Genome wide association mapping and genomic selection for agronomic and disease traits in soybean
Full text
Figure
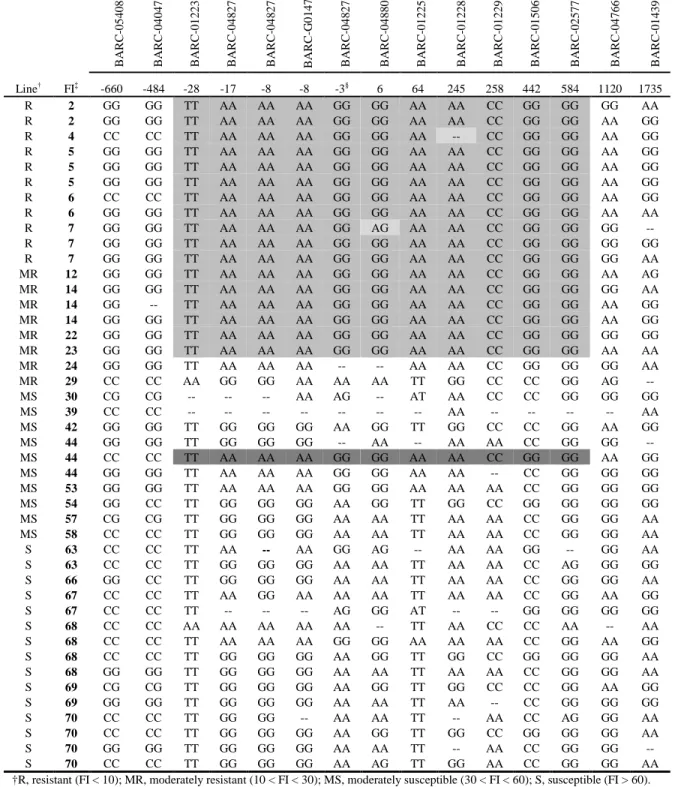
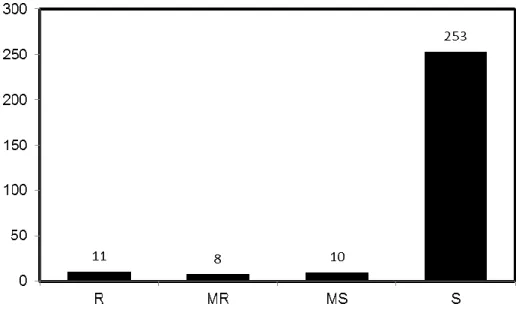
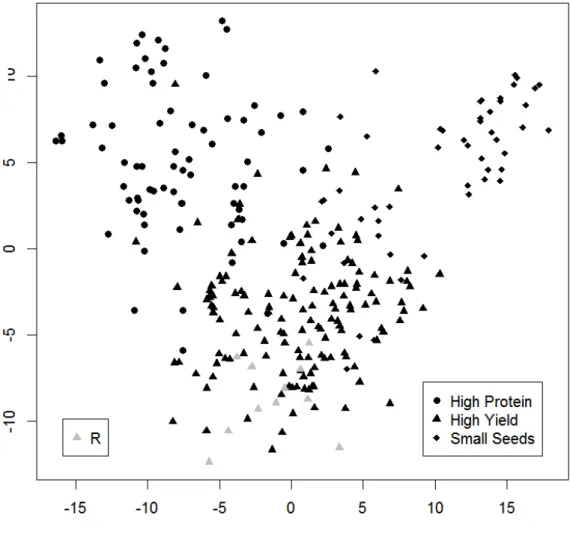
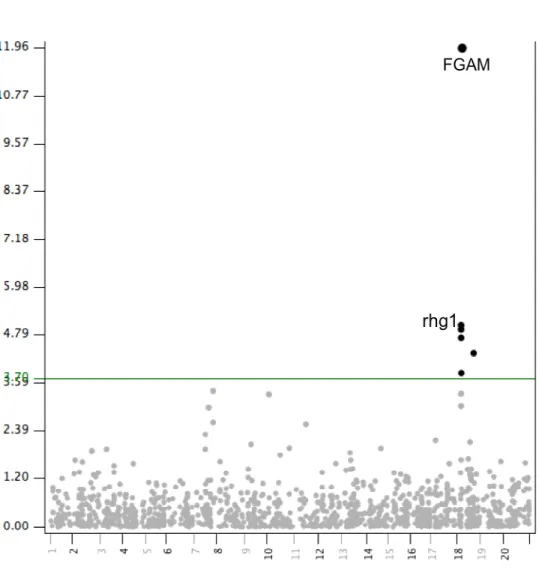
Outline
Related documents
pyogenes strains collected from sterile clinical sites during 2012 were submitted to the NML from provincial and territorial public health laboratories for surveillance or
In addi- tion, the PTFSs are better able to explain trend-following funds’ returns than standard buy-and-hold benchmark returns on major asset classes, as well as benchmarks used by
In a speech to the DPP’s national convention in October 2001, president Chen Shui-bian linked the policies of the single-member dual-ballot system and halving of the seats in
Both Rochon and Warren said they do not recall seeing many mainstream media reports about the process to adopt a child from China nor to they remember seeing any mainstream
The study will analyze the four dimensions (industrial relations, vocational training, interfirm relations, and corporate governance) of the French variety of capitalism and how
62. Among other prominent ethics scholars, Fred Zacharias may stand out in the degree to which and the consistency with which he referenced the lawyer’s duty to abide by the “sprit”
Some prior studies have proposed using P2P approaches for the signaling phase of VoIP communication. However, a P2P approach may increase the call setup time, an important
We note, however, that the expressions linking changes in the skill premium to changes in domestic expenditure shares di¤er from those in the Ricardian model developed in the paper,
