Copyright © 1999, American Society for Microbiology. All Rights Reserved.
Mycobacterium malmoense-Specific Nested PCR Based on a
Conserved Sequence Detected in Random Amplified
Polymorphic DNA Fingerprints
JUHA KAUPPINEN,
1* RAUNO MA
¨ NTYJA¨RVI,
1ANDMARJA-LEENA KATILA
2Department of Clinical Microbiology, University of Kuopio, FIN-70211 Kuopio,
1and Department of
Clinical Microbiology, Kuopio University Hospital, FIN-70210 Kuopio,
2Finland
Received 3 September 1998/Returned for modification 7 December 1998/Accepted 13 February 1999
Mycobacterium malmoense is an opportunistic human pathogen of increasing clinical importance. Since it is
difficult to detect and identify the organism by conventional techniques, it was decided to seek a nucleic acid
amplification method specific for M. malmoense. The method was based on detection of a conserved band in
random amplified polymorphic DNA (RAPD) fingerprints of 45 M. malmoense strains. This band was a
1,046-bp product which was proven to be M. malmoense specific in dot blot hybridization analysis with a panel
of mycobacterial strains belonging to 39 other species. The fragment was sequenced, and oligonucleotide
primers were synthesized to evaluate the specificity of the PCR. Two primer pairs were found to be specific and
sensitive in the nested PCR that was developed. All 49 M. malmoense strains analyzed produced a PCR product
of the expected size. In contrast, no strains belonging to the other mycobacterial species tested produced
amplicons with these primers under specified reaction conditions. The results of the electrophoresis were
confirmed by the hybridization with the M. malmoense-specific oligonucleotide probe. This method could be
applied to the analysis of clinical or environmental samples, permitting the rapid detection of M. malmoense.
Mycobacterium malmoense is a potential human pathogen
which causes infections similar to those caused by the
Myco-bacterium avium complex (9, 14). It has steadily increased in
clinical importance in the Nordic countries and Scotland (3, 7,
20), and recently, it has also been isolated from clinical samples
in other countries (32). M. malmoense has also been detected
in natural environments (11, 23, 26). M. malmoense is a difficult
species to isolate (8, 12, 13), and its identification may also be
problematic by conventional identification tests (34). Analytic
techniques based on lipid profiles or gene sequencing have
been found to be more reliable for its identification (21, 28,
33).
Several PCR-based methods have been developed for the
detection and/or identification of atypical mycobacteria. In
these techniques, a number of primers may be used (multiplex
PCR [18]) or a selected conserved region of the genome is
amplified, and the recognition of the species is based on the
detection of differences in some bases in the amplified
se-quence. In the latter case, the final species identification is
based on the analysis of the amplicon by nucleic acid
sequenc-ing (17, 28), restriction endonuclease digestion (4, 19, 35), or
nucleic acid hybridization (29, 30).
The genome of M. malmoense is poorly characterized. No
published information on M. malmoense-specific genes is
avail-able. The data published so far comprise data for
species-specific portions of the genome that are only a few bases in
length (6, 28). One useful way to discover longer
species-specific parts in a sequence of a microbe whose genomic data
are unknown is to perform fingerprint profile analysis of
sev-eral strains belonging to the same species (22). We applied this
method to the analysis of M. malmoense for species-specific
sequences which could be used as targets for an M.
malmoense-specific PCR.
MATERIALS AND METHODS
Strains.The following Mycobacterium type strains were included in the study:
M. agri ATCC 27406 (American Type Culture Collection [ATCC], Rockville,
Md.), M. aichiense ATCC 27280, M. asiaticum ATCC 25276, M. austroafricanum ATCC 33464, M. avium ATCC 15769, M. branderi ATCC 51788 and ATCC 51789, M. celatum I ATCC 51130, M. celatum II ATCC 51131, M. chitae ATCC 19627, M. chubuense ATCC 27278, M. cookii ATCC 49103, M. diernhoferi ATCC 19340, M. fallax ATCC 35219, M. flavescens ATCC 14474, M. fortuitum ATCC 35754, ATCC 6841, and ATCC 6842, M. gastri ATCC 15754, M. gordonae ATCC 14470, M. hiberniae ATCC 49874, M. interjectum ATCC 51457, M. intermedium ATCC 51848, M. intracellulare ATCC 13950, M. kansasii ATCC 12478, M.
komossense ATCC 33013, M. marinum ATCC 927, M. malmoense ATCC 29571, M. neoaurum ATCC 25795, M. nonchromogenicum ATCC 19530, ATCC 19533,
and ATCC 35783, M. peregrinum ATCC 14467, M. phlei ATCC 12298, M. pulveris ATCC 35154, M. scrofulaceum ATCC 19981 and ATCC 35792, M. shimoidei ATCC 27962, M. simiae ATCC 25275, M. smegmatis ATCC 35797, M. sphagni ATCC 33027, M. szulgai ATCC 35799, M. terrae ATCC 15755, M. triviale ATCC 23291, M. tuberculosis ATCC 25177 and ATCC 27294 (H37Rv), and M. xenopi ATCC 19250. M. bovis BCG 965/Gothenburg (vaccine strain) was a gift from M. Riddel, University of Gothenburg, Gothenburg, Sweden. Two strains, M.
chelo-nae QC500/91 and M. haemophilum QC190/91, that were distributed in an
Australian quality control scheme were gifts from A. Marzec, Austin Hospital, Melbourne, Australia. The following clinical isolates from the culture collection of the Department of Clinical Microbiology, Kuopio University Hospital, were included: M. bovis BCG (one strain), M. chelonae subsp. abscessus (one strain), and M. malmoense (39 Finnish and 6 British strains) (10). In addition, three Finnish environmental isolates of M. malmoense that originated from stream water were included (11).
The strains were stored at270°C in Middlebrook 7H9 broth (Difco Labora-tories, Detroit, Mich.). For the present study, the strains were grown on Middle-brook 7H11 agar with MiddleMiddle-brook OADC (oleic acid, albumin, dextrose, cata-lase) enrichment (Difco Laboratories). If necessary, the identity of a strain was reconfirmed by fatty acid analysis by gas-liquid chromatography complemented with a panel of biochemical tests previously described in detail (10, 31).
For DNA extraction, all mycobacteria except M. haemophilum were grown in Middlebrook 7H9 broth at 37°C; M. haemophilum was grown in hemin-supple-mented broth at 30°C. The bacterial cells were harvested by centrifugation at 3,0003g for 15 min, washed once with TE buffer (10 mM Tris, 1 mM EDTA),
and resuspended in 1 to 3 ml of TE buffer, depending on the size of the cell pellet. The suspensions were stored at220°C until they were processed further.
DNA extraction.The chromosomal DNA of the mycobacteria was isolated as
described previously (16). In brief, the mycobacterial cells were lysed with
Na-* Corresponding author. Mailing address: Department of Clinical
Microbiology, University of Kuopio, P.O. Box 1627, FIN-70211
Kuo-pio, Finland. Phone: 358-17-162708. Fax: 358-17-162705. E-mail: Juha
[email protected].
1454
on May 15, 2020 by guest
http://jcm.asm.org/
garse protease (Sigma, St. Louis, Mo.) and lysozyme (Sigma). The lysis was followed by phenol-chloroform extractions, RNase (Sigma) digestion, and etha-nol precipitation. The DNA concentrations were quantified both with the GeneQuant DNA/RNA calculator (Pharmacia LKB Biochrom Ltd., Cambridge, United Kingdom) and by agarose gel electrophoresis by comparison of a sample band to the bands of HindIII-digested bacteriophage lambda DNA (New En-gland Biolabs, Inc., Beverly, Mass.). The DNA stock was stored at 4°C, and the diluted DNA was stored at220°C.
RAPD-PCR.Random amplified polymorphic DNA PCR analysis
(RAPD-PCR) of M. malmoense has been described in detail in a publication describing our previous study in which 45 strains were amplified with selected 10-mer primers (15). In the RAPD-PCR, the genomic DNA is amplified under opti-mized conditions by using a short, nonspecific oligonucleotide primer. In the present study, other Mycobacterium species were fingerprinted by the same method with the OPA2 primer (Operon Technologies, Inc., Alameda, Calif.).
Fragment extraction from agarose gel. A DNA product representing a
1,046-bp band that was present in all 45 M. malmoense strains was cut as a slice from the fingerprint of M. malmoense ATCC 29571 obtained in an agarose gel after electrophoresis. The DNA fragment was purified by a phenol extraction method described elsewhere in detail (27).
Cloning and sequencing.The fragment (named MA2/6) was cloned into
SmaI-digested pBluescript II SK (Stratagene, La Jolla, Calif.) by the standard method (27). The insert was digested with restriction endonucleases SalI and PvuII, and the 557-bp SalI-PvuII fragment was recloned into SalI- and PvuII-digested pBluescript II SK. The cloned inserts were sequenced with an ALF automatic DNA sequencer with the Auto Read Sequencing kit (Pharmacia Biotech, Sol-lentuna, Sweden) with the primers for the T3 and T7 sequences of pBluescript II SK. The sequence segments were aligned to constitute the complete sequence. Analyses of the DNA sequences were performed with the GeneJockey II com-puter program (Biosoft, Cambridge, United Kingdom).
Dot blot hybridization.Dot blot hybridizations of mycobacterial chromosomal
DNA were carried out by modifying the published protocol (2). One microgram of EcoRI-digested chromosomal DNA was applied onto each dot on an Immo-bilon-N membrane (Millipore, Bedford, Mass.) which had been prewetted for 2 s in ethanol and saturated in 33SSC (13SSC is 0.15 M NaCl plus 0.015 M sodium citrate). The DNA was denaturated by incubating the membrane for 10 min on two stacked blotting papers (Schleicher & Schuell GmbH, Dassel, Germany) saturated in an alkaline solution (0.5 M NaOH, 1 M NaCl). The neutralization was performed in the same way by using a neutralization solution (1 M Tris-HCl, 2 M NaCl [pH 7.5]). For DNA binding, the dried membrane was baked for 1 h at 80°C according to the instructions of the membrane manufacturer.
The MA2/6 fragment and the 16S rRNA gene (rDNA) probe (16) were chosen as probes for the dot blot hybridization analyses. The probes were32P labelled
with the Rediprime kit (Amersham, Buckinghamshire, United Kingdom) with Redivue [a-32P]dCTP AA 0005 (Amersham) according to the instructions on the
insert in the Rediprime package. The specific activities of the probes were 43 108cpm/mg for the MA2/6 fragment and 23108cpm/mg for the 16S rDNA
probe.
The membrane was incubated in the prehybridization solution at 65°C for 1 h before hybridization. There was 150 kcpm of labelled probe per ml of hybrid-ization solution, and the hybridhybrid-ization was allowed to proceed for 18 h at 65°C. To visualize the label, the membrane was exposed to Hyperfilm MP (Amer-sham). If the membrane was to be reprobed, the attached probe was stripped off by incubating the membrane in 0.1 M NaOH for 30 min and in 0.13SSC–0.2 M Tris-HCl (pH 8.0) containing 0.5% sodium dodecyl sulfate for 45 min at room temperature.
PCRs.To prevent carryover contamination, treatment of the strains,
extrac-tion of DNA, preparaextrac-tion of PCR mixtures, amplificaextrac-tion, and analysis of the amplicons were each done in separate areas. Filter-barrier pipette tips were used at all stages. In the amplification control reactions with 16S rRNA gene primers and nested amplifications with the MA2/6 primers, the PCR mixtures (total volume, 50 ml) contained 13 DynaZyme DNA polymerase reaction buffer (Finnzymes Oy, Espoo, Finland), 0.3mM primers (synthesized by Medprobe, Oslo, Norway), 200mM (each) dATP, dCTP, dGTP, and dTTP (Promega, Madison, Wis.), 1 to 2 ng of genomic DNA (or 1ml from the product of the first reaction to the second reaction of the nested PCR), and 2 U of DynaZyme DNA polymerase (Finnzymes Oy).
The primers for the amplification control reactions were 16S-A (59-GGATG ACGGCCTTCGGGTTG-39), which complements bases 366 to 385, and 16S-B (59-GACAGCCATGCACCACCTGC-39), which consists of bases 993 to 1012 in the M. malmoense 16S rRNA gene sequence (GenBank accession no. X52930) (25). The primers for the first reaction of the M. malmoense-specific nested PCR were MAL-1A (59-CGACGAGACACCCCCAGC-39), which complements bases 12 to 29, and MAL-1B (59-TCAGCCAGCGGTGTAGC-39), which con-sists of bases 988 to 1004; and the primers of the second reaction were MAL-2A (59-CAGTCAGCGAGGAAGTCGGGAATC-39), which complements bases 72 to 95, and 59-end biotinylated primer MAL-2B-biot (59-GGGTTCATCATTCC GCTGGGCTAC-39), which consists of bases 482 to 505 in the M. malmoense MA2/6 sequence (GenBank accession no. AF085175).
The amplification programs were optimized for a GeneAmp PCR System 9600 apparatus (Perkin-Elmer, Norwalk, Conn.). Control PCRs with 16S rRNA gene primers were performed by a program which included predenaturation at 98°C
for 3 min, a first 15 cycles consisting of 96°C for 20 s, 54°C for 35 s, and 72°C for 50 s, and a subsequent 25 cycles consisting of 95°C for 25 s, 58°C for 35 s, and 72°C for 45 s; after these cycles, there was an extra extension step at 72°C for 10 min. In the M. malmoense-specific nested PCR, the first program included pre-denaturation at 98°C for 3 min and 25 cycles consisting of 96°C for 25 s, 63°C for 50 s, and 72°C for 1 min, and the second one included 25 cycles consisting of 96°C for 20 s, 65°C for 30 s, and 72°C for 40 s and, finally, an extra extension step at 72°C for 10 min. After the program was run, the reaction tubes were held at 4°C until agarose gel electrophoresis was run.
Agarose gel electrophoresis.The PCR products were run on a 1.3% agarose
(FMC Bioproducts, Rockland, Maine) gel containing 0.5mg of ethidium bromide (Sigma) per ml and 13Tris-acetate-EDTA buffer. The gel was photographed under UV light.
Hybridization of nested PCR product.The nested PCR products were
hybrid-ized with 59-end digoxigenin-labelled oligonucleotide probe MAL-p-dig (59-AG CTGTTCGTGGGTATCAGAGAGTTA-39), which complements bases 160 to 185 (synthesized by Medprobe, Oslo, Norway) in the M. malmoense MA2/6 sequence, in microplate wells by a method described elsewhere (1), with slight modifications. After the amplification reaction with the M. malmoense-specific primers MAL-2A and MAL-2B-biot, 15ml of the PCR product was applied to the well of a streptavidin-coated microplate (Labsystems, Helsinki, Finland) in 185ml of working solution consisting of 1 M phosphate-buffered saline, 1% bovine serum albumin, and 0.1% Tween 20 (pH 7.4). The biotinylated amplifi-cation product was immobilized onto the well for 18 h at 4°C. For denaturation of the PCR product, the well was first washed once with 200ml of 0.1 N NaOH, and after that, 200ml of 0.1 N NaOH was added and the plate was incubated for 5 min at room temperature. After two washing steps with the working solution, the hybridization with the digoxigenin-labelled oligonucleotide probe MAL-p-dig was carried out in 150ml of hybridization solution consisting of 63SSC, 0.1% sodium dodecyl sulfate, 5 mM EDTA, 103Denhardt solution (which contains 0.2% Ficoll 400, 0.2% polyvinylpyrrolidone, and 0.2% bovine serum albumin) for 4 h at 56°C. After hybridization, the well was washed twice with working solution. The hybridized digoxigenin-labelled oligonucleotide probe was detected by adding to each well 150ml of anti-digoxin monoclonal antibody clone DI-22 conjugated with alkaline phosphatase (Sigma) at a dilution of 1:1,000 in working solution. After incubation for 1 h at room temperature, the well was washed two times. To detect the attached anti-digoxin antibody, 150ml of substrate solution, prepared from Sigma Fast-pNPP substrate tablets (Sigma), was added. The substrate was incubated for 30 min at room temperature. The reaction was stopped by adding 40ml of 3 N NaOH per well. The absorbance was measured at 405 nm with a Multiskan plus photometric reader (Labsystems).
Nucleotide sequence accession number.The nucleotide sequence reported
here has been submitted to the GenBank nucleotide sequence database under accession no. AF085175.
RESULTS
The 45 M. malmoense strains exhibited a variety of
RAPD-PCR patterns when the strains were analyzed with the OPA2
10-mer primer. However, one band of 1,046 bp was conserved
and was present in the fingerprint of every M. malmoense
strain. The band of similar size was uncommon in the other
mycobacterial species tested. An example of the fingerprint
profiles is shown in Fig. 1.
The existence of the MA2/6 sequence in the genomes of
mycobacteria other than M. malmoense was estimated by dot
blot hybridization analysis of the set of the mycobacterial
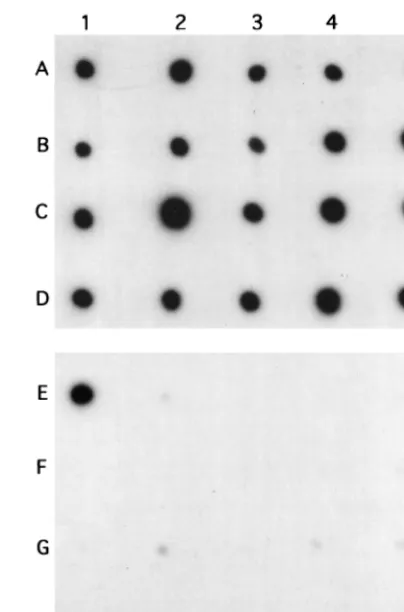
strains selected. Only M. malmoense DNA exhibited a distinct
positive result (Fig. 2). On the same membranes, all strains
studied produced a positive result by the control dot blot
hy-bridization assay, carried out with the labelled 16S rDNA
frag-ment. On this basis, the MA2/6 fragment was regarded as a
potential candidate for an M. malmoense-specific fragment. No
high degrees of nucleotide sequence homologies were found in
homology searches when the nucleotide sequence of MA2/6
was compared with those present in GenBank version 110.0.
On the basis of the sequencing data for the MA2/6 fragment
(Fig. 3), several primers were designed for PCR. The M.
mal-moense specificity of the primers were evaluated by combining
the sense and antisense primers in different combinations. The
primers found to be most suitable are described in Materials
and Methods. Primers MAL-1A and MAL-1B produced a
band of the expected size of 993 bp only from M. malmoense at
60°C or above. In contrast, a band was also obtained from M.
tuberculosis, M. kansasii, and M. fortuitum if the annealing
on May 15, 2020 by guest
http://jcm.asm.org/
temperature was adjusted to below 56°C (data not shown). The
band produced from M. tuberculosis and M. kansasii had a size
of approximately 1.3 kbp, whereas the band from M. fortuitum
was equal in size to the M. malmoense band. The PCR products
amplified with primers MAL-1A and MAL-1B from M.
tuber-culosis, M. kansasii, and M. fortuitum were partially sequenced.
No homologies with the M. malmoense sequence were
ob-served (data not shown).
Nested PCR, consisting of reactions first with primers
MAL-1A and MAL-1B and second with primers MAL-2A and
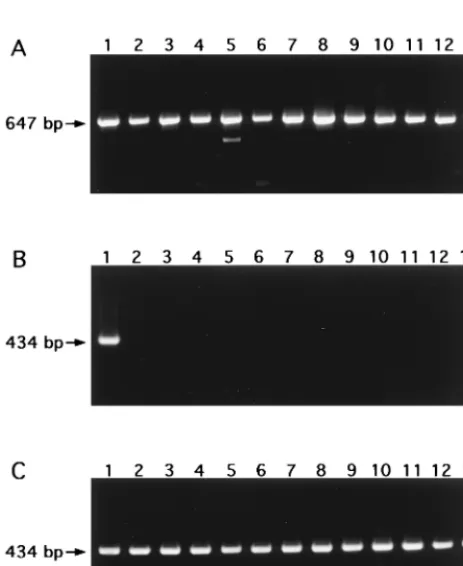
MAL-2B-biot, was highly specific for M. malmoense. Among
the mycobacterial species included in this analysis, only the 48
M. malmoense strains produced a band of 434 bp in the nested
PCR. No bands of other sizes were detected in the lanes
containing M. malmoense. The lanes containing other
Myco-bacterium species were free of any bands. A subset of the
results is presented in Fig. 4. In hybridization analysis of the
nested PCR products with the digoxigenin-labelled
oligonucle-otide probe MAL-p-dig, all M. malmoense strains with the
exception of one clinical isolate gave an absorbance value of
0.85 to 1.13; the absorbance value for the clinical isolate that
was the exception was 0.58. In contrast, the absorbance values
for the other Mycobacterium species were less than 0.09,
indi-cating negative results. The mean value for the negative
con-trol reactions (PCRs without sample DNA) was 0.02.
Control amplifications carried out with the primers for the
16S rRNA gene confirmed that the bacterial PCR templates
also amplified those strains that remained negative with the M.
malmoense-specific primers. All Mycobacterium strains
pro-duced a band of approximately 647 bp in the control reactions.
The sensitivity of the nested PCR was estimated by using a
dilution series of M. malmoense chromosomal DNA and the
DNA of the plasmid containing the MA2/6 fragment. Both 4 fg
of chromosomal DNA and 12 ag of plasmid DNA could be
detected, indicating the limit of detection to represent one
target molecule in a reaction tube. There was no difference in
the detection limit by electrophoresis or hybridization. A
slightly visible band by gel electrophoresis gave an absorbance
reading of 0.50 by hybridization analysis.
DISCUSSION
The clinical significance of mycobacterial species such as M.
malmoense is likely to be underestimated; its detection in
[image:3.612.330.532.71.377.2]clin-ical samples is particularly difficult due to its exceptionally slow
rate of growth and the need for special culture conditions (8,
12, 13). We have previously confirmed that modifications to
culture media increase the rates of detection of such species
(12). Biochemical identification of M. malmoense is laborious,
and some uncommon Mycobacterium species may easily be
incorrectly identified as M. malmoense (34). Nucleic acid
am-plification methods offer an alternative approach to the
detec-tion and identificadetec-tion of microbes whose identities are difficult
to confirm by conventional methods. Several PCR protocols
have been used for the identification of atypical mycobacteria.
In most cases, identification has been based on the detection of
differences in conserved sequences after amplification with
genus-specific primers. This also applies to M. malmoense, at
least by selected methods (6, 24, 28). In the present study, we
wanted to develop a rapid technique that was also potentially
suitable for the direct detection of M. malmoense from clinical
or environmental samples. The specific PCR method makes it
FIG. 1. RAPD fingerprint patterns of the Mycobacterium species obtained with primer OPA2. Lanes 1 to 4, M. malmoense; lane 5, M. gastri; lane 6, M.
scrofulaceum; lane 7, M. asiaticum; lane 8, M. avium; lane 9, M. intracellulare; lane
10, M. xenopi; lane 11, M. szulgai; lane 12, M. kansasii; lane 13, M. haemophilum; lane 14, M. marinum; lane 15, M. tuberculosis; lane 16, M. celatum I; lane 17, M.
celatum II. The 1,046-bp fragment of M. malmoense is indicated by an arrow. On
[image:3.612.57.294.73.215.2]the basis of RAPD, this fragment was analyzed further as a candidate for a species-specific probe for M. malmoense. Numbers on the left are in kilobase pairs.
FIG. 2. Dot blot analysis of EcoRI-digested mycobacterial DNA with the
32P-labelled 16S rDNA probe (rows A to D) and the MA2/6 fragment (rows E to
H). A1 and E1, M. malmoense ATCC 29571; A2 and E2, M. tuberculosis ATCC 25177; A3 and E3, M. tuberculosis ATCC 27294 (H37Rv); A4 and E4, M. avium ATCC 15769; A5 and E5, M. intracellulare ATCC 13950; B1 and F1, M. bovis BCG vaccine strain; B2 and F2, M. bovis BCG clinical isolate; B3 and F3, M.
kansasii ATCC 12478; B4 and F4, M. fortuitum ATCC 6841; B5 and F5, M. for-tuitum ATCC 6842; C1 and G1, M. marinum ATCC 927; C2 and G2, M. asiati-cum ATCC 25276; C3 and G3, M. xenopi ATCC 19250; C4 and G4, M. nonchro-mogenicum ATCC 19530; C5 and G5, M. nonchrononchro-mogenicum ATCC 35783; D1
and H1, M. simiae ATCC 25275; D2 and H2, M. phlei ATCC 12298; D3 and H3,
M. flavescens ATCC 14474; D4 and H4, M. smegmatis ATCC 35797; D5 and H5, M. fallax ATCC 35219.
on May 15, 2020 by guest
http://jcm.asm.org/
possible to preliminarily identify M. malmoense DNA within a
working day. With the hybridization of the amplicon, the
method takes 2 days. All M. malmoense strains tested,
includ-ing an ATCC strain and 48 clinical or environmental isolates,
produced the expected PCR product. Furthermore, the results
verified the high degree of species specificity of the MA2/6
fragment. None of the 43 other mycobacterial species tested
produced amplicons if the specific PCR conditions were used.
We performed the amplification reactions with the 16S rRNA
gene primers as DNA or inhibition controls. The sensitivity of
the nested PCR used was high under the experimental
condi-tions used.
There are several ways to find species-specific areas in
mi-crobial genomic nucleic acids. The specific sites close to or
within a conserved part of the genome, such as the 16S rRNA
genes, are potential targets that are mostly found by
sequenc-ing genomes of several species. It is possible to use primers
which anneal to the genomes of many species if further
iden-tification is done by analysis of the amplified sequences by
restriction endonuclease digestion, oligonucleotide
hybridiza-tion, or sequencing. All of these applications can reveal a
difference of even a single base.
We used an approach in which the RAPD fingerprint
pro-files of M. malmoense strains and some closely related
myco-bacterial species were compared. The basis of the present
study was the detection in the M. malmoense strains of
con-served bands which were not present in the other species. One
amplification product of this kind was found to be M.
mal-moense specific, and it was further analyzed as a suitable
can-didate for targeting by nested PCR analysis. To ensure the
species specificity of fragment MA2/6, it was extracted from an
agarose gel and used as a probe in a hybridization analysis in
which the template DNA was derived from several M.
mal-moense strains and other closely related species. The primer
candidates were selected from the sequence of the fragment by
using the appropriate computer program, and the M.
mal-moense-specific primers were found experimentally.
Negative hybridization results with the other species tested
suggested that the MA2/6 fragment was a potential candidate
as an amplification target. The stringency in the dot blot
hy-bridization was relatively high due to the hyhy-bridization
tem-perature and the salt concentration used. Therefore, sequences
closely related to the MA2/6 probe could have been detected.
In addition, the MA2/6 sequence did not show a high degree of
nucleotide sequence homology with any of the sequences in
data banks. With the selected primers, some PCR products
were obtained from mycobacterial species other than M.
mal-moense, but only at the low annealing temperatures that favor
nonspecific binding. The specificity and sensitivity of the PCR
were increased by using two nested primer pairs.
[image:4.612.58.290.331.614.2]By combining the developed nested PCR with a sample
processing method such as that of Eisenach et al. (5), it could
be used not only for the identification of M. malmoense from
cultures but also for the direct detection of M. malmoense from
samples without the need for preceding isolation of the
organ-ism. This would allow the detection of the organism in both
clinical samples and suspected environmental reservoirs. Since
the identification is based on the detection of a specific
se-quence with species-specific primers instead of the detection of
minor differences in the sequence flanked by the binding sites
FIG. 3. Nucleotide sequence of fragment MA2/6 in the genome of M. malmoense ATCC 29571. The sequence has been submitted to the GenBank nucleotide sequence database under accession no. AF085175.FIG. 4. Agarose gel electrophoresis after PCR with 16S rRNA gene primers 16S-A and 16S-B (A) and nested PCR with M. malmoense-specific primers MAL-1A and MAL-1B, and primers MAL-2A and MAL-2B-biot (B). Lane 1,
M. malmoense ATCC 29571; lane 2, M. tuberculosis ATCC 27294 (H37Rv); lane
3, M. tuberculosis ATCC 25177; lane 4, M. bovis BCG vaccine strain; lane 5, M.
avium ATCC 15769; lane 6, M. intracellulare ATCC 13950; lane 7, M. scrofula-ceum ATCC 19981; lane 8, M. szulgai ATCC 35799; lane 9, M. gastri ATCC
15754; lane 10, M. marinum ATCC 927; lane 11, M. kansasii ATCC 12478; lane 12, M. fortuitum ATCC 6841; lane 13, M. xenopi ATCC 19250. (C) Nested PCR with M. malmoense-specific primers MAL-1A and MAL-1B, and primers MAL-2A and MAL-2B-biot. Lane 1, M. malmoense ATCC 29571; lanes 2, 4, 8, 10, and 11, Finnish clinical isolates of M. malmoense; lanes 3, 5 to 7, and 9, British clinical isolates of M. malmoense; lanes 12 and 13, Finnish environmental isolates of M. malmoense.
on May 15, 2020 by guest
http://jcm.asm.org/
of the universal primers, the results can be interpreted quickly
and easily. The sensitivity of the amplification reaction was
excellent. When the sample collection and preparation are
optimized, M. malmoense could be reliably detected in the
course of a few days. This requires further evaluation with
clinical and environmental specimens.
We conclude that the nested PCR described here is a
spe-cific method for the identification of M. malmoense and that
the RAPD-PCR is a useful tool in searches for species-specific
genomic fragments.
ACKNOWLEDGMENTS
We thank Eila Pelkonen for technical assistance.
This study was supported by the Memorial Foundation of Maud
Kuistila and the Finnish Antituberculosis Foundation.
REFERENCES
1. Ackermann, L., I. T. Harvima, J. Pelkonen, V. Ritama¨ki-Salo, A.
Naukkari-nen, R. J. Harvima, and M. Horsmanheimo.Mast cells of psoriatic skin are
strongly positive for interferon-gamma. Br. J. Dermatol., in press. 2. Andersson, M. L. M., and B. D. Young. 1985. Quantitative filter
hybridisa-tion, p. 73–111. In B. D. Hames and S. J. Higgins (ed.), Nucleic acid hybrid-isation: a practical approach. IRL Press, Oxford, United Kingdom. 3. Bollert, F. G., B. Watt, A. P. Greening, and G. K. Crompton. 1995.
Non-tuberculous pulmonary infections in Scotland: a cluster in Lothian? Thorax
50:188–190.
4. De Beenhouwer, H., Z. Liang, P. De Rijk, C. Van Eekeren, and F. Portaels. 1995. Detection and identification of mycobacteria by DNA amplification and oligonucleotide-specific capture plate hybridization. J. Clin. Microbiol.
33:2994–2998.
5. Eisenach, K. D., M. D. Cave, and J. T. Crawford. 1993. PCR detection of
Mycobacterium tuberculosis, p. 191–196. In D. H. Persing, T. F. Smith, F. C.
Tenover, and T. J. White (ed.), Diagnostic molecular microbiology: princi-ples and applications. American Society for Microbiology, Washington, D.C. 6. Glennon, M., M. G. Cormican, U. Ni Riain, M. Heginbothom, F. Gannon,
and T. Smith.1996. A Mycobacterium malmoense-specific DNA probe from
the 16S/23S rRNA intergenic spacer region. Mol. Cell. Probes 10:337–345. 7. Henriques, B., S. E. Hoffner, B. Petrini, I. Juhlin, P. Wahlen, and G.
Kal-lenius.1994. Infection with Mycobacterium malmoense in Sweden: report of
221 cases. Clin. Infect. Dis. 18:596–600.
8. Hoffner, S. E., B. Henriques, B. Petrini, and G. Kallenius. 1991.
Mycobac-terium malmoense: an easily missed pathogen. J. Clin. Microbiol. 29:2673–
2674.
9. Horsburgh, C. R., Jr. 1996. Epidemiology of disease caused by nontubercu-lous mycobacteria. Semin. Respir. Infect. 11:244–251.
10. Katila, M. L., E. Brander, E. Jantzen, R. Huttunen, and L. Linkosalo. 1991. Chemotypes of Mycobacterium malmoense based on glycolipid profiles. J. Clin. Microbiol. 29:355–358.
11. Katila, M. L., E. Iivanainen, P. Torkko, J. Kauppinen, P. Martikainen, and
P. Va¨a¨na¨nen.1995. Isolation of potentially pathogenic mycobacteria in the
Finnish environment. Scand. J. Infect. Dis. Suppl. 98:9–11.
12. Katila, M. L., and J. Mattila. 1991. Enhanced isolation of MOTT on egg media of low pH. APMIS 99:803–807.
13. Katila, M. L., J. Mattila, and E. Brander. 1989. Enhancement of growth of
Mycobacterium malmoense by acidic pH and pyruvate. Eur. J. Clin.
Micro-biol. Infect. Dis. 8:998–1000.
14. Katila, M. L., T. Viljanen, and E. Brander. 1991. Mycobacterium malmoense infections. Z. Erkr. Atmungsorgane. 176:159–161.
15. Kauppinen, J., R. Ma¨ntyja¨rvi, and M. L. Katila. 1994. Random amplified polymorphic DNA genotyping of Mycobacterium malmoense. J. Clin. Micro-biol. 32:1827–1829.
16. Kauppinen, J., J. Pelkonen, and M. L. Katila. 1994. RFLP analysis of
Mycobacterium malmoense strains using ribosomal RNA gene probes: an
additional tool to examine intraspecies variation. J. Microbiol. Methods
19:261–267.
17. Kirschner, P., B. Springer, U. Vogel, A. Meier, A. Wrede, M. Kiekenbeck,
F. C. Bange, and E. C. Bottger.1993. Genotypic identification of
mycobac-teria by nucleic acid sequence determination: report of a 2-year experience in a clinical laboratory. J. Clin. Microbiol. 31:2882–2889.
18. Kox, L. F., H. M. Jansen, S. Kuijper, and A. H. Kolk. 1997. Multiplex PCR assay for immediate identification of the infecting species in patients with mycobacterial disease. J. Clin. Microbiol. 35:1492–1498.
19. Kox, L. F., J. van Leeuwen, S. Knijper, H. M. Jansen, and A. H. Kolk. 1995. PCR assay based on DNA coding for 16S rRNA for detection and identifi-cation of mycobacteria in clinical samples. J. Clin. Microbiol. 33:3225–3233. 20. Lamden, K., J. M. Watson, G. Knerer, M. J. Ryan, and P. A. Jenkins. 1996. Opportunist mycobacteria in England and Wales: 1982 to 1994. Commun. Dis. Rep. CDR Rev. 6:R147–R151.
21. Luquin, M., V. Ausina, F. Lopez Calahorra, F. Belda, M. Garcia Barcelo, C.
Celma, and G. Prats.1991. Evaluation of practical chromatographic
proce-dures for identification of clinical isolates of mycobacteria. J. Clin. Microbiol.
29:120–130.
22. Martinez Murcia, A. J., and F. Rodriguez Valera. 1994. The use of arbitrarily primed PCR (AP-PCR) to develop taxa specific DNA probes of known sequence. FEMS Microbiol. Lett. 124:265–269.
23. Portaels, F., L. Larsson, and P. A. Jenkins. 1995. Isolation of Mycobacterium
malmoense from the environment in Zaire. Tubercle Lung. Dis. 76:160–162.
24. Rogall, T., T. Flohr, and E. C. Bo¨ttger. 1990. Differentiation of
Mycobacte-rium species by direct sequencing of amplified DNA. J. Gen. Microbiol.
136:1915–1920.
25. Rogall, T., J. Wolters, T. Flohr, and E. C. Bo¨ttger. 1990. Towards a phylog-eny and definition of species at the molecular level within the genus
Myco-bacterium. Int. J. Syst. Bacteriol. 40:323–330.
26. Saito, H., H. Tomioka, K. Sato, H. Tasaka, and S. Dekio. 1994.
Mycobacte-rium malmoense isolated from soil. Microbiol. Immunol. 38:313–315.
27. Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
28. Soini, H., E. C. Bottger, and M. K. Viljanen. 1994. Identification of myco-bacteria by PCR-based sequence determination of the 32-kilodalton protein gene. J. Clin. Microbiol. 32:2944–2947.
29. Takewaki, S., K. Okuzumi, I. Manabe, M. Tanimura, K. Miyamura, K.
Nakahara, Y. Yazaki, A. Ohkubo, and R. Nagai.1994. Nucleotide sequence
comparison of the mycobacterial dnaJ gene and PCR-restriction fragment length polymorphism analysis for identification of mycobacterial species. Int. J. Syst. Bacteriol. 44:159–166.
30. Taylor, T. B., C. Patterson, Y. Hale, and W. W. Safranek. 1997. Routine use of PCR-restriction fragment length polymorphism analysis for identification of mycobacteria growing in liquid media. J. Clin. Microbiol. 35:79–85. 31. Torkko, P., M. Suutari, S. Suomalainen, L. Paulin, L. Larsson, and M. L.
Katila.1998. Separation among species of Mycobacterium terrae complex by
lipid analyses: comparison with biochemical tests and 16S rRNA sequencing. J. Clin. Microbiol. 36:499–505.
32. Tortoli, E., C. Piersimoni, A. Bartoloni, C. Burrini, A. P. Callegaro, G.
Caroli, D. Colombrita, A. Goglio, A. Mantella, C. P. Tosi, and M. T.
Simo-netti.1997. Mycobacterium malmoense in Italy: the modern Norman
inva-sion? Eur. J. Epidemiol. 13:341–346.
33. Valero-Guille´n, P., F. Martı´n-Luengo, L. Larsson, J. Jimenez, I. Juhlin, and
F. Portaels.1988. Fatty and mycolic acids of Mycobacterium malmoense.
J. Clin. Microbiol. 26:153–154.
34. Wayne, L. G., and H. A. Sramek. 1992. Agents of newly recognized or infrequently encountered mycobacterial diseases. Clin. Microbiol. Rev. 5:1– 25.
35. Zolg, J. W., and S. Philippi Schulz. 1994. The superoxide dismutase gene, a target for detection and identification of mycobacteria by PCR. J. Clin. Microbiol. 32:2801–2812.