)
C4—C3—C2 116.4 (4) C8—C14—C13 113.0 (4)
C12—C4—C3 121.2 (4) C8—C14—C9 114.7 (4)
C12—C4—O2 111.6 (3) C13—C14—C9 107.2 (4)
C3—C4—O2 127.0 (4) C8—C14—H14 107.2
O2—C5—C6 107.0 (3) C13—C14—H14 107.2
O2—C5—C13 105.9 (3) C9—C14—H14 107.2
C6—C5—C13 114.5 (3) C16—C15—C13 110.7 (4)
O2—C5—H5 109.8 C16—C15—H15A 109.5
supporting information
sup-8
Acta Cryst. (2006). E62, o255–o257
C13—C5—H5 109.8 C16—C15—H15B 109.5
O3—C6—C7 121.3 (5) C13—C15—H15B 109.5
O3—C6—C5 119.7 (4) H15A—C15—H15B 108.1
C7—C6—C5 118.9 (4) N1—C16—C15 111.7 (4)
C8—C7—C6 122.7 (5) N1—C16—H16A 109.3
C8—C7—H7 118.6 C15—C16—H16A 109.3
C6—C7—H7 118.6 N1—C16—H16B 109.3
C7—C8—C14 123.1 (4) C15—C16—H16B 109.3
C7—C8—H8 118.4 H16A—C16—H16B 107.9
C14—C8—H8 118.4 N1—C17—H17A 109.5
N1—C9—C10 116.2 (4) N1—C17—H17B 109.5
N1—C9—C14 105.5 (4) H17A—C17—H17B 109.5
C10—C9—C14 112.7 (4) N1—C17—H17C 109.5
N1—C9—H9 107.3 H17A—C17—H17C 109.5
C10—C9—H9 107.3 H17B—C17—H17C 109.5
C14—C9—H9 107.3 O1—C18—H18A 109.5
C11—C10—C9 115.0 (4) O1—C18—H18B 109.5
C11—C10—H10A 108.5 H18A—C18—H18B 109.5
C9—C10—H10A 108.5 O1—C18—H18C 109.5
C11—C10—H10B 108.5 H18A—C18—H18C 109.5
C9—C10—H10B 108.5 H18B—C18—H18C 109.5
O2—C5—C6—C7 −118.7 (4) C12—C13—C14—C8 70.3 (5) C13—C5—C6—O3 176.0 (4) C12—C13—C14—C9 −56.9 (5) C13—C5—C6—C7 −1.6 (5) C15—C13—C14—C8 −168.6 (4) O2—C5—C13—C12 29.7 (4) C15—C13—C14—C9 64.1 (5) O2—C5—C13—C14 144.8 (4) C5—C13—C15—C16 172.0 (3) O2—C5—C13—C15 −88.2 (4) C12—C13—C15—C16 62.1 (4) C6—C5—C13—C12 −87.9 (4) C14—C13—C15—C16 −56.7 (5) C6—C5—C13—C14 27.1 (5) C13—C15—C16—N1 52.0 (5)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
C18—H18A···O1i 0.96 2.75 3.285 (6) 116
C10—H10A···O2ii 0.97 2.62 3.423 (5) 140
C14—H14···O3iii 0.98 2.66 3.389 (6) 131
C5—H5···O3iii 0.98 2.70 3.260 (5) 117