Determination of Trace Elements in High-Purity Molybdenum
by Solid-Phase Extraction/ICP-MS
Shin-ichi Hasegawa, Hitoshi Yamaguchi, Katsura Yamada and Takeshi Kobayashi
National Institute for Materials Science, Tsukuba 305-0047, Japan
We attempted a simple pretreating method consisting of solid-phase extraction using bonded silica gel with benzenesulfonic acid (SCX) as the solid-phase sorbent to determine trace elements in pure molybdenum samples by means of inductively coupled plasma mass spectrometry (ICP-MS). Molybdenum was anionized by adding hydrogen peroxide solution to a sample decomposed with acid, and separated from cation trace impurities that had been kept in the chemically bonded silica gel of the ion-exchange type. The target elements retained in the solid phase were eluted with a small amount of dilute nitric acid. In this method, some trace elements, such as Be, Al, Mg, Cr, Mn, Fe, Co, Ni, Cu, Zn, Ga, Cd, In, Tl, Pb and Bi were determined by ICP-MS using the eluate. The detection limitations (3), as follows: Be 0.028, Al 2.64, Mg 1.9, Cr 0.20, Mn 0.13, Fe 3.85, Co 0.019, Ni 0.48, Cu 0.084, Zn 0.25, Ga 0.092, Cd 0.014, In 0.059, Tl: 0.027, Pb 0.044 and Bi 0.012 ng/g (ppb). (Received September 26, 2003; Accepted January 19, 2004)
Keywords: inductively coupled plasma mass spectrometry, solid-phase extraction, bonded silica as a solid-phase sorbent, beryllium, aluminum, magnesium, chromium, manganese, iron, cobalt, nickel, copper, zinc, gallium, cadmium, indium, thallium, lead and bismuth, trace analysis, pure molybdenum
1. Introduction
Molybdenum is used for functional materials such as electronic materials. Its high purification potential has been promoted to eliminate the impact of impurities contained in elements. Therefore, a trace analytical method for the impurities is strongly desired. So far, Glow Discharge-Mass
Spectrometry (GD-MS),1,2) Graphite Furnace-Atomic
Ab-sorption Spectrometry (GF-AAS),3–5) Inductively Coupled
Plasma-Atomic Emission Spectrometry (ICP-AES), and Inductively Coupled Plasma-Mass Spectrometry (ICP-MS) have been reported for determining quantities of trace elements contained in highly purified molybdenum. In principle, GD-MS allows quantification of almost all ele-ments from major ones to extremely small traces of impurities. The samples used in GD-MS are formed into pins or disks because this is a solid analysis. In contrast, in GF-AAS, ICP-AES and ICP-MS, the measured solutions are usually decomposed with acid or alkali. Although GF-AAS is widely used because of its super sensitivity, it is used for single elements. Therefore it requires a long time to determine quantities of multiple elements. On the other hand, ICP-AES can determine quantities of many elements simultaneously and is applicable to many elements. How-ever, when the matrix and molybdenum exist together in a sample solution, spectra-interference and physical interfer-ence occur and adversely affect the analyses. Molybdenum is
then separated by co-precipitation separation6,7) or
ion-exchange separation.4,5) ICP-AES has poorer detection
sensitivity than GF-AAS and ICP-MS. ICP-MS is used to determine many elements simultaneously and offers excel-lent in detection sensitivity. This method can also be applied to samples in which the matrix also exists. However, in that case, ion scattering or the space charge reduces sensitivity. Analyses are influenced by a clogging skimmer cone or memory effect when concentration of the matrix is high. For
that reason, pretreatments such as ion-exchange separation8)
are usually required to ameliorate those problems.
We succeeded in applying the solid phase extraction method as a pretreatment for ICP-MS: a chemically bonded silica gel was employed to extract chemicals for quantitative trace analyses of a small amount of the elements contained in
high-purity samples of both iron9,10)and aluminum.11,12)
This method is not harmful to humans or the environment like organic solutions; moreover, it is a very simple separation method. Therefore, we attempted to introduce the solid phase extraction method to this study using
chemically bonded silica gel to extract chemicals.13)
Separation by ion-exchange resin is usually included in a solid phase extraction method. However, chemically bonded silica gel was used to extract chemicals in this study. This extraction method enables rapid separation of a large amount of sample solutions with less use of extraction chemicals (0.25–1 g) because, compared to ion-exchange resin, this
type of silica gel has smaller granules (ca. 15–100mm)
offering greater surface area. Also, this material produces less abnormal absorption than ion exchange resin. Consequently, solutions can be separated with less eluent. In turn, that enables considerable enrichment. So far, chemically bonded silica gel has been used to separate and concentrate organic
compounds in various fields.14–18) However, this is the first
study to report application of this method to trace analyses of metallic ingredients.
2. Experiments
2.1 Equipment
The apparatuses used were ELAN 6000 for ICP-MS analysis (Perkin-Elmer, USA), 5100 PC for AAS analysis, and OPTIMA 3300DV for ICP-AES analysis (Perkin-Elmer, USA), to evaluate the conditions for separation and quantify residual molybdenum in the solutions measured. The light source of AAS was a hollow cathode molybdenum lamp (Perkin-Elmer, USA). The GL-SPE absorption manifold system (GL Science, Japan) was employed to perform solid extraction.
2.2 Reagents
For molybdenum solution (concentration: 50 kg/m3
(50 mg/ml), 25 g of the powdered molybdenum that was highly purified (99.99%: Spex Industries, USA or
Rareme-taric, Japan) was placed in a 500 cm3 beaker made from 4
ethylene fluoride resin (PTFE), and decomposed with 5 cm3
of nitric acid and 20 cm3 of hydrogen peroxide. After
cooling, the solution was moved to a 500 cm3 polyethylene
volumetric flask and diluted with water to a constant volume. The standard solution employed was composed of many elements, or the Custom Multi-element Standard XSTC-13 (Spex CertiPrep, USA)(Ag, Al, As, Ba, Be, Bi, Ca, Cd, Co, Cr, Cs, Cu, Fe, Ga, Hg, In, K, Li, Mg, Mn, Na, Ni, Pb, Rb, Se,
Sr, Th, Tl, V, Zn, U: 0.01 kg/m3 (10mg/ml) for each
element). Whenever it was used, each element was diluted to
0.005 kg/m3 with water (31-element mixed solution). The
Standard Solution for Atomic Absorption (Cica-Merck, Japan)(Be, Al, Mg, Cr, Mn, Fe, Co, Ni, Cu, Zn, Ga, Cd, In,
Tl, Pb and Bi: 1 kg/m3(1 mg/ml) for each element) was also
used, and each element was mixed and diluted to 0.005 kg/
m3 (16-element mixed solution). Residual molybdenum in
the solution measured was quantified using the molybdenum
solution (1 kg/m3), namely, the Standard Solution for
Atomic Absorption (Cica-Merck, Japan). Bond Elut SCX
(granule diameter 40mm: VARIAN, USA) was employed for
solid phase extraction chemicals; when it was used, a proper
quantity was injected into the 10 cm3 Bond Elut Reservoir
(VARIAN, USA). Polyethylene filters were installed on top and bottom of the extraction chemicals. The reagents used for the conditioning columns were sodium hydroxide (pro analysis, GR for analysis, Merck, Germany), acetonitrile of super-high grade reagent (Cica-Merck, Japan), and hydro-chloric acid of TAMAPURE-AA100 (Tama Kagaku, Japan). TAMAPURE-AA100 (Tama Kagaku, Japan) was also used
for hydrogen peroxide (35 mass%), nitric acid (68 mass%) and ammonia solution (20 mass%). The water used was refined by an apparatus for producing super-pure water, MILLI-Q SP TOC (Millipore, USA), and refined further using a distilled water production apparatus by a non-boiling technique (Fujiwara Seisakusho, Japan).
2.3 Experimental procedures
A molybdenum solution (50 kg/m3; 2 cm3) was removed
into a 100 cm3PTFE beaker and added to a 31-element mixed
solution or a 16-element mixed solution. It was diluted to
50 cm3with water prior to adjusting its pH with an ammonia
solution. Then it was passed through a conditioned solid-phase-extraction column (SCX) to maintain target elements in the extraction chemicals. After rinsing with water several times, target elements were separated by letting a dilute nitric acid eluent pass through the extraction chemicals. Then, several rinses were performed. The eluent and rinse water were moved into a volumetric flask and kept at constant
volume of 100 cm3. This solution was measured for the
conditions shown in Table 1 by ICP-AES or ICP-MS. Residual molybdenum of this solution was measured for the conditions shown in Table 1 by AAS.
Concentrations of the elements were calculated by a calibration curve after several blank solutions obtained were prepared through the experimental procedures. To each blank
solution was added 0–1 cm3of the 16-element mixed solution
(0.005 kg/m3for each element), stage-by-stage. Then it was
removed to a 100 cm3 volumetric flask maintaining constant
volume.
Extraction chemicals were subjected to conditioning. First,
5 cm3acetonitrile and water of 10 cm3were poured out; this
was repeated three times. Subsequently, 15 cm3of 0.1 kmol/
[image:2.595.44.562.508.789.2]m3 sodium hydroxide solution, together with 10 cm3 water,
Table 1 Operating conditions of AAS, ICP-AES and ICP-MS. AAS conditions
Wavelength (nm) Mo (313.3)
Light source 30 mA (Hollow cathode lamp)
Spectral band-width 0.7 nm
Oxidant gas (N2O) 6.0 l min1
Fuel gas (C2H2) 7.8 l min1
ICP-AES conditions
RF Power 1.3 kW
Plasma gas (Ar) 15.0 l min1
Auxiliary gas (Ar) 0.5 l min1
Carrier gas (Ar) 0.8 l min1
Wavelength (nm) Be(313.107), Mg(279.077), Al(308.215), Cr(283.563), Mn(257.610), Fe(238.204), Co(228.616), Ni(221.648), Cu(324.752), Zn(213.857), Ga(294.364), Cd(214.440), In(230.606), Tl(190.801), Pb(220.353), Bi(223.061) ICP-MS conditions
RF Power 1.0 kW
Plasma gas (Ar) 15.0 l min1
Auxiliary gas (Ar) 1.2 l min1
Carrier gas (Ar) 0.68 l min1
then 15 cm3 of 0.1 kmol/m3 hydrochloric acid, and finally
10 cm3 water were poured onto the chemicals.
3. Results and Discussion
3.1 Effect of pH at separation
Molybdenum was anionized and could not be retained in the solid-phase extraction chemicals of the cation exchange because the sample was decomposed with hydrochloric acid and nitric acid and then added to a hydrogen peroxide solution. In contrast, as most impurities in the sample were cations, they were retained with the extraction chemicals. We separated them under optimum conditions using a 31-element mixed solution; then we measured them by ICP-MS. Among the 18 target elements — Ag, Be, Al, Mg, Cr, Mn, Fe, Co, Ni, Cu, Zn, Ga, Cd, In, Tl, Pb, V, and Bi — recovery rates of over 90% were obtained from 16 of them (Be, Al, Mg, Cr, Mn, Fe, Co, Ni, Cu, Zn, Ga, Cd, In, Tl, Pb, and Bi). Optimum conditions to separate the 16 target elements are described below.
The molybdenum solution (50 kg/m3) of 2 cm3 and the
10 cm3 of the 16-element solution, which were adjusted
previously, were moved into PTFE beakers of 8 (A). The molybdenum solution was put into another eight beakers by itself (B). These solutions were adjusted to pH 0.01, 0.1, 0.2, 0.3, 0.5, 1.0, 1.5, 2.0 in turn, using nitric acid or ammonia solution. They were passed through the solid phase extraction columns that had been conditioned to retain the target elements; then the columns were rinsed several times. The eluent was then passed through the columns and several rinses were conducted. Before the extraction chemicals or cation exchange-type SCX of 0.5 g were used, they were
injected into a 10 cm3Bond Elut Reservoir. The eluent used
was 10 cm3of 2 kmol/m3nitric acid. The eluent and the rinse
water were transferred to a volumetric flask for constant
volume of 100 cm3 (A0) (B0). Meanwhile, the acid and
hydrogen peroxide solution were put into another beaker, adjusted to pH 2.0 with ammonia solution, and then subjected to solid-phase extraction following the procedures. The eluent and the rinse water obtained were added to a
16-element mixed solution of 10 cm3and diluted to 100 cm3(C).
After these solutions were measured by ICP-AES, the recovery rate in each pH area was evaluated based on the
measurement resultfðA0B0Þ=C100g, as shown in Fig. 1.
The recovery rate of Cr was 94% at pH 1.0–3.5, but the other elements exhibited recovery rates of almost 100% at pH 1.5– 2.0. As a result, pH at separation was determined as 2.0. Residual molybdenum in these solutions was quantified in each pH area by AAS. Results indicated that molybdenum of
approximately 0.0001 kg/m3 was observed in all the areas
shown in the figure, suggesting that this amount would have no effect on ICP-MS measurement.
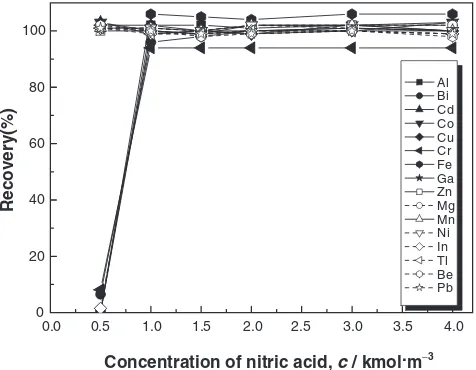
3.2 Concentration of eluent
We examined the eluent for separating the target elements retained in the extraction chemicals. Nitric acid selected for the eluent, considering the impact on ICP-MS, was evaluated in its optimum concentration and the amount used. The 16-element mixed solution was added to the molybdenum
solution moved to a 100 cm3PTFE beaker and adjusted to pH
2.0 using ammonia water. Six molybdenum solutions prepared with this procedure were passed through the solid phase extraction columns (SCX 0.5 g) to retain target elements in the solid phase. After several rinses with water,
the target elements were separated by passing 10 cm3 nitric
acid, which was adjusted to 0.5, 1.0, 1.5, 2.0, 3.0, and 4 kmol/
m3, through the six columns. Several water rinses were
performed; then the eluent and the rinse water were moved to
a 100 cm3 volumetric flask for constant volume. Other
solutions were also prepared: the eluent that separated only
molybdenum and the solution of 100 cm3that extracted only
acid and hydrogen peroxide solution and to which was added the 16-element mixed solution. These solutions were meas-ured by ICP-AES, and the relationship between the concen-trations of the eluents and recovery rates was evaluated, as shown in Fig. 2. All the elements separated with 1.5–4 kmol/
m3eluent exhibited recovery rates of 94–106%. As a result,
2 kmol/m3nitric acid of 10 cm3was selected as the eluent.
3.3 Selection and amount of solid-phase extraction chemicals
Chemically bonded silica gel used for solid-phase-extrac-tion chemicals is classified into three types based on its
0.0 0.5 1.0 1.5 2.0
0 20 40 60 80 100
Concentration of molybdenum,
c / 10 − 3kg . m − 3 0 0.2 0.4 0.6 0.8 1.0 Al Bi Cd Co Cu Cr Fe Ga Zn Mg Mn Ni In Tl Be Pb Mo Recovery(%) pH
Fig. 1 Effect of pH on the recovery of analytes.
0.0 1.0 2.0 3.0 4.0
0 20 40 60 80 100
Concentration of nitric acid, c / kmol.m−3
Recovery(%) Al Bi Cd Co Cu Cr Fe Ga Zn Mg Mn Ni In Tl Be Pb
0.5 1.5 2.5 3.5
[image:3.595.310.546.74.233.2] [image:3.595.309.547.581.766.2]functional group: octadecyl and octyl in the reversed phase type; cyanopropyl and diol in the normal phase type; and benzylsulfonic acid and trimethylaminopropyl in the ion exchange type. Molybdenum is anionized, forming oxoacid in the presence of hydrogen peroxides. Silica gel, employed to retain only target cation elements in solid-phase-extraction chemicals in this study, was the chemically bonded type. Alternatively, we used Bond Elut SCX belonging to the cation exchange type of the benzylsulfonic acid functional group. Because the amount of the extraction chemicals is related to the retention ability and treatment time of the target elements, the optimum amount used in solid-phase extraction was examined at the levels of 0.25, 0.5, 0.75, and 1.0 g. A sample of 0.100 g contains a very small quantity of target elements: 0.25 g of the extraction chemicals demonstrated that it is possible to attain the goal. However, under the assumption that a sample contains large quantities of target elements, the amount of solid-phase-extraction chemicals used was modified to 0.5 g.
3.4 Detection limit
The 16-element mixed solution (10 cm3) was added to the
blank solution and solid-phase extraction was performed. The detection limit was calculated as three times the standard
deviation () of ion intensity (n¼10) for the mass of each
element of the background, in terms of concentration.
Considering that the amount of eluent used was 10 cm3 of
2 kmol/m3 nitric acid, based on 3.2, the measured solution
can be enriched. Therefore, the lower limit of quantification can be improved.
4. Results of Quantitative Analyses
4.1 Analytical methods
Prior to heating and decomposition of the 0.100 g sample, a
small amount of water, 5 cm3 hydrochloric acid, and 5 cm3
nitric acid were added in a 100 cm3 beaker (PTFE). Heating
was continued until the acids and water were vaporized and the sample was completely dried. Salts were dissolved with
heating after the sample was cooled and 10 cm3 hydrogen
peroxide was added. The sample was cooled again, diluted to
50 cm3with water and adjusted to pH 2.0 using an ammonia
solution. The solution was passed through a solid-phase-extraction column that was conditioned to retain the target elements in the extraction chemicals. The solid-phase-extraction chemicals were rinsed several times and the rinse
water was disposed of. The eluent, 10 cm3 of 2 kmol/m3
nitric acid, was then passed through the extraction column to separate the target elements. After several rinses with water, the eluent and rinse water were moved to a volumetric flask
to maintain a constant volume of 100 cm3. The solution
introduced to ICP-MS was measured for conditions shown in Table 1. Concentrations of the target elements were calcu-lated using a calibration curve. No sample was used to produce the calibration curve, but the 16 element-mixed
solution of 0–10 cm3was added to a blank solution obtained
through the stage-by-stage procedures; then the mixture was
diluted to a constant volume of 100 cm3.
4.2 Results of analyses
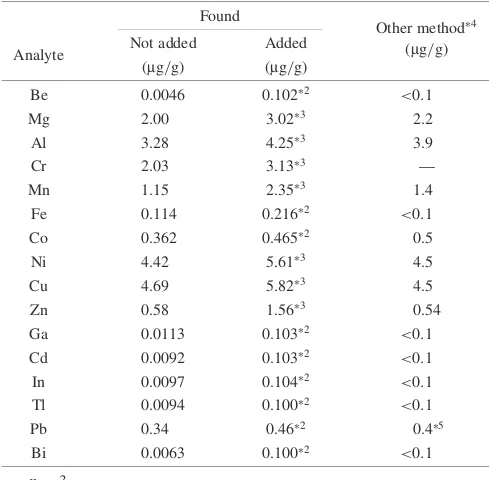
Highly purified molybdenum (99.99%; Spex Industries Inc., USA) was quantified by this method. The value of molybdenum was compared with those of the molybdenum
samples produced by adding either 0.01mg or 0.1mg of each
element to molybdenum because there was no known quantitative value for the element in this sample. Table 2 presents results and their quantitative values, along with those for the above-mentioned samples that were analyzed by the other methods (GF-AAS and ICP-AES). Satisfactory results were obtained in quantitative values of the molybde-num samples to which each analytical element was added.
5. Conclusion
To establish a rapid trace-quantification scheme for elements contained in highly purified molybdenum, we studied the most suitable conditions for separating the elements by the solid-phase-extraction method, as a pretreat-ment for ICP-MS. We employed chemically bonded silica gels as extraction chemicals. They belonged to the functional group of benzylsulfonic acid. The target elements retained in
the chemicals were eluted with 10 cm3 of 2 kmol/m3 nitric
acid. Quantities of the obtained target elements were determined by ICP-MS.
In conclusion, highly sensitive quantification was estab-lished for the 16 trace elements in highly purified molybde-num, or Be, Al, Mg, Cr, Mn, Fe, Co, Ni, Cu, Zn, Ga, Cd, In,
Tl, Pb and Bi, with the following detection limitations (3):
[image:4.595.304.549.94.334.2]Be 0.028, Al 2.64, Mg 1.99, Cr 0.20, Mn 0.13, Fe 3.85, Co 0.019, Ni 0.48, Cu 0.084, Zn 0.25, Ga 0.092, Cd 0.014, In 0.059, Tl: 0.027, Pb 0.044, and Bi 0.012 ng/g (ppb).
Table 2 Results of the recovery of analytes in high purity molybdenum by ICP-MS.
Found
Other method4
Analyte Not added Added (mg/g)
(mg/g) (mg/g)
Be 0.0046 0.1022 <0:1
Mg 2.00 3.023 2.2
Al 3.28 4.253 3.9
Cr 2.03 3.133 —
Mn 1.15 2.353 1.4
Fe 0.114 0.2162 <0:1
Co 0.362 0.4652 0.5
Ni 4.42 5.613 4.5
Cu 4.69 5.823 4.5
Zn 0.58 1.563 0.54
Ga 0.0113 0.1032 <0:1
Cd 0.0092 0.1032 <0:1
In 0.0097 0.1042 <0:1
Tl 0.0094 0.1002 <0:1
Pb 0.34 0.462 0.45
Bi 0.0063 0.1002 <0:1
n¼3
1SpeX molybdenum powder.
2analytes added 0.01mg (equivalent to the 0.1m/g in a sample).
3analytes added 0.1mg (equivalent to the 1mg/g in a sample).
4Ref. 5), determined by ICP-AES or GF-AAS.
REFERENCES
1) T. Mizota, T. Nakamura and K. Iwasaki: BUNSEKI KAGAKU41
(1992) 425–431.
2) M. Saito, F. Hirose and H. Okochi: Anal. Sci.11(1995) 695–697. 3) S. Hasegawa, T. Kobayashi, K. Ide, H. Okochi and R. Hasegawa: J.
Japan Inst. Metals58(1994) 23–29.
4) H. Yamaguchi, T. Kobayashi, K. Yamada and H. Okochi: BUNSEKI KAGAKU39(1990) 19–24.
5) F. Mogi, K. Itoh, N. Okamoto, M. Narita and M. Fujine: Denki Seiko59
(1988) 263–270.
6) O. Kujirai, M. Kori, K. Yamada and H. Okochi: Anal. Sci.6(1990) 379–383.
7) O. Kujirai, K. Yamada, M. Kori and H. Okochi:Fresenius’ Z. Anal. Chem.339(1991) 133–136.
8) T. Kawamura: BUNSKI KAGAKU37(1988) 585–588.
9) S. Hasegawa, T. Kobayashi, K. Sato, S. Igarashi and K. Naito: J. Japan
Inst. Metals63(1999) 1497–1502.
10) S. Hasegawa, K. Ide, T. Kobayashi, K. Sato, S. Igarashi and K. Naito: Tetsu-to-Hagane89(2003) 958–961.
11) S. Hasegawa, K. Sato, K. Ide, T. Kobayashi, S. Igarashi and K. Naito: J. Japan Inst. Metals64(2000) 1212–1217.
12) S. Hasegawa, H. Yamaguchi, S. Itoh, K. Ide and T. Kobayashi: BUNSEKI KAGAKU52(2003) 533–537.
13) K. C. Van Horne: Sorbent extraction technology, (Analytichem International, Inc., Harbor City, 1985).
14) G. Lensmeyer: Clin. Chem.31(1985) 196–201. 15) P. Kao: Clin. Chem.30(1984) 56–61.
16) G. Schieffer, G. Wheeler and C. Cimino: J. Liquid Chromato.7(1984) 2659–2669.
17) M. D. Mu¨ller: Anal. Chem.59(1987) 617–623.