Copyright © 2004, American Society for Microbiology. All Rights Reserved.
Identification of Clinically Relevant Viridans Group Streptococci by
Sequence Analysis of the 16S-23S Ribosomal DNA Spacer Region
Chao Chien Chen,
1Lee Jene Teng,
2and Tsung Chain Chang
3*
Department of Medical Technology, Buddhist Tzu Chi General Hospital, Hualien,1School ofMedical Technology, National Taiwan University College of Medicine, Taipei,2and Department of Medical Technology, School of Medicine, National Cheng Kung University, Tainan,3Taiwan, Republic of China
Received 15 August 2003/Returned for modification 15 December 2003/Accepted 2 March 2004
The feasibility of sequence analysis of the 16S-23S ribosomal DNA (rDNA) intergenic spacer (ITS) for the identification of clinically relevant viridans group streptococci (VS) was evaluated. The ITS regions of 29
reference strains (11 species) of VS were amplified by PCR and sequenced. These 11 species wereStreptococcus
anginosus,S. constellatus,S. gordonii,S. intermedius,S. mitis,S. mutans,S. oralis,S. parasanguinis,S. salivarius,
S. sanguinis, andS. uberis. The ITS lengths (246 to 391 bp) and sequences were highly conserved among strains within a species. The intraspecies similarity scores for the ITS sequences ranged from 0.98 to 1.0, except for
the score forS. gordoniistrains. The interspecies similarity scores for the ITS sequences varied from 0.31 to
0.93. Phylogenetic analysis of the ITS regions revealed that evolution of the regions of some species of VS is not parallel to that of the 16S rRNA genes. One hundred six clinical isolates of VS were identified by the Rapid ID 32 STREP system (bioMe´rieux Vitek, Marcy l’Etoile, France) and by ITS sequencing, and the level of disagreement between the two methods was 18% (19 isolates). Most isolates producing discrepant results could be unambiguously assigned to a specific species by their ITS sequences. The accuracy of using ITS sequencing
for identification of VS was verified by 16S rDNA sequencing for all strains except strains ofS. oralisandS.
mitis, which were difficult to differentiate by their 16S rDNA sequences. In conclusion, identification of species of VS by ITS sequencing is reliable and could be used as an alternative accurate method for identification of VS.
Viridans group streptococci (VS) are the most common eti-ologic agents of subacute infective endocarditis and are capa-ble of causing a variety of pyogenic infections (9). VS can be divided into five major groups on the basis of the 16S ribo-somal DNA (rDNA) sequences: (i) the mutans group, (ii) the salivarius group, (iii) the anginosus group (also called the mil-leri group), (iv) the sanguinis group, and (v) the mitis group (9).
VS can be isolated as part of the normal flora of the respi-ratory, genital, and alimentary tracts. However, species of VS are playing an increasing role in infections among immuno-compromised patients (33). The clinical significance of VS may differ between species, and sometimes it is important to iden-tify the individual species associated with diseases (18). Jacobs et al. (19) studied 104 isolates of VS recovered from blood
cultures and found thatStreptococcus oralisand S. mitiswere
the two species most frequently isolated from patients in the hematology unit, whereas species of the anginosus group were the most common species isolated from the general hospital population. Chang et al. (7) found that VS accounted for 9% of all cases of culture-proven bacterial meningitis in adults, and more than 80% of VS causing meningitis were species of the anginosus group. These data provided further insight into the search for the link between species and the diseases that they can cause.
The antimicrobial susceptibilities of VS isolated from
pa-tients with significant infections in Taiwan revealed that
high-level penicillin resistance (MICs ⱖ 4 g/ml) was frequently
found in isolates ofS. oralis(35%) andS. mitis(20%).
Mac-rolide resistance also occurred most frequently in S. oralis
isolates (55%) but in none ofS. mutansisolates (35). However,
Tracy et al. (38) found that there were no differences in
anti-microbial susceptibilities among species (S. anginosus,S.
con-stellatus, and S. intermedius) of the anginosus group. Those investigators expressed the opinion that it was unnecessary to identify VS to the species level and that identification of an infecting organism as a member of the anginosus group might be sufficient for patient management, but whether the same can be said for other VS remains to be established (38).
Nearly 40 conventional tests have been used to differentiate species of VS, but phenotypic tests do not allow an unequivocal identification of some species of this heterogeneous group of bacteria (1, 10, 23). This is because variability in a common phenotypic trait among strains is common (4, 17, 22). Species of S. mutans strains and strains of the anginosus and mitis groups are the most problematic to differentiate (11, 18, 32).
Several molecular methods have been developed for the identification of VS to the species level (5, 11, 12, 18, 21, 29, 36). The targets include the rRNA genes (6, 8, 18, 20, 30), the tRNA gene intergenic spacer (ITS) (12), and the genes
encod-ingD-alanine–D-alanine ligase (11), manganese-dependent
su-peroxide dismutase (sodAint) (26), and heat shock proteins
(groESL) (34). A variety of techniques, including DNA hybrid-ization (18, 21, 30, 32), PCR product length polymorphism analysis (12), arbitrarily primed PCR (29), and comparative DNA sequence analysis, have been used for molecular diag-nosis (6, 8, 26, 36). Compared with the lengths of 16S rDNA
(⬇1.5 kb) (27) andgroESL(⬇2 kb) (36), the ITS lengths of VS
* Corresponding author. Mailing address: Department of Medical Technology, School of Medicine, National Cheng Kung University, 1 University Rd., Tainan 701, Taiwan, Republic of China. Phone: 886-6-2353535, ext. 5790. Fax: 886-6-2363956. E-mail: [email protected] .edu.tw.
2651
on May 15, 2020 by guest
http://jcm.asm.org/
are relatively short (⬍400 bp). In addition,S. mitisandS. oralis
cannot be differentiated by analyzing the groESL (36) and
sodAintgenes (26), since intraspecies sequence variations are
higher than interspecies variations.
The ITS separating the 16S and 23S rDNAs has been sug-gested to be a good candidate for use in molecular assays for bacterial species identification (3, 39) and strain typing (15). Sequences of the ITS regions have been found to have low levels of intraspecies variation and high levels of interspecies divergence (40). The aim of this study was to investigate the feasibility of using the ITS sequences to identify 11 clinically relevant species of VS.
MATERIALS AND METHODS
Bacterial strains. Twenty-nine type or reference strains of VS, includingS. anginosus,S. constellatus,S. gordonii,S. intermedius,S. mitis,S. mutans,S. oralis,
S. parasanguinis,S. salivarius,S. sanguinis, andS. uberis, were used in this study (Table 1). Most of these strains were obtained from the American Type Culture Collection (ATCC; Manassas, Va.).S. intermediusNCDO 2227 was obtained from the National Collection of Food Bacteria, Reading, United Kingdom. In addition, type strains ofS. agalactiae(ATCC 13813),S. pneumoniae(ATCC 33400),S. pyogenes(ATCC 14289), andEnterococcus faecalis(ATCC 19433) and two reference strains ofS. pneumoniae(ATCC 27336 and ATCC 49169) were also used for comparison of their ITS sequences with those of VS. A total of 106 clinical isolates of VS were obtained from National Taiwan University Hospital (Taipei, Taiwan) and National Cheng Kung University Medical Center (Tainan, Taiwan). Most of the clinical isolates were recovered from blood cultures and deep abscesses (e.g., brain abscesses) and were phenotypically identified with the Rapid ID 32 STREP system (bioMe´rieux Vitek, Marcy l’Etoile, France).
DNA preparation.The boiling method described by Millar et al. (25) was used to extract DNA from the bacteria. Briefly, one colony from pure cultures was suspended in 20l of sterilized water and heated at 100°C for 15 min in a heating block. After centrifugation in a microcentrifuge (6,000⫻g for 3 min), the supernatant containing bacterial DNA was stored at⫺20°C for further use.
Amplification of ITS regions and nucleotide sequence determination.The bacterium-specific universal primers 13BF (5⬘-GTGAA TACGT TCCCG GGC CT-3⬘) (27) and 6R (5⬘-GGGTT YCCCC RTTCR GAAAT-3⬘) (15) (where Y is C or T and R is A or G) were used to amplify a DNA fragment that encompassed a small portion of the 16S rDNA region, the ITS, and a small portion of the 23S rDNA region. The 5⬘end of primer 13BF is located at position 1371 of the 16S rDNA, and the 5⬘end of primer 6R is located at position 108 downstream of the 5⬘end of the 23S rDNA (Escherichia colinumbering). PCR was performed with 5l (1 to 5 ng) of template DNA in a total reaction volume of 50l consisting of 10 mM Tris-HCl (pH 8.3), 50 mM KCl, 1.5 mM MgCl2, 0.8 mM deoxyribonucleoside triphosphates (0.2 mM each), 0.1M (each) primer, 1 U ofTaqDNA polymerase, and 50l of a mineral oil overlay. The PCR program consisted of 8 cycles of denaturation (94°C for 2 min), annealing (55°C for 1 min), and extension (72°C for 1 min), followed by 30 cycles of denaturation (94°C for 1 min), annealing (60°C for 1 min), and extension (72°C for 1 min) and a final extension step at 72°C for 3 min. An OmniGen thermal cycler (Hybaid Limited, Winsford, United Kingdom) was used for PCR.
The PCR products were purified with a PCR-M Clean Up kit (Viogene, Taipei, Taiwan) and were sequenced on a model 377 sequencing system (Applied Biosystems, Taipei, Taiwan) with a BigDye Terminator Cycle Sequencing kit (version 3.1; Applied Biosystems). After sequencing of the PCR products, por-tions of the 16S and 23S rDNA regions were removed to obtain the full ITS sequences. For all VS studied, the 5⬘ends of the ITS sequences were CTAAGG, whereas the 3⬘ends of the ITS sequences were AATAA. The similarity score for the ITS sequence of a strain was obtained by comparing its sequence with that of the type strain of the same species, and the PileUp command of the Wisconsin Genetics Computer Group (GCG) package (version 10.3; Accelrys Inc., San Diego, Calif.) was used for calculation. The PrettyBox program of the GCG package was used to align multiple ITS sequences.
[image:2.603.300.540.96.621.2]Phylogenetic analysis.Multiple-sequence alignment of the ITS sequences was carried out with the CLUSTAL X program (version 1.63b) (37), and an unrooted phylogenetic tree was constructed by using the neighbor-joining method (31) listed in the MEGA (molecular evolutionary genetic analysis; version 2.1) ana-lytical software package (24). For neighbor-joining analysis, the distance between the sequences was calculated by using Kimura’s two-parameter model. The
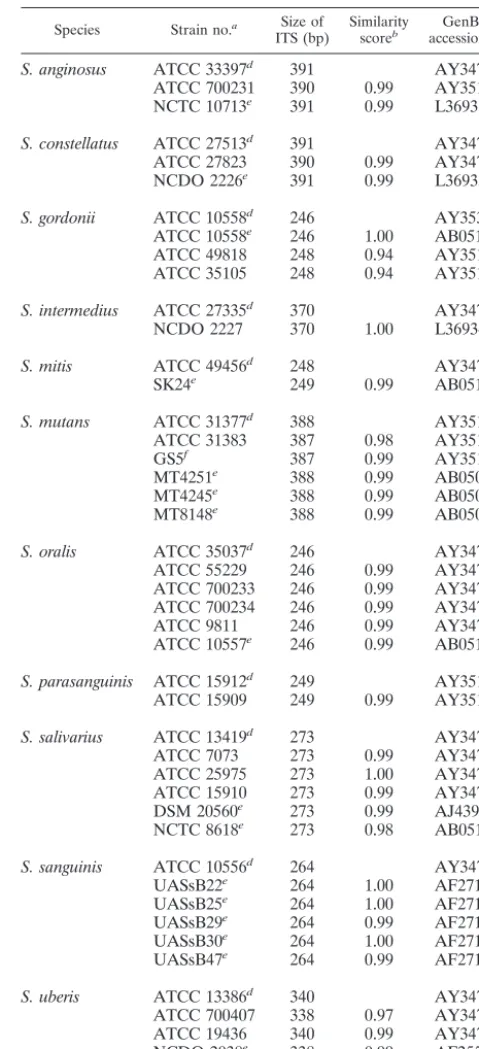
TABLE 1. Intraspecies similarity scores for the 16S-23S rDNA ITSs of different species of VS
Species Strain no.a Size of
ITS (bp) Similarityscoreb accession no.GenBank c
S. anginosus ATCC 33397d 391 AY347541
ATCC 700231 390 0.99 AY351328 NCTC 10713e 391 0.99 L36931
S. constellatus ATCC 27513d 391 AY347545
ATCC 27823 390 0.99 AY347546 NCDO 2226e 391 0.99 L36933
S. gordonii ATCC 10558d 246 AY353081
ATCC 10558e 246 1.00 AB051019 ATCC 49818 248 0.94 AY351328 ATCC 35105 248 0.94 AY351326
S. intermedius ATCC 27335d 370 AY347549
NCDO 2227 370 1.00 L36934
S. mitis ATCC 49456d 248 AY347550
SK24e 249 0.99 AB051020
S. mutans ATCC 31377d 388 AY351323
ATCC 31383 387 0.98 AY351324
GS5f 387 0.99 AY351312
MT4251e 388 0.99 AB050990
MT4245e 388 0.99 AB050988
MT8148e 388 0.99 AB050989
S. oralis ATCC 35037d 246 AY347551
ATCC 55229 246 0.99 AY347553 ATCC 700233 246 0.99 AY347554 ATCC 700234 246 0.99 AY347555
ATCC 9811 246 0.99 AY347568
ATCC 10557e 246 0.99 AB051018
S. parasanguinis ATCC 15912d 249 AY351320
ATCC 15909 249 0.99 AY351319
S. salivarius ATCC 13419d 273 AY347562
ATCC 7073 273 0.99 AY347561
ATCC 25975 273 1.00 AY347564 ATCC 15910 273 0.99 AY347563 DSM 20560e 273 0.99 AJ439458 NCTC 8618e 273 0.98 AB051016
S. sanguinis ATCC 10556d 264 AY347565
UASsB22e 264 1.00 AF271353
UASsB25e 264 1.00 AF271355
UASsB29e 264 0.99 AF271351
UASsB30e 264 1.00 AF271354
UASsB47e 264 0.99 AF271352
S. uberis ATCC 13386d 340 AY347566
ATCC 700407 338 0.97 AY347567 ATCC 19436 340 0.99 AY347538 NCDO 2038e 338 0.99 AF255657 ATCC 27958e 340 0.99 U39768
aDSM, Deutsche Sammlung von Mikroorganismen, Braunschweig, Germany;
NCDO, National Collection of Food Bacteria, Reading, United Kingdom; NCTC, National Collection of Type Cultures, Public Health Laboratory, Lon-don, United Kingdom.
bThe type strains were used as the basis for calculation of similarity scores. cThe ITS sequences (GenBank accession numbers) ofS. agalactiaeATCC
13813 (AY347539), S. pneumoniaeATCC 33400 (AY347557), ATCC 27336 (AY347556), ATCC 49169 (AY347558),S. pyogenesATCC 14289 (AY347560), andE. faecalisATCC 19433 (AY351321) were also submitted to GenBank.
dType strain.
eStrains for which ITS sequences are available in the GenBank database. fClinical isolate.
on May 15, 2020 by guest
http://jcm.asm.org/
robustness of the neighbor-joining method was statistically evaluated by boot-strap analysis with 1,000 bootboot-strap samples.
Identification of clinical isolates of VS by ITS sequencing.A total of 106 clinical isolates were tested to evaluate the feasibility of using ITS sequencing for VS identification. These isolates were first identified with the Rapid ID 32 STREP system (bioMe´rieux Vitek). The ITS sequence of a clinical isolate was compared to those of the type strains (Table 1), and a species name was given according to the highest similarity score obtained. Clinical isolates for which the Rapid ID 32 STREP system and ITS sequence analysis produced discrepant identifications were further identified according to their 16S rDNA sequences.
Species identification by 16S rDNA sequencing.Primer pair 8FPL (5⬘-GTTT GATCCTGGCTCAG-3⬘) and 1492RPL (5⬘-GGTTACCTTGTTACGACTT-3⬘) was used for PCR amplification (27) of the nearly complete length of the 16S rDNA of clinical isolates for which the Rapid ID 32 STREP system and ITS sequencing produced discrepant identifications. The amplicons were cycle se-quenced in both directions by using the two primers described above and an additional primer, primer 1055r (5⬘-CACGAGCTGACGACAGCCAT-3⬘). The sequences determined were compared to known 16S rRNA gene sequences in the National Center for Biotechnology Information database by using the BLAST algorithm.
Nucleotide sequence accession numbers.The ITS sequences of 29 reference strains and 1 clinical isolate (a total of 30 sequences) of VS and several strep-tococcal species were submitted to GenBank. The accession numbers are listed in Table 1.
RESULTS
Amplification and sequencing of the ITS fragments. The ITS fragments of 11 species (29 reference strains) of clinically rel-evant VS were amplified by PCR with primers 13BF and 6R. A single amplicon was observed for each species (Fig. 1). After sequencing and removal of the partial 16S and 23S rDNA portions, the exact lengths and sequences of the ITSs were
obtained. S. oralis had the shortest ITS fragment (246 bp),
followed byS. mitis(248 to 249 bp), whereasS. constellatusand
S. anginosusyielded the longest ITS fragments (390 to 391 bp). Intraspecies variations in ITS lengths were within 3 bp (Table 1).
Sequence similarities of ITS regions. Generally, the
in-traspecies similarity scores were very high, ranging from 0.97 to
1.0 (Table 1). However, an exception was found forS. gordonii;
two of the three reference strains (strains ATCC 49818 and ATCC 35105) had similarity scores of only 0.94 each when their sequences were compared with that of the type strain (ATCC 10558). In addition to the 11 species of VS, the ITS accession numbers of several commonly encountered
strepto-coccal species and E. faecalisare also listed in footnotec of
Table 1. Some ITS sequences of VS available in GenBank are also included in Table 1 to facilitate intraspecies comparisons. Table 1 demonstrates the high degree of intraspecies conser-vation of both the lengths and the sequences of the ITS re-gions.
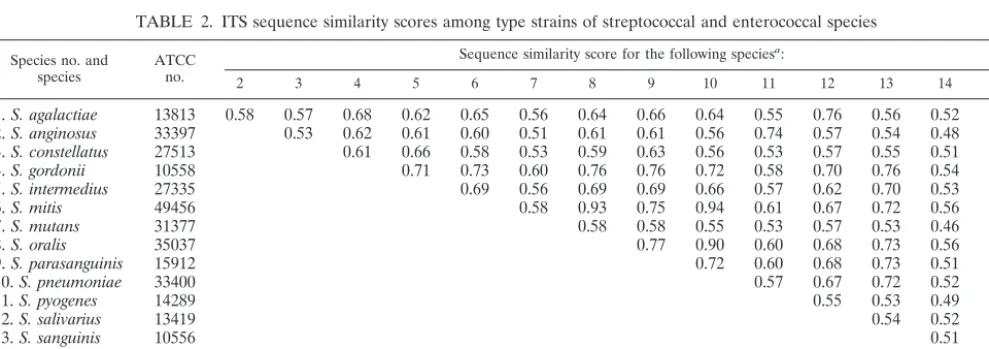
Table 2 shows the results of pairwise comparisons of the ITS sequences between type strains of any two given species. In
addition to the 11 species of VS, type strains ofS. agalactiae,S.
pyogenes,S. pneumoniae, andE. faecaliswere also included for comparison. Table 2 reveals a high level of interspecies diver-gence of the ITS sequences. Among the type strains of VS, the
similarity scores ranged from 0.51 (S. mutans versusS.
angino-sus) to 0.93 (S. mitisversusS. oralis). However, a high similarity
score (0.94) between type strains ofS. pneumoniaeandS. mitis
was also observed (Table 2). In general, the interspecies ITS similarity scores were less than 0.80.
Table 3 shows the results of intra- and interspecies
compar-FIG. 1. Amplification of VS type strains with primers 13BF and 6R and separation of the PCR products by 2% agarose gel electrophore-sis. Lanes: 1,S. mitis; 2,S. oralis; 3,S. constellatus; 4,S. intermedius; 5,
[image:3.603.45.282.70.181.2]S. salivarius; 6,S. anginosus; 7,S. mutans; 8,S. sanguinis; 9,S. gordonii; 10,S. parasanguis; 11,S. uberis; M, 100-bp ladder.
TABLE 2. ITS sequence similarity scores among type strains of streptococcal and enterococcal species
Species no. and
species ATCCno.
Sequence similarity score for the following speciesa:
2 3 4 5 6 7 8 9 10 11 12 13 14 15
1.S. agalactiae 13813 0.58 0.57 0.68 0.62 0.65 0.56 0.64 0.66 0.64 0.55 0.76 0.56 0.52 0.35
2.S. anginosus 33397 0.53 0.62 0.61 0.60 0.51 0.61 0.61 0.56 0.74 0.57 0.54 0.48 0.35
3.S. constellatus 27513 0.61 0.66 0.58 0.53 0.59 0.63 0.56 0.53 0.57 0.55 0.51 0.36
4.S. gordonii 10558 0.71 0.73 0.60 0.76 0.76 0.72 0.58 0.70 0.76 0.54 0.32
5.S. intermedius 27335 0.69 0.56 0.69 0.69 0.66 0.57 0.62 0.70 0.53 0.34
6.S. mitis 49456 0.58 0.93 0.75 0.94 0.61 0.67 0.72 0.56 0.36
7.S. mutans 31377 0.58 0.58 0.55 0.53 0.57 0.53 0.46 0.35
8.S. oralis 35037 0.77 0.90 0.60 0.68 0.73 0.56 0.35
9.S. parasanguinis 15912 0.72 0.60 0.68 0.73 0.51 0.35
10.S. pneumoniae 33400 0.57 0.67 0.72 0.52 0.35
11.S. pyogenes 14289 0.55 0.53 0.49 0.33
12.S. salivarius 13419 0.54 0.52 0.36
13.S. sanguinis 10556 0.51 0.37
14.S. uberis 13386 0.34
15.E. faecalis 19433
aThe species numbers correspond to the species numbers identified on the left.
on May 15, 2020 by guest
http://jcm.asm.org/
[image:3.603.48.543.542.716.2]isons of the ITS sequences between strains ofS. mitis,S. oralis, andS. pneumoniae. It was noted thatS. pneumoniaecould not
be differentiated fromS. mitisby the similarity scores for the
ITS sequences. For example, the similarity score between the
type strains ofS. pneumoniaeATCC 33400 andS. mitisATCC
49456 (0.99) was higher than the intraspecies similarity score
betweenS. mitisATCC 49456 andS. mitisSK24 (0.98).
The ITS sequences ofS. mitisand S. oralisalso have high
similarity scores. The intraspecies similarity scores for the ITS
regions ranged from 0.98 to 1.0 and from 0.99 to 1.0 forS. mitis
and S. oralisstrains, respectively (Table 3). However, the in-terspecies similarity scores between strains of the two species (0.93 to 0.95) were consistently lower than those observed for
strains within each species. Therefore,S. mitisstrains could be
effectively differentiated from S. oralisstrains on the basis of
their ITS sequences. Moreover, it was interesting that all six
reference strains ofS. oralishad a single deletion at ITS
nu-cleotide positions 199 and 217 ofS. mitis; however, the
nucle-otides were A and T at these two positions in the type strain of
S. mitis(ATCC 49456) andS. mitisSK24 (GenBank accession no. AB051020), respectively. Another important characteristic
that could be used to differentiateS. mitisfromS. oraliswas
that the ITS lengths inS. mitis(248 to 249 bp) were consistently
2 to 3 bp longer than those inS. oralis(246 bp) (Table 1).
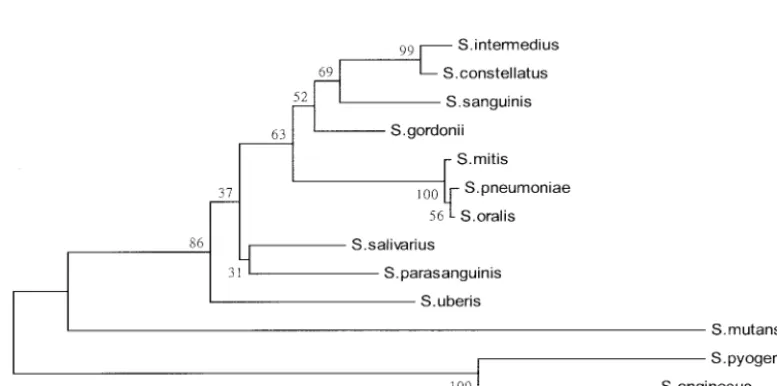
Phylogenetic relationships. The phylogenetic relationships
derived from the ITS sequences of the 11 type strains of VS
and the type strains of S. pneumoniae and S. pyogenes are
presented in Fig. 2. The phylogenetic tree constructed revealed
that strains of the mitis group (S. mitis and S. oralis) and S.
pneumoniaewere highly related and grouped together.
How-ever, it was noted thatS. parasanguinisdid not cluster with the
other two species (S. gordoniiandS. sanguinis) constituting the
sanguinis group. In addition,S. anginosuswas genetically
[image:4.603.44.543.80.235.2]sep-arated fromS. constellatusandS. intermediusof the anginosus
TABLE 3. Intraspecies and interspecies ITS sequence similarity scores among strains ofS. mitis, S. oralis,andS. pneumoniae
Species no. and
species ATCCno.
Sequence similarity score for the following speciesa:
2 3 4 5 6 7 8 9 10 11 12 13
1.S. mitis 49456b 0.98 0.94 0.95 0.94 0.95 0.94 0.94 0.95 0.95 0.99 0.95 1.00
2.S. mitis SK24c 0.93 0.93 0.93 0.93 0.93 0.93 0.93 0.94 0.99 0.94 0.99
3.S. oralis 49296 1.00 1.00 1.00 0.99 0.99 1.00 0.90 0.93 0.90 0.95
4.S. oralis 9811 1.00 1.00 0.99 0.99 1.00 0.89 0.93 0.89 0.94
5.S. oralis 10557c 1.00 0.99 0.99 1.00 0.90 0.93 0.90 0.95
6.S. oralis 35037b 0.99 0.99 1.00 0.89 0.93 0.89 0.94
7.S. oralis 55229 1.00 0.99 0.90 0.93 0.90 0.95
8.S. oralis 700233 0.99 0.90 0.93 0.90 0.95
9.S. oralis 700234 0.89 0.93 0.89 0.94
10.S. pneumoniae M60763c 0.93 0.99 0.95
11.S. pneumoniae 33400b 0.93 0.98
12.S. pneumoniae 27336 0.95
13.S. pneumoniae 49169
aThe species numbers correspond to the species numbers identified on the left. bType strains.
cStrains that were not obtained from ATCC but whose ITS sequences were available in the GenBank database.
FIG. 2. Phylogenetic relationship of various species of VS,S. pneumoniae, andS. pyogeneson the basis of the ITS sequences. The phylogenetic tree was generated by the neighbor-joining method within the MEGA package. The numbers at the nodes are the percentages of occurrence in 1,000 bootstrapped resamplings.
on May 15, 2020 by guest
http://jcm.asm.org/
[image:4.603.98.487.497.690.2]group and from the other nine species of VS in the phyloge-netic tree.
Identification of clinical isolates of VS by ITS sequencing.A
total of 106 clinical isolates were analyzed to validate ITS sequencing for identification of VS. Overall, the species iden-tities obtained with the Rapid ID 32 STREP system and by ITS sequence analysis showed 82% (87 isolates) agreement. Con-cordant identifications were obtained for the following species
(with the numbers of isolates indicated in parentheses): S.
anginosus(n⫽11),S. constellatus(n⫽12),S. gordonii(n⫽2),
S. intermedius(n⫽12),S. mitis(n⫽9),S. mutans(n⫽8),S. oralis(n⫽11),S. parasanguinis(n⫽1),S. salivarius(n⫽11),
S. sanguinis(n⫽8), andS. uberis(n⫽2). It was interesting
that all clinical isolates ofS. mitisandS. oralisanalyzed in this
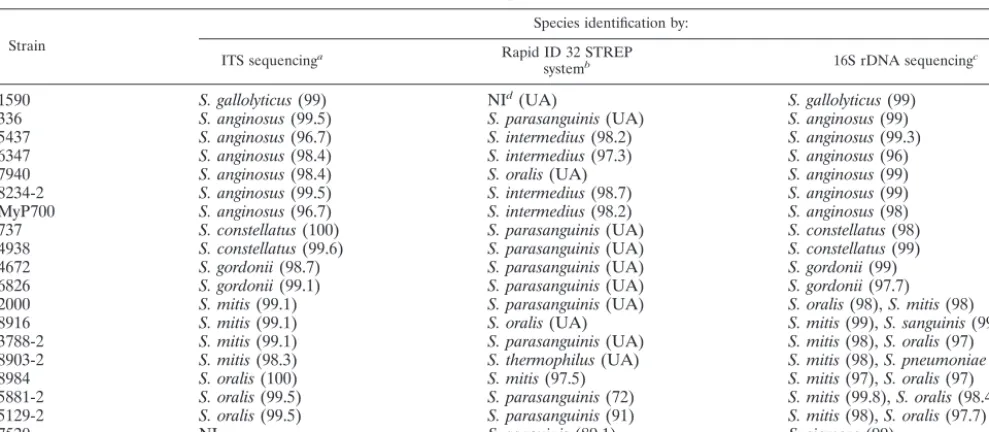
study possessed the same characteristics as the reference strains, i.e., either the presence or the absence of A and T residues at ITS nucleotide positions 199 and 217, respectively. The 19 (18%) clinical isolates for which the Rapid ID 32 STREP system and ITS sequencing produced discrepant iden-tifications are listed in Table 4. Strains that produced discrep-ant results were usually given an “unacceptable profile” or low identification percentages with the Rapid ID 32 STREP sys-tem. Conversely, the ITS sequence identities were greater than 96.7% for all strains with discrepant results (Table 4), as com-parisons were made between these strains and the respective type strains. The strains for which discrepant identifications
tended to be produced were species of the anginosus group (S.
anginosus, S. constellatus, andS. intermedius), species of the
mitis group (S. mitisandS. oralis), andS. gordonii(Table 4).
Nine discrepant results were caused by the identification ofS.
parasanguinis strains with the Rapid ID 32 STREP system.
Four strains phenotypically identified asS. intermediuswith the
Rapid ID 32 STREP system were determined to beS.
angino-susby the present molecular approach.
The 19 strains with discrepant results were further identified
by 16S rDNA sequencing. Except for strains ofS. mitisandS.
oralis, the identities of all 11 strains of other species identified by ITS sequencing were confirmed by sequencing of their 16S
rDNA (Table 4). Among the seven strains identified asS. mitis
or S. oralis by ITS sequencing, their 16S rDNA sequences
always matched those of bothS. mitisandS. oralis with very
high scores by use of database searches with the BLAST pro-gram. The ITS sequence of strain 1590 had a similarity of 99%
with that ofS. gallactolyticusATCC 9809, whose ITS sequence
we sequenced and submitted to GenBank (GenBank accession no. AY347542). The 16S rDNA sequence of strain 1590 also
had 99% identity with that ofS. gallactolyticusATCC 43143 in
the GenBank database. Strain 7520 was not identified by ITS
sequencing; however, it was identified as S. sanguinis and S.
siemenswith the Rapid ID 32 STREP system and by 16S rDNA sequencing, respectively. Because the 16S rDNA sequence of
strain 7520 had a 99% identity with that ofS. siemens(strain
HK; GenBank accession no. AF432856), this strain was
con-sidered to beS. siemens.
DISCUSSION
[image:5.603.46.541.89.305.2]This study has described a method that could identify clin-ically relevant VS to the species level. The method consists of (i) a PCR with two universal primers for amplification of a fragment encompassing the ITS sequence and partial regions of the 16S and 23S rDNAs, (ii) sequencing of the amplicon, and (iii) comparison of the ITS sequence with those of the type and reference strains of VS. The whole procedure can be
TABLE 4. Clinical isolates of VS for which discrepant identifications were produced with the Rapid ID 32 STREP system and by ITS sequence analysis
Strain
Species identification by:
ITS sequencinga Rapid ID 32 STREP
systemb 16S rDNA sequencingc
1590 S. gallolyticus(99) NId(UA) S. gallolyticus(99)
336 S. anginosus(99.5) S. parasanguinis(UA) S. anginosus(99)
5437 S. anginosus(96.7) S. intermedius(98.2) S. anginosus(99.3)
6347 S. anginosus(98.4) S. intermedius(97.3) S. anginosus(96)
7940 S. anginosus(98.4) S. oralis(UA) S. anginosus(99)
8234-2 S. anginosus(99.5) S. intermedius(98.7) S. anginosus(99)
MyP700 S. anginosus(96.7) S. intermedius(98.2) S. anginosus(98)
737 S. constellatus(100) S. parasanguinis(UA) S. constellatus(98)
4938 S. constellatus(99.6) S. parasanguinis(UA) S. constellatus(99)
4672 S. gordonii(98.7) S. parasanguinis(UA) S. gordonii(99)
6826 S. gordonii(99.1) S. parasanguinis(UA) S. gordonii(97.7)
2000 S. mitis(99.1) S. parasanguinis(UA) S. oralis(98),S. mitis(98)
8916 S. mitis(99.1) S. oralis(UA) S. mitis(99),S. sanguinis(99)
3788-2 S. mitis(99.1) S. parasanguinis(UA) S. mitis(98),S. oralis(97)
8903-2 S. mitis(98.3) S. thermophilus(UA) S. mitis(98),S. pneumoniae(98)
8984 S. oralis(100) S. mitis(97.5) S. mitis(97),S. oralis(97)
5881-2 S. oralis(99.5) S. parasanguinis(72) S. mitis(99.8),S. oralis(98.4)
5129-2 S. oralis(99.5) S. parasanguinis(91) S. mitis(98),S. oralis(97.7)
7520 NI S. sanguinis(89.1) S. siemens(99)
aThe values in parentheses are the percent identities of the strains studied compared to the sequence of the corresponding type strain. bThe values in parentheses are the percent identities obtained with the Rapid ID 32 STREP system. UA, unacceptable profile. cThe values in parentheses are the percent 16S rDNA sequence identities with the sequences available in GenBank. dNI, not identified.
on May 15, 2020 by guest
http://jcm.asm.org/
finished within 24 h if isolated colonies are available. This method might be useful under conditions that necessitate iden-tification of VS to the species level. It provides an accurate alternative for the delineation of some species of VS that are difficult to identify.
PCR amplification of the ITS regions depends on the occur-rence of highly conserved regions in the flanking termini of the 16S and 23S rRNA genes. The primers used in this study (primers 13BF and 6R) also could efficiently amplify the ITS regions of nutritionally variant streptococci and streptococci other than VS by using the PCR conditions specified here (data not shown). Whiley et al. (40) found that some atypical strains of the anginosus group produced PCR products whose lengths were different from those of “normal” strains. How-ever, most of these atypical strains were eventually found to be misclassified, as further examined by DNA-DNA reassociation experiments (40).
Pairwise comparison of two given species of VS revealed a lower level of sequence similarity between their ITS regions (Table 2) than between their 16S rDNA sequences (20). These results indicate that the ITS region might constitute a more discriminative target sequence than 16S rDNA for the differ-entiation of closely related species of VS. It was found that considerable variation in both the lengths and the sequences of the ITS regions can occur between species (14). The ITS re-gion has been suggested to be a suitable target from which useful taxonomic information for bacteria can be derived, par-ticularly with regard to identification to the species level (3, 16, 28, 34, 39) and genotyping (13, 14). A limitation for bacterial identification based on ITS sequencing is the limited availabil-ity of ITS sequences; however, the numbers of ITS region sequences available in public databases has increased rapidly in recent years.
16S rDNA sequencing (sequence length, approximately 1.5 kb) is well established as a standard method for the identifi-cation of species, genera, and families of bacteria (2). How-ever, the ITS lengths of VS are relatively short (246 to 391 bp) (Table 1), and sequencing of the ITS region would be easier and more straightforward. In this study, each of the 11 clini-cally important species of VS produced a single PCR product that facilitated direct sequencing (Fig. 1).
In this study, four strains identified asS. intermediuswith the
Rapid ID 32 STREP system were actually S. anginosus, as
revealed by their ITS and 16S rDNA sequences. However,
strains identified as S. parasanguinis with the Rapid ID 32
STREP system could be S. anginosus, S. constellatus, or S.
gordonii(Table 4). Jacobs et al. (19) reported that bloodstream
isolates ofS. anginosusare frequently identified asS.
interme-diuswith the Rapid ID 32 STREP system.
ThegroESL(36) andsodAint(26) genes have been used for differentiation of species of VS and streptococci, respectively. Since sequence heterogeneity of these two genes exists among different species, highly degenerate primers were used to am-plify the two genes from different species. The disadvantage of using highly degenerate primers is that the annealing temper-ature of the PCR must be low enough to ensure effective pairing of the primers and the template DNA. However, under low-stringency conditions, the chances of producing aberrant
PCR products might increase. The drawback of using the
so-dAint gene for the identification of streptococci is that the
intraspecies sequence divergence may vary greatly, depending upon the species considered. Therefore, the identification of species can be made only by determining the positions of their
sodAintsequences on the phylogenetic tree constructed from
type strains (26). In addition,S. mitisand S. oraliscannot be
differentiated by sequence analysis of the groESL (36) and
sodAint(26) genes, since intraspecies sequence variations are
greater than interspecies variations.
Kikuchi et al. (21) found that among the 17 strains identified asS. mitiswith the Rapid ID 32 STREP system, the results for only 3 strains were in agreement with the results of DNA-DNA hybridization. Jacobs et al. (19) reported that bloodstream
isolates identified asS. oralisare mainly those previously
iden-tified as S. mitis. The 16S rDNA sequence identities of type
strains ofS. oralis(ATCC 35037) andS. mitis(ATCC 49456)
are greater than 99% (20). After pairwise comparisons of the
16S rDNA sequences of six reference strains ofS. mitis(ATCC
903, ATCC 6249, ATCC 15914, ATCC 49456, NCTC 12261,
and NCTC 3165) and four reference strains ofS. oralis(ATCC
700233, ATCC 9811, ATCC 35037, and NCTC 11427) avail-able in the GenBank database, it was found that the intraspe-cies sequence variations (0.93 to 1.0) were higher than the interspecies sequence variations (0.96 to 1.0). Therefore,
iden-tification of S. mitis and S. oralison the basis of 16S rDNA
sequence analysis may not be reliable. This phenomenon was clearly demonstrated in the present study (Table 4). An anal-ysis with the BLAST algorithm showed that the 16S rDNA
sequences of strains identified as S. mitis by their ITS
se-quences always matched the 16S rRNA gene sese-quences of both
S. mitisandS. oraliswith high degrees of identity. This was also
true forS. oralisstrains.
However,S. mitiscould easily be differentiated fromS. oralis
by ITS sequence comparison (Table 3) or sequence alignment. The intraspecies ITS similarity scores for both species were higher than the interspecies ITS similarity scores (Table 3). In
addition, all seven reference strains ofS. oralishad one
dele-tion at ITS nucleotide posidele-tions 199 and 217, and this charac-teristic could be used as a fingerprint to differentiate species of
S. oralisandS. mitis. However, the intraspecies ITS divergence ofS. mitisandS. pneumoniaewas higher than the interspecies divergence of the two species (Table 3). Therefore, simple biochemical tests such as optochin susceptibility and bile
sol-ubility tests should be used to differentiateS. pneumoniaefrom
S. mitis; both tests are positive forS. pneumoniae, whereasS. mitisis optochin resistant and bile insoluble (9).
From the phylogenetic tree constructed by use of the ITS
sequences, S. parasanguinis and S. anginosusdid not cluster
with their respective groups (the sanguinis group and the
an-ginosus group, respectively). However,S. mitis,S. oralis, andS.
pneumoniaewere related and grouped together (Fig. 2). These three species were also grouped together when the genes
en-codinggroESL(36),sodAint(26), and 16S rRNA (6, 20) were
analyzed. Many bootstrap values shown in the phylogenetic tree (Fig. 2) were less than 70%; therefore, the ITS region may not be a good target for constructing a phylogenetic tree for species of VS. This implies that there exists an unparallel evolution between the ITS region and the 16S rRNA gene that is essential for the survival of all microorganisms.
In conclusion, identification of species of VS by ITS
on May 15, 2020 by guest
http://jcm.asm.org/
quencing seems to be reliable and could be used as an accurate alternative for the identification of VS.
ACKNOWLEDGMENTS
This project was supported by a grant (grant NSC 88-2314-B006-078) from the National Science Council and in part by a grant from the Department of Medical Research of the National Taiwan University Hospital, Taipei, Taiwan.
We thank S. Liu for help with constructing the phylogenetic tree and S. K. Tung for assistance with 16S rDNA sequencing.
REFERENCES
1. Ahmet, Z., M. Warren, and E. T. Houang.1995. Species identification of members of theStreptococcus millerigroup isolated from the vagina by ID 32 Strep system and differential phenotypic characteristics. J. Clin. Microbiol.
33:1592–1595.
2. Amann, R. I., W. Ludwig, and K.-H. Schleifer.1995. Phylogenetic identifi-cation and in situ detection of individual microbial cells without cultivation. Microbiol. Rev.59:143–169.
3. Barry, T., G. Colleran, M. Glennon, L. K. Dunican, and F. Gannon.1991. The 16S/23S ribosomal spacer region as a target for DNA probes to identify eubacteria. PCR Methods Appl.1:51–56.
4. Beighton, D., J. M. Hardie, and A. Whiley.1991. A scheme for the identifi-cation of viridans streptococci. J. Med. Microbiol.35:367–372.
5. Bentley, R. W., and J. A. Leigh.1995. Development of PCR-based hybrid-ization protocol for identification of streptococcal species. J. Clin. Microbiol.
33:1296–1301.
6. Bentley, R. W., J. A. Leigh, and M. D. Collins.1991. Intrageneric structure ofStreptococcusbased on comparative analysis of small-subunit rRNA se-quences. Int. J. Syst. Bacteriol.41:487–494.
7. Chang, W. N., J. J. Wu, C. R. Huang, Y. C. Tsai, C. C. Chien, and C. H. Lu.
2002. Identification of viridans streptococcal species causing bacterial men-ingitis in adults in Taiwan. Eur. J. Clin. Microbiol. Infect. Dis.21:393–396. 8. Clarridge, J. E., III, S. M. Attorri, Q. Zhang, and J. Bartell.2001. 16S ribosomal DNA sequence analysis distinguishes biotypes ofStreptococcus bovis:Streptococcus bovisbiotype II/2 is a separate genospecies and the predominant clinical isolate in adult males. J. Clin. Microbiol.39:1549–1552. 9. Facklam, R.2002. What happened to the streptococci: overview of
taxo-nomic and nomenclature changes. Clin. Microbiol. Rev.15:613–630. 10. Flynn, C. E., and K. L. Ruoff.1995. Identification ofStreptococcus milleri
group isolates to the species level with a commercially available rapid test. J. Clin. Microbiol.33:2704–2706.
11. Garnier, F., G. Gerbaud, P. Courvalin, and M. Galimand.1997. Identifica-tion of clinically relevant viridans group streptococci to the species level by PCR. J. Clin. Microbiol.35:2337–2341.
12. Gheldre, Y. D., P. Vandamme, H. Goossens, and M. J. Struelens.1999. Identification of clinically relevant viridans streptococci by analysis of trans-fer DNA intergenic spacer length polymorphism. Int. J. Syst. Bacteriol.
49:1591–1598.
13. Gurtler, V.1993. Typing ofClostridium difficilestrains by PCR-amplification of variable length 16S-23S rDNA spacer regions. J. Gen. Microbiol.139:
3089–3097.
14. Gurtler, V., and H. D. Barrie.1995. Typing ofStaphylococcus aureusstrains by PCR-amplification of variable-length 16S-23S rDNA spacer regions: char-acterization of spacer sequences. Microbiology141:1255–1265.
15. Gurtler, V., and V. A. Stanisich.1996. New approaches to typing and iden-tification of bacteria using the 16S-23S rDNA spacer region. Microbiology
142:3–16.
16. Hamid, M. E., A. Roth, O. Landt, R. M. Kroppenstedt, M. Goodfellow, and H. Mauch.2002. Differentiation betweenMycobacterium farcinogenesand
Mycobacterium senegalensestrains based on 16S-23S ribosomal DNA internal transcribed spacer sequences. J. Clin. Microbiol.40:707–711.
17. Hillman, J. D., S. W. Andrews, S. Painetr, and P. Stashenko.1989. Adap-tative changes in a strain ofStreptococcus mutansduring colonization of the human oral cavity. Microb. Ecol. Health Dis.2:231–239.
18. Jacobs, J. A., C. S. Schot, A. E. Bunschoten, and L. M. Schouls.1996. Rapid species identification of “Streptococcus milleri” strains by line blot hybridiza-tion: identification of a distinct 16S rRNA population closely related to
Streptococcus constellatus.J. Clin. Microbiol.34:1717–1721.
19. Jacobs, J. A., H. C. Schouten, E. E. Stobberingh, and P. B. Soeters.1995. Viridans streptococci isolated from the bloodstream. Relevance of species identification. Diagn. Microbiol. Infect. Dis.22:267–273.
20. Kawamura, Y., X. Hou, F. Sultana, H. Miura, and T. Ezaki.1995. Determi-nation of 16S rRNA sequences ofStreptococcus mitisandStreptococcus gordoniiand phylogenetic relationships among members of the genus Strep-tococcus.Int. J. Syst. Bacteriol.45:406–408.
21. Kikuchi, K., T. Enari, K. Totsuka, and K. Shimizu.1995. Comparison of phenotypic characteristics, DNA-DNA hybridization results, and results with a commercial rapid biochemical and enzymatic reaction system for identifi-cation of viridans group streptococci. J. Clin. Microbiol.33:1215–1222. 22. Kilian, M., L. Mikkelsen, and J. Henrichsen.1989. Taxonomic studies of
viridans streptococci: description of Streptococcus gordonii sp. nov. and emended descriptions ofStreptococcus sanguis(White and Niven 1946),
Streptococcus oralis(Bridge and Sneath 1982), andStreptococcus mitis (An-drewes and Horder 1906). Int. J. Syst. Bacteriol.39:471–484.
23. Kilpper-Ba¨lz, R., B. L. Williams, R. Lutticken, and K. H. Schleifer.1984. Relatedness of “Streptococcus melleri” with Streptococcus anginosus and
Streptococcus constellatus.Syst. Appl. Microbiol.5:494–500.
24. Kumar, S., K. Tamura, and M. Nei.1993. MEGA: molecular evolutionary genetic analysis, version 1.01. The Pennsylvania State University, University Park.
25. Millar, B. C., X. Jiru, J. E. Moore, and J. A. Earle.2000. A simple and sensitive method to extract bacterial, yeast and fungal DNA from blood culture material. J. Microbiol. Methods42:139–147.
26. Poyart, C., G. Quesne, S. Coulon, P. Berche, and P. Trieu-Cuot.1998. Identification of streptococci to species level by sequencing the gene encod-ing the manganese-dependent superoxide dismutase. J. Clin. Microbiol.36:
41–47.
27. Relman, D. A.1993. Universal bacterial 16S rDNA amplification and se-quencing, p. 489–495.InD. H. Persing, T. F. Smith, F. C. Tenover, and T. J. White (ed.), Diagnostic molecular microbiology. American Society for Mi-crobiology, Washington, D.C.
28. Roth, A., M. Fischer, M. E. Hamid, S. Michalke, W. Ludwig, and H. Mauch.
1998. Differentiation of phylogenetically related slowly growing mycobacte-ria based on 16S-23S rRNA gene internal transcribed spacer sequences J. Clin. Microbiol.36:139–147.
29. Rudney, J. D., and C. J. Larson.1999. Identification of oral mitis group streptococci by arbitrary primed polymerase chain reaction. Oral Microbiol. Immunol.14:33–42.
30. Saarela, M., S. Alaluusua, T. Takei, and S. Asikainen.1993. Genetic diver-sity within isolates of mutans streptococci recognized by an rRNA gene probe. J. Clin. Microbiol.31:584–587.
31. Saitou, N., and M. Nei.1987. The neighbor-joining method: a new method for reconstructing phylogenetic tree. Mol. Biol. Evol.4:406–425. 32. Schmidhuber, S., W. Ludwig, and K. H. Schleifer.1988. Construction of a
DNA probe for the specific identification ofStreptococcus oralis.J. Clin. Microbiol.26:1042–1044.
33. Shenep, J. L.2000. Viridans-group streptococcal infections in immunocom-promised hosts. Int. J. Antimicrob. Agents14:129–135.
34. Suffys, P. N., A. da Silva Rocha, M. de Oliveira, C. E. Dias Campos, A. M. Werneck Barreto, F. Portaels, L. Rigouts, G. Wouters, G. Jannes, G. van Reybroeck, W. Mijs, and B. Vanderborght.2001. Rapid identification of mycobacteria to the species level using INNO-LiPA mycobacteria, a reverse hybridization assay. J. Clin. Microbiol.39:4477–4482.
35. Teng, L. J., P. R. Hsueh, S. W. Ho, and K. T. Luh.1998. Antimicrobial susceptibility of viridans group streptococci in Taiwan with an emphasis on the high rates of resistance to penicillin and erythromycin inStreptococcus oralis.J. Antimicrob. Chemother.41:621–627.
36. Teng, L.-J., P.-R. Hsueh, J.-C. Tsai, P.-W. Chen, J.-C. Hsu, H.-C. Lai, C.-N. Lee, and S.-W. Ho.2002.groESL sequence determination, phylogenetic analysis, and species differentiation for viridans group streptococci. J. Clin. Microbiol.40:3172–3178.
37. Thompson, J. D., T. J. Gibson, F. Plewniak, F. Jeanmougin, and D. G. Higgins.1997. The CLUSTAL X Windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res.25:4876–4882.
38. Tracy, M., A. Wanahita, Y. Shuhatovich, E. A. Goldsmith, J. E. Clarridge, and D. M. Musher.2001. Antibiotic susceptibilities of genetically character-izedStreptococcus milleri group strains. Antimicrob. Agents Chemother.
45:1511–1514.
39. Uemori, T., K. Asada, I. Kato, and R. Harasawa.1992. Amplification of the 16S-23S spacer region in rRNA operons of mycoplasmas by the polymerase chain reaction. Syst. Appl. Microbiol.15:181–186.
40. Whiley, R. A., B. Duke, J. M. Hardie, and L. M. C. Hall.1995. Heterogeneity among 16S-23S rRNA intergenic spacers of species within the “Streptococcus millerigroup.” Microbiology141:1461–1467.