Acta Cryst.(2002). E58, o1157±o1158 DOI: 10.1107/S1600536802017373 Chen, Guo and Zhou C8H13NO3
o1157
organic papers
Acta Crystallographica Section E Structure Reports Online
ISSN 1600-5368
Ethyl (
E
)-3-acetamido-2-butenoate
Xuanhua Chen,aRongwei Guob*
and Zhongyuan Zhoub
aDepartment of Chemistry, Central China
Normal University, Wuhan, People's Republic of China, Department of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong, andbDepartment of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study
T= 294 K
Mean(C±C) = 0.003 AÊ
Rfactor = 0.053
wRfactor = 0.153
Data-to-parameter ratio = 19.3
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2002 International Union of Crystallography Printed in Great Britain ± all rights reserved
The crystal structure determination of the title compound, C8H13NO3, shows that it is the E isomer. An NÐH O
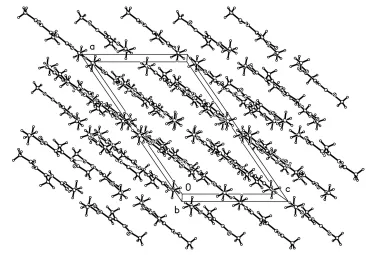
intermolecular hydrogen bond is observed and is responsible for the formation of in®nite chains stretching along thebaxis of the crystal.
Comment
The title compound, (I), is one of the products obtained from ethyl 3-amine-2-butenoate on re¯ux with acetic anhydride for a period of 24 h. This prochiral ole®n is a model substrate studied in the asymmetric hydrogenation reaction (Hackler & Wickiser, 1985; Lubellet al., 1991). The structure determina-tion of (I) was conducted in order to obtain more stereo-chemical information about the behaviour of these kinds of substrates in hydrogenation reactions.
The crystal structure of (I) (Fig. 1) shows that the molecule is nearly planar. The angles C3ÐC4ÐC5 [124.71 (19)] and
C4ÐC3ÐC8 [125.27 (18)] are wider and N1ÐC3ÐC8
[112.12 (16)] narrower than the value of 120. This results in a
close mutual repulsion between the methyl group on C3 and the carbonyl group on C4 (Table 1). The molecules in the crystal structure are interconnected by NÐH O hydrogen bonding (Table 2). As shown in the packing diagram (Fig. 2), the NÐH O hydrogen bonds link the molecules along theb
axis.
Experimental
The title compound was synthesized according the literature method (Zhuet al., 1999) A crystal suitable for X-ray analysis was grown slowly from a mixture of ethyl acetate and hexane at room temperature.1H NMR (400 MHz, acetone-d6, p.p.m.):1.24 (t, J=
7.1 Hz, 3H), 2.06 (s, 3H), 2.33 (s, 3H), 4.09 (q, J= 7.1 Hz, 2H), 6.89 (d, J= 1 Hz, 1H), 8.79 (br, 1H).
Crystal data C8H13NO3 Mr= 171.19
Monoclinic,C2=c a= 20.006 (4) AÊ b= 9.545 (2) AÊ c= 11.922 (2) AÊ
= 124.479 (4)
V= 1876.7 (6) AÊ3 Z= 8
Dx= 1.212 Mg mÿ3
MoKradiation Cell parameters from 2174
re¯ections
= 1±27.5
= 0.09 mmÿ1 T= 294 (2) K Plate, colourless 0.340.280.10 mm
Data collection
Siemens SMART CCD area-detector diffractometer
'and!scans
Absorption correction: multi-scan (SADABS; Sheldrick, 1996) Tmin= 0.969,Tmax= 0.991 6283 measured re¯ections
2162 independent re¯ections 1053 re¯ections withI> 2(I) Rint= 0.035
max= 27.6 h=ÿ22!25 k=ÿ12!12 l=ÿ15!15 Re®nement
Re®nement onF2 R[F2> 2(F2)] = 0.053 wR(F2) = 0.153 S= 1.05 2162 re¯ections 112 parameters
H-atom parameters constrained w= 1/[2(F
o2) + (0.07P)2]
whereP= (Fo2+ 2Fc2)/3
(/)max< 0.001
max= 0.20 e AÊÿ3
min=ÿ0.16 e AÊÿ3
Table 1
Selected geometric parameters (AÊ,).
O1ÐC2 1.220 (2)
O2ÐC5 1.200 (2)
N1ÐC2 1.358 (2)
N1ÐC3 1.398 (2)
C3ÐC4 1.333 (3)
C3ÐC8 1.495 (3)
C4ÐC5 1.463 (3)
O1ÐC2ÐN1 122.88 (17)
O1ÐC2ÐC1 121.88 (17)
C4ÐC3ÐC8 125.27 (18)
N1ÐC3ÐC8 112.12 (16)
C3ÐC4ÐC5 124.71 (19)
O2ÐC5ÐC4 128.1 (2)
O3ÐC5ÐC4 109.46 (17)
C3ÐN1ÐC2ÐO1 2.3 (3)
C8ÐC3ÐC4ÐC5 0.2 (3)
C6ÐO3ÐC5ÐO2 ÿ1.5 (3)
C6ÐO3ÐC5ÐC4 178.83 (17)
C3ÐC4ÐC5ÐO3 ÿ171.69 (19)
C5ÐO3ÐC6ÐC7 ÿ178.91 (19)
Table 2
Hydrogen-bonding geometry (AÊ,).
DÐH A DÐH H A D A DÐH A
N1ÐH1A O1i 0.86 2.10 2.943 (2) 165
Symmetry code: (i)1
2ÿx;yÿ12;12ÿz.
H atoms were included in the riding-model approximation with
Uisovalues equal toUeqof the atom to which they are bound. Data collection: SMART (Siemens, 1995); cell re®nement:
SMART; data reduction:SAINT(Siemens, 1995) andSHELXTL-NT
(Siemens, 1995); program(s) used to solve structure: SHELXS97 (Sheldrick, 1997); program(s) used to re®ne structure:SHELXL97 (Sheldrick, 1997); molecular graphics:SHELXTL-NT; software used to prepare material for publication:SHELXTL-NT.
The authors thank the Hong Kong Polytechnic University ASD Fund for ®nancial support of this study.
References
Hackler, R. E. & Wickiser, D. I. (1985). Br. Patent No. GB 2141712. Lubell, W. D., Kitamura, M. & Noyori, R. (1991).Tetrahedron Asymmetry,2,
543±554.
Sheldrick, G. M. (1996).SADABS. University of GoÈttingen, Germany. Sheldrick, G. M. (1997). SHELXL97 and SHELXS97. University of
GoÈttingen, Germany.
Siemens (1995).SMART(Version 5.0),SAINT(Version 5.0) and SHELXTL-NT(Version 5.10). Siemens Analytical X-ray Instruments Inc., Madison, Wisconsin, USA.
Zhu, G. X., Chen, Z. G. & Zhang, X. M. (1999).J. Org. Chem.64, 6907±6910.
Figure 2
Packing diagram for (I). The hydrogen bonds are indicated by dashed lines.
Figure 1
supporting information
sup-1
Acta Cryst. (2002). E58, o1157–o1158supporting information
Acta Cryst. (2002). E58, o1157–o1158 [doi:10.1107/S1600536802017373]
Ethyl (
E
)-3-acetamido-2-butenoate
Xuanhua Chen, Rongwei Guo and Zhongyuan Zhou
S1. Comment
The title compound, (I), is one of the products obtained from ethyl 3-amine-2-butenoate on reflux with acetic anhydride
for a period of 48 h. This prochiral olefin is a model substrate studied in the asymmetric hydrogenation reaction (Hackler
et al., 1985; Lubell et al., 1991). The structure determination of (I) was conducted in order to obtain more stereochemical
information about the behaviour of these kinds of substrates in hydrogenation reactions.
The crystal structure of (I) (Fig. 1) shows that the molecule is nearly planar. The angles C3—C4—C5 [124.71 (19)°]
and C4—C3—C8 [125.27 (18)°] are wider and N1—C3—C8 [112.12 (16)°] narrower than the value of 120°. This results
in a close mutual repulsion between the methyl group on C3 and the carbonyl group on C4 (Table 1). The molecules in
the crystal structure are interconnected by N—H···O hydrogen bonding (Table 2). As shown in the packing diagram (Fig.
2), the N—H···O hydrogen bond links the molecules along b axis.
S2. Experimental
The title compound was synthesized according theliterature (Zhu et al., 1999) A crystal suitable for X-ray analysis was
grown slowly from a mixture of ethyl acetate and hexane at room temperature. 1H NMR (400 MHz, acetone-d
6, p.p.m.): δ
1.24 (t, J = 7.1 Hz, 3H), 2.06 (s, 3H), 2.33 (s, 3H), 4.09 (q, J = 7.1 Hz, 2H), 6.89 (d, J = 1 Hz, 1H), 8.79 (br, 1H).
S3. Refinement
H atoms were included in the riding-model approximation with Uiso values equal to Ueq of the atom to which they were
Figure 1
[image:4.610.117.483.102.357.2]The molecular structure of (I), showing ellipsoids at the 30% probability level (Siemens, 1995).
Figure 2
Packing diagram for (I). The hydrogen bonds are indicated by dashed lines.
(E)-Ethyl 3-acetamido-2-butenoate
Crystal data
C8H13NO3
Mr = 171.19
Monoclinic, C2/c a = 20.006 (4) Å
b = 9.545 (2) Å
c = 11.922 (2) Å
β = 124.479 (4)°
V = 1876.7 (6) Å3
Z = 8
F(000) = 736
Dx = 1.212 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 2174 reflections
θ = 1–27.5°
µ = 0.09 mm−1
T = 294 K Plate, colorless 0.34 × 0.28 × 0.10 mm
Data collection
Siemens SMART CCD area-detector diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
φ and ω scans
Absorption correction: empirical (using intensity measurements)
(SADABS; Sheldrick, 1996)
Tmin = 0.969, Tmax = 0.991
6283 measured reflections 2162 independent reflections 1053 reflections with I > 2σ(I)
Rint = 0.035
θmax = 27.6°, θmin = 3.4°
h = −22→25
k = −12→12
supporting information
sup-3
Acta Cryst. (2002). E58, o1157–o1158Refinement
Refinement on F2
Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.053
wR(F2) = 0.153
S = 1.05 2162 reflections 112 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained
w = 1/[σ2(F
o2) + (0.07P)2]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001
Δρmax = 0.20 e Å−3
Δρmin = −0.16 e Å−3
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
O1 0.24031 (10) 0.98771 (15) 0.21996 (17) 0.0769 (5)
O2 0.43183 (11) 0.88023 (17) 0.06239 (18) 0.0906 (6)
O3 0.40086 (9) 1.09940 (14) 0.08209 (14) 0.0692 (5)
N1 0.27942 (10) 0.77021 (15) 0.20365 (16) 0.0551 (5)
H1A 0.2724 0.6830 0.2123 0.066*
C1 0.19104 (15) 0.7954 (2) 0.2785 (3) 0.0819 (8)
H1B 0.1506 0.8600 0.2661 0.123*
H1C 0.2267 0.7718 0.3732 0.123*
H1D 0.1652 0.7120 0.2267 0.123*
C2 0.23847 (12) 0.8610 (2) 0.2317 (2) 0.0564 (6)
C3 0.33135 (12) 0.79732 (19) 0.16286 (19) 0.0518 (5)
C4 0.34191 (12) 0.9254 (2) 0.1309 (2) 0.0592 (6)
H4 0.3128 0.9984 0.1356 0.071*
C5 0.39601 (13) 0.9594 (2) 0.0889 (2) 0.0623 (6)
C6 0.45348 (14) 1.1505 (2) 0.0444 (2) 0.0752 (7)
H6A 0.4350 1.1172 −0.0457 0.090*
H6B 0.5084 1.1174 0.1085 0.090*
C7 0.45111 (17) 1.3072 (3) 0.0457 (3) 0.0932 (9)
H7A 0.3961 1.3385 −0.0140 0.140*
H7B 0.4830 1.3443 0.0155 0.140*
H7C 0.4728 1.3392 0.1365 0.140*
C8 0.37290 (14) 0.6677 (2) 0.1617 (2) 0.0690 (6)
H8A 0.4154 0.6928 0.1509 0.104*
H8B 0.3344 0.6087 0.0874 0.104*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
O1 0.1022 (13) 0.0467 (9) 0.1292 (14) 0.0032 (8) 0.0939 (12) 0.0030 (8)
O2 0.1093 (14) 0.0831 (11) 0.1347 (15) 0.0115 (10) 0.1024 (14) 0.0118 (10)
O3 0.0809 (11) 0.0683 (10) 0.0891 (11) −0.0093 (8) 0.0667 (10) 0.0011 (7)
N1 0.0636 (11) 0.0454 (8) 0.0789 (11) −0.0032 (8) 0.0538 (10) −0.0026 (8)
C1 0.1037 (19) 0.0548 (12) 0.141 (2) −0.0015 (12) 0.1015 (19) 0.0013 (13)
C2 0.0624 (13) 0.0481 (12) 0.0771 (14) −0.0016 (10) 0.0505 (12) −0.0029 (10)
C3 0.0489 (11) 0.0585 (12) 0.0567 (12) −0.0010 (9) 0.0350 (11) −0.0044 (9)
C4 0.0662 (14) 0.0564 (12) 0.0786 (14) −0.0033 (10) 0.0552 (13) −0.0036 (10)
C5 0.0644 (14) 0.0699 (14) 0.0705 (14) −0.0014 (11) 0.0489 (13) 0.0012 (11)
C6 0.0704 (16) 0.0911 (17) 0.0808 (16) −0.0113 (13) 0.0529 (14) 0.0127 (13)
C7 0.103 (2) 0.0963 (19) 0.0912 (18) −0.0282 (15) 0.0613 (18) 0.0042 (14)
C8 0.0747 (15) 0.0631 (13) 0.0928 (16) 0.0095 (11) 0.0615 (14) 0.0032 (11)
Geometric parameters (Å, º)
O1—C2 1.220 (2) C3—C8 1.495 (3)
O2—C5 1.200 (2) C4—C5 1.463 (3)
O3—C5 1.345 (2) C4—H4 0.9300
O3—C6 1.444 (2) C6—C7 1.496 (3)
N1—C2 1.358 (2) C6—H6A 0.9700
N1—C3 1.398 (2) C6—H6B 0.9700
N1—H1A 0.8600 C7—H7A 0.9600
C1—C2 1.485 (3) C7—H7B 0.9600
C1—H1B 0.9600 C7—H7C 0.9600
C1—H1C 0.9600 C8—H8A 0.9600
C1—H1D 0.9600 C8—H8B 0.9600
C3—C4 1.333 (3) C8—H8C 0.9600
C5—O3—C6 116.38 (17) O2—C5—C4 128.1 (2)
C2—N1—C3 129.64 (16) O3—C5—C4 109.46 (17)
C2—N1—H1A 115.2 O3—C6—C7 107.52 (19)
C3—N1—H1A 115.2 O3—C6—H6A 110.2
C2—C1—H1B 109.5 C7—C6—H6A 110.2
C2—C1—H1C 109.5 O3—C6—H6B 110.2
H1B—C1—H1C 109.5 C7—C6—H6B 110.2
C2—C1—H1D 109.5 H6A—C6—H6B 108.5
H1B—C1—H1D 109.5 C6—C7—H7A 109.5
H1C—C1—H1D 109.5 C6—C7—H7B 109.5
O1—C2—N1 122.88 (17) H7A—C7—H7B 109.5
O1—C2—C1 121.88 (17) C6—C7—H7C 109.5
N1—C2—C1 115.24 (17) H7A—C7—H7C 109.5
C4—C3—N1 122.61 (17) H7B—C7—H7C 109.5
C4—C3—C8 125.27 (18) C3—C8—H8A 109.5
N1—C3—C8 112.12 (16) C3—C8—H8B 109.5
supporting information
sup-5
Acta Cryst. (2002). E58, o1157–o1158C3—C4—H4 117.6 C3—C8—H8C 109.5
C5—C4—H4 117.6 H8A—C8—H8C 109.5
O2—C5—O3 122.39 (18) H8B—C8—H8C 109.5
C3—N1—C2—O1 2.3 (3) C6—O3—C5—O2 −1.5 (3)
C3—N1—C2—C1 −177.4 (2) C6—O3—C5—C4 178.83 (17)
C2—N1—C3—C4 −7.3 (3) C3—C4—C5—O2 8.7 (4)
C2—N1—C3—C8 171.98 (19) C3—C4—C5—O3 −171.69 (19)
N1—C3—C4—C5 179.42 (18) C5—O3—C6—C7 −178.91 (19)
C8—C3—C4—C5 0.2 (3)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
N1—H1A···O1i 0.86 2.10 2.943 (2) 165