Development and Validation of a Dissolution Method for Desloratadine Coated
Tablets
Brunna Ricci Falcão, Letícia de Melo Teixeira, Francine Zachow Philippsen, Tiago Rafael Sausen*
1Curso de Farmácia, Pontifícia Universidade Católica do Paraná, campus Toledo, 85902–532, Brazil
Article Information Received 01 November 2016 Received in revised form 27 Jan 2017 Accepted 29 Jan 2017
Abstract
The aim of this work was the development and validation of a dissolution methodology for
desloratadine-coated tablets by spectrophotometry on ultraviolet (UV). 0.1 M hydrochloric
acid (HCl), pH 4.5-citrate buffer and pH 6.8 phosphate buffer were tested as dissolution
medium. In addition, influences of apparatus, and rotation speed were evaluated. After an UV
scan spectrum from 500 to 200 nm, to determine the maximum wavelength absorbance,
samples were analyzed by UV visible spectrophotometric method. The parameters selected
were 0.1 M HCl as dissolution medium, using paddles as apparatus at 50 rpm, with analysis
at wavelength of 280 nm. The method was validated as per ICH guidelines, and the results
showed that the dissolution methodology for desloratadine-coated tablets with 0.1 M HCl as
dissolution medium, using paddles as apparatus at 50 rpm, with analysis at wavelength of
280 nm, with sampling points at 5, 10, 15 and 30 minutes is specific, linear, precise and
accurate and could be applied for quality control of desloratadine tablets, since there is no
official monograph. Keywords:
Desloratadine, Dissolution, Spectrophotometry Corresponding Author: E-mail : [email protected] Mob.: +554532778600
1 Introduction
The dissolution tests allow to obtain information regarding the
drug biological availability, being recognized also as an
important tool to pharmaceutical companies on development
and quality control, because helps on the research and
development to guide the best formulation and ensure the
lot-to-lot quality of pharmaceutical dosage forms or for justifying
post-approval product changes such as in the formulation and the
manufacturing process1-9.
Dissolution is the only test that addresses product performance,
therefore when developing a dissolution methodology, the
knowledge related to solubility, permeability, and
pharmacokinetics of the drug should be considered and the test
must be capable to display the maximum discriminative profile
and, also to allow to detect any deviation in quality standards
initially proposed. The results from dissolution assays provides
important information, including to establish in vitro/in vivo
correlation2, 7, 10-14.
Methodology validation enables to know limitations and
reliability of measurements performed. Per official codex,
validation process is essential to ensure that the analytical
method is suitable for the intended purpose, demonstrating
reliable information through parameters such as specificity,
linearity, accuracy and precision suitable for analysis2, 15-21. Desloratadine,
(8Chloro-11-piperidin-4-ylidene-6,11-diydro-5H-benzo [5,6]cyclohepta[1,2-b]pyridine), an off white powder,
slightly soluble in water e very soluble in ethanol and propylene
glycol, is used to relief of symptoms of seasonal allergic rhinitis,
perennial (non-seasonal) allergic rhinitis by blocking the action
of histamine, acting as an antihistamine drug, is commercially
available in Brazil by the brand name of Desalex® as coated tablets with a labeled amount of 5 mg, and also available as 0.5
mg/mL syrup. Desloratadine is classified, according to
Biopharmaceutical Classification System (BCS) as a Class I
drug21-23.
UK Journal of Pharmaceutical and Biosciences
Available at www.ukjpb.com
Therefore, since there’s no official method describe in
pharmacopoeias, this paper describes the development and
validation of a dissolution test for desloratadine coated-tablets
using a simple, fast and inexpensive ultraviolet method. The
development and validation were carried out in compliance with
the International Conference on Harmonization17. 2 Materials and Methods
All chemicals and reagents were of analytical reagent grade.
Desloratadine reference substance (99.82 %) was kindly
donated by the pharmaceutical company Prati–Donaduzzi
(Toledo, Brazil). The reference drug product (Desalex®), labelled as containing 5 mg of desloratadine and the following
excipients (dibasic calcium phosphate, microcrystalline
cellulose, starch, talc, hypromellose, titanium dioxide,
polyethylene glycol, white wax and carnauba wax) were
obtained commercially.
Equipment and instruments used in the present study were analytical scale (Gehaka, AG–200 model), dissolution test
apparatus (Nova Ética, 301-6 AUT model), ultrasonic bath
(Químis, Q355D model) and UV spectrophotometer (UV-1600
Pró–Análise).
2.1 Methodology
2.1.1 Maximum wavelength absorption determination
Standard solutions with 5.56 μg/mL of desloratadine were
prepared in the following medium: 0.1 M hydrochloric acid
(HCl), pH 4.5-acetate buffer and pH 6.8 phosphate buffer. The
samples were submitted to an UV scan spectrum between 500
to 200 nm to determinate desloratadine maximum wavelength
absorption.
2.1.2 Dissolution test conditions
Dissolution testing of tablets was perfomed with the reference
drug product (Desalex® 5 mg), to define the method conditions.
Initially, to determinate the more discrimating dissolution
medium, using paddles (USP apparatus II) at a stirring speed of
100 rpm, a volume of 900 mL of the following dissolution media,
pre-heated to 37 °C ± 0.5 °C, were tested: 0.1 M HCl; pH
4.5-citrate buffer and pH 6.8–phosphate buffer. Manual sampling
aliquots of 20.0 mL were withdrawn at 5, 10, 15, 30 and 60
minutes, filtered in a Millex® 0.45 µm filter and analyzed on a
UV/VIS Spectrophotometer (280 nm). There was no sample
dilution nor medium replacement after the sampling. The
desloratadine standard solution was prepared to obtain a final
concentration of 5.56 μg/mL.
With the results of the dissolution medium, tests to determinate
the apparatus (paddles and baskets) and rotation speed (50
and 100 rpm for paddles; 75 and 100 rpm for baskets) were
performed using as dissolution medium that one with better
results, maintained at 37 °C ± 0.5 ºC. Again, samples of 20.0
mL were withdrawn manually at 5, 10, 15, 30, 45 and 60
minutes, filtered (0.45 µm Millex® filter) and analyzed on a UV/VIS Spectrophotometer (280 nm), along with a desloratine
standard solution in 5.56 μg/mL final concentration.
To perform the filter evaluation, tests were performed in the
mentioned dissolution mediums and the samples withdrawn
were not filtered. Filtration of dissolution samples is usually
necessary to prevent undissolved drug particles from entering
the analytical sample and removes insoluble excipients that
otherwise cause high background or turbidity. The filter
evaluation is necessary to verify whether it can be used in the
dissolution test without adsorption of the drug into the filter24. 2.1.3 Validation
To demonstrate the method’s suitability for use as a dissolution
test, it was validated based on specificity, linearity, precision
and accuracy parameter16, 25. 2.1.3.1 Specificity
Placebo samples of desloratadine, a mix of the excipients from
the desloratadine reference drug tablets (Desalex®) were prepared in their usual compositions, according to literature 22, 26
. The placebo samples were transferred to different vessels
(n=6) with 900 mL of 0.1 M HCl as dissolution medium at 37 °C
± 0.5 °C and stirred for 30 minutes at 50 rpm using a paddle
(USP apparatus II). Aliquots of these solutions were filtered
through a Millex® filter and analyzed by the UV method (280 nm).
2.1.3.2 Linearity
Desloratadine stock solutions at 55.55 mg/mL, using 0.1 M HCl
as solvent, were prepared. Aliquots of this solution were
transferred to volumetric flasks to obtain final concentrations of
1.11; 2.78; 4.17; 5.56; 6.94 and 8.33 µg/mL. Each solution was
prepared in triplicate. The linearity was evaluated by linear
regression analysis, which was calculated by the least squares
regression method and analysis of variance (ANOVA).
2.1.3.3 Precision
Precision was evaluated on two levels, repeatability and
intermediate precision. The evaluation of intermediate precision
(inter-day precision) of the dissolution test was performed on
three different days. The repeatability was evaluated on the
same day for intra-day precision in six vessels used for the
dissolution test, in a concentration of 5.56 µg/mL. The relative
standard deviation (RSD) from the results was calculated.
2.1.3.4 Accuracy
Accuracy was determinate by the recovery percentage of a
known amount of desloratadine reference substance added to a
placebo solution. A recovery study was conducted by adding
known amounts of the desloratadine stock solution to the
UK J Pharm & Biosci, 2017: 5(1); 14 the nominal assay (5 mg). Each concentration was prepared in
triplicate and analyzed by the UV method at 280 nm.
3 Results and Discussion
3.1 Dissolution method development
To determine which wavelength desloratadine shows UV absorption, an UV scan spectrum between 500–200 nm was
performed. The results demonstrated that desloratadine showed
a maximum absorption at 280 nm (Figure 1).
Fig. 1: UV spectrum of desloratadine solution at 5.56 µg/mL in 0.1 M HCl
During the development of a dissolution methodology, different
parameters were evaluated. As dissolution method
development for immediate release drugs must be tested in
dissolution mediums ranging physiological pH (1.2 to 6.8), the
dissolution medium selected 0.1 M HCl, pH 4.5-citrate buffer
and pH 6.8–phosphate buffer were evaluated to determinate
which medium showed better discriminative dissolution profile
27
. The results (Table 1 and Figure 2) demonstrated that pH
4.5-citrate buffer and pH 6.8–phosphate buffer as dissolution
medium showed dissolution profiles very similar, with an initial
dissolution around 70 % (71.64 % and 14.72 %, respectively).
However, both medium doesn’t allowed total desloratadine
dissolution, since the final drug dissolution was 74.76 % for pH
4.5-citrate buffer and 80.79 % for pH 6.8-phosphate buffer.
The dissolution medium where desloratadine showed complete
dissolution, was 0.1 M HCl. Also, the dissolution profile at five
points (5, 10, 15, 30 and 60 minutes) showed that the drug
dissolved more than 85% in 15 minutes (Table 1 and Figure 2),
classifying desloratadine as very rapidly dissolving drug. As
desloratadine is an antihistamine drug used to relieve allergy
symptoms, it must be ready to act in the body around 30
minutes after the drug intake22, a rapidly drug dissolution is required for immediate-release dosage forms16,26. This results, corroborates with the fact that desloratadine is classified as a
Class I drug (high solubility and high permeability) in
Biopharmaceutical Classification System (BCS)16. The results indicated that 0.1 M HCl was selected as dissolution medium.
Table 1: Percentage of desloratadine tablets in 0.1 M HCl, pH 4.5-acetate buffer and pH 6.8-phosphate buffer as dissolution mediums
Time 0.1 M
HCl
pH 4.5-acetate buffer
pH 6.8-phosphate
buffer
5 min 90.56% 74.72% 74.72%
10 min 95.35% 74.35% 78.88%
15 min 97.69% 78.74% 81.06%
30 min 98.84% 79.16% 84.98%
60 min 100.75% 83.79% 87.14%
Fig. 2: Dissolution profiles of desloratadine tablets in 0.1 M HCl, pH 4.5-acetate buffer and pH 6.8-phosphate buffer as dissolution mediums
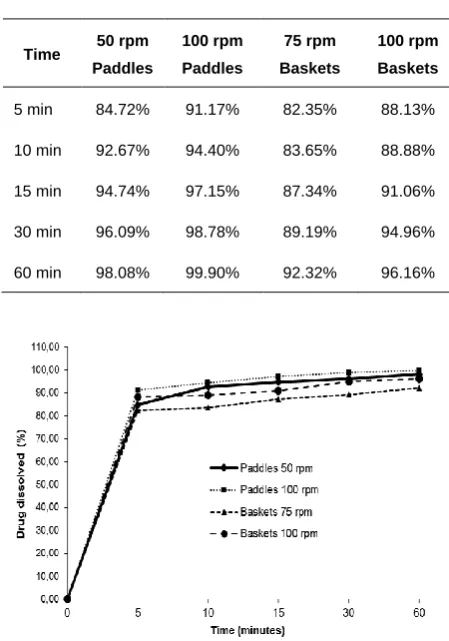
After the dissolution medium was determinate, apparatus type
and speed were tested. The most common dissolution
conditions are basket (USP apparatus I) at 75 or 100 rpm as
rotation speed and paddles (USP apparatus II) in a rotation of
50 or 100 rpm 13. According with Table 2 and Figure 3, the use
of baskets as apparatus doesn’t allowed a complete
desloratadine dissolution, since the drug dissolved at the end of
the test was 92.32 % to baskets as 75 rpm and 96.15 % to
baskets as 100 rpm.
Meanwhile, paddles apparatus, from both speeds (50 rpm and
100 rpm), showed dissolution profiles very similar, with a fast
dissolution at the first sampling point (around 90 % of drug
dissolved). At the end of the test, both paddles at 50 rpm and at
100 rpm showed a good drug dissolution. Thus, since there was
no difference between the amount of desloratadine dissolved
with paddles in both speeds, the conditions selected were
paddles (USP apparatus II) in a rotation of 50 rpm, because
there is no need for a higher apparatus speed to provide a good
drug dissolution profile.
Regarding the sampling time, as the results from samples
with values higher than 99 % of drug dissolved, the time of the
dissolution test was set on 30 minutes, because since there was
no more drug to be dissolved after 30 minutes of test, there’s no
reason to keep the test performing for 30 minutes longer.
Table 2: Percentage of dissolved desloratadine tablets in paddles (50 and 100 rpm) and baskets (75 and 100 rpm) using 0.1 M HCl as dissolution medium
Time 50 rpm
Paddles
100 rpm Paddles
75 rpm Baskets
100 rpm Baskets
5 min 84.72% 91.17% 82.35% 88.13%
10 min 92.67% 94.40% 83.65% 88.88%
15 min 94.74% 97.15% 87.34% 91.06%
30 min 96.09% 98.78% 89.19% 94.96%
60 min 98.08% 99.90% 92.32% 96.16%
Fig. 3: Dissolution profile of desloratadine tablets in paddles (50 and 100 rpm) and baskets (75 and 100 rpm) using 0.1 M HCl as dissolution medium.
Based on the results, is possible to determinate that a good
dissolution method for desloratadine-coated tablets is using 900
mL 0.1 M HCl as dissolution medium with paddles at 50 rpm,
samples withdraw at 5, 10, 15 and 30 minutes, filtered and
analyzed at spectrophotometer at 280 nm.
3.2 Dissolution method validation
For the developed dissolution method meets the requirements
of the analytical applications, ensuring results reliability, it
should be validated.
To the results of specificity, analysis of desloratadine placebo
solutions showed that the UV method suffer no interference
from the formulation of the tablet evaluated, demonstrating to be
specific (Figure 4). Since there was no interference from the
excipients with the selected wavelength (280 nm), UV can be
used to quantify desloratadine. Analysis by UV are usually used
in quality control of pharmaceuticals because most of drugs
absorbed energy in UV region, and is a method, which does not
require a complex or expensive equipment, and there is no
need for toxic solvents 19. In addition, by using UV method, results can be obtained faster, analysis is simpler and fewer
solvents are used, making this valuable in routine analysis 16, 20,
26, 29
.
Fig. 4: UV spectrum of desloratadine and excipients solutions for specificity.
To assess linearity, three calibration curves of desloratadine
were constructing and plotted graphically as concentration
(µg/mL) versus absorbance. The results showed a good
correlation coefficient (R2: 0.9994) in the studied concentration range (1.11; 2.78; 4.17; 5.56; 6.94 and 8.33 µg/mL). Also, the
representative linear equation was y = 0,0432x + 0,0019 and
the data were validated by means of the analysis of variance
(ANOVA), which demonstrated significant linear regression and
no significant linearity deviation (p < 0.01). According to the
results, linearity was proved because an appropriate linear
correlation was found since the obtained correlation coefficients
showed values higher that 0.99 16, 24, 30.
The method showed precision by measuring the repeatability
and intermediate precision on the concentration of 5.56 µg/mL.
The results from intra-day precision showed a relative standard
deviation (RSD) of 0.82, 0.71 and 1.58 % from each of the three
days and an RSD of 1.04 % for inter-day precision. As RSD
values were lower than 5 %, the results indicated the good
precision of this method 16, 18, 25.
The accuracy of the analytical procedure, the accordance
between the accepted value and the value found 15, was demonstrated by the recovery of known amounts of
desloratadine in the dissolution vessels. In the present study,
three concentrations were evaluated (2.78, 5.56 and 8.25
µg/mL) and each concentration was measured three times. The
average recovery percentage found was 101.37 % with a RSD
of 0.90 %, indicating method’s accuracy. As the measured recovery is typically 95–105 % 26
, the results indicated good
accuracy of the method. The recovery percentage was
UK J Pharm & Biosci, 2017: 5(1); 16 4 Conclusions
A discriminative dissolution method to evaluate desloratadine
tablets was successfully developed and demonstrate to be an
easy, fast and simple method. The conditions allowing
dissolution determination were 900 mL of 0.1 M HCl as
dissolution medium at 37.0 ± 0.5 ºC, using USP type II
apparatus (paddles) at 50 rpm and analysis by
spectrophotometric detection in a wavelength of 280 nm. The
spectrophotometric method was validated and showed to be
specific, linear, precise and accurate. The developed method is
suitable for its purpose and could be applied in routine quality
control of desloratadine tablets since there is no official
monograph using spectrophotometric method for this drug in the
pharmacopoeias.
5 Acknowledgements
The authors would like to thanks Fundação Araucária for the
research’s scholarship to the first author.
6 Conflict of interests
No conflict of interest has been declared by the authors.
7 Author’s contributions
Research was designed by BRF and TRS. BRF carried out
literature review and BRF, LMT and FZP carried out data
acquisition/analysis/interpretation and manuscript preparation.
BRF worked out the final draft of the manuscript and TRS, the
corresponding author, review manuscript draft and all the
authors approved the final manuscript.
8 References
1. Grady LT. Third generation dissolution testing: Dissolution
as a batch phenomenon. Drug. Inf. J. 1996; 30: 1063-70.
2. Marcolongo R. Dissolução de medicamentos:
fundamentos, aplicações, aspectos regulatórios e
perspectivas na área farmacêutica. São Paulo: USP/
Faculdade de Ciências Farmacêuticas, 2003.
3. Sausen TR, Pavei C, Silva APC, Fialho S, Mayorga P.
Development of Clozapine Tablets by Direct Compression – Analysis of Pharmaceutical Equivalence by Dissolution
Profiles. Lat. Am. J. Pharm. 2010; 29(5): 719-24.
4. Kulkarni AP, Mohd S, Zahi Z, Deghan MHG. Development
and Validation of a Dissolution Method for Pioglitazone
Tablets. Dissol Tech. 2012; 19(4): 36-47.
5. Garcia, CV, Paim CS, Steppe M, Schapoval EES.
Development and validation of a dissolution test for
rabeprazole sodium in coated tablets. J. Pharm Biomed
Anal. 2006; 41(3): 833-837.
6. Martins, MT, Paim, CS, Steppe, M. Development of a
dissolution test for lamotrigine in tablet form using an
ultraviolet method. Braz. J. Pharm. Scien. 2010; 46(2):
179-186.
7. Dressman, J. Evolution of Dissolution Media Over the
Last Twenty Years. Dissol Tech. 2014; 21: 6-10.
8. Fortunato, D. Dissolution Method Development for
Immediate Release Solid Oral Dosage Forms. Dissol
Tech. 2005; 12(3): 12-14.
9. Marroum, PJ. History and Evolution of the Dissolution
Test. Dissol Tech. 2014; 21: 11-16.
10. Rossi, RC, Dias CL, Bajerski L, Bergold AM, Froehlich
PE. Development and application of a validated HPLC
method for the analysis of dissolution samples of
levothyroxine sodium drug products. J. Pharm, Biomed.
Anal. 2011; 54(3): 439-444.
11. Shah, R., Patel S, Patel H, Pandey S, Shah S, Shah
D. Development and validation of dissolution method for
carvedilol compression-coated tablets. Braz. J. Pharm.
Sci. 2011; 47(4): 899-906.
12. Martin GP, Gray VA. General considerations for
dissolutions methods: Development, validation and
transfer. J. Valid. Tech. 2011; 17(1): 8-11.
13. FDA. Guidance for Industry. Dissolution testing of
immediate release solid oral dosage forms. Rockville, Md:
Center for Drug Evaluation and Research, 1997.
14. Manadas R., Pina ME, Veiga F. Dissolution studies in
vitro as a prognostic tool for oral absorption of modified
release pharmaceutical dosage forms. Rev. Bras. Cien. Farm. 2002; 38(4): 375–399
15. Brito NM, Junior OPA, Polese L, Ribeiro ML. Validação de
métodos analíticos: estratégia e discussão. Rev. Ecol.
Amb. 2003; 13: 129-156.
16. Brasil. Agência Nacional de Vigilância Sanitária.
Resolução Específica nº 899 de 29 de maio de 2003.
17. ICH. International Conference on Harmonization of
Technical Requirements for Registration of
Pharmaceuticals for Human Use: Guideline on Validation
of Analytical Procedures Q2 (R1); 2005.
18. FDA. Analytical Procedures and Methods Validation for
Drugs and Biologics. Guidance for Industry. Rockville,
Md: Center for Drug Evaluation and Research; 2015.
19. FIP. Guidelines for dissolution testing of solid oral
products (Final draft, 1995).
20. Fonseca, LB. Desenvolvimento e validação de método de
dissolução aplicado a suspensões orais de nimesulida.
21. USP. The United States Pharmacopoeia. Rockville, USA:
United States Pharmacopoeial Convention. 38th Ed; 2015.
22. Goodman, A. The Pharmacological Basis of Therapeutics.
Elmsford, New York: McGraw-Hill; 2011.
23. Amidon GL, Lennernas H. Shah VP, Crison JR. A
theoretical basis for a biopharmaceutic drug classification:
the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 1995; 12: 413–420.
24. USP. The United States Pharmacopeia and National
Formulary USP 37–NF 32; The United States
Pharmacopoeial Convention, Inc.: Rockville, MD; 2014.
25. ICH. International Conference on Harmonization of
Technical Requirements for Registration of
Pharmaceutical for Human Use. Validation of Analytical
Procedures: Methodology: ICH4. ICH Harmonized
Tripartite Guideline; 1996.
26. Kibbe A. Handbook of Pharmaceutical Excipients. 6th ed. Washington, DC: American Pharmaceutical Association;
2009.
27. Gibaldi M. Biopharmaceutics and Clinical
Pharmacokinetics. 4th ed. Philadelphia: Lea & Febiger; 1991.
28. Farmacopéia Brasileira. 5a ed. Brasília, DF. Anvisa; 2010. 29. Silva WM, Santos FR, Batistuti JP. Validação de um
método analítico para quantificação de ácido fólico por
espectrofotometria-UV. Braz. J. Food. Nutr. 2013; 24
275–282
30. ICH. International Conference on Harmonization of
Technical Requirements for Registration of
Pharmaceutical for Human use. Validation of Analytical
Procedures: Methodology: Q2B. ICH Harmonized