Copyright 0 1989 by the Genetics Society of America
DNA Polymorphism in a Subdivided Population: The Expected Number
of
Segregating Sites in the Two-Subpopulation Model
Fumio Tajima
Department of Biology, Kyushu University, Hakozaki, Fukuoka 812, Japan
Manuscript received March 29, 1989
Accepted for publication June 3, 1989
ABSTRACT
Using the two subpopulation model, the expected numbers of segregating sites in a number of DNA sequences randomly sampled from a subdivided population were examined for several types of population subdivisions. It is shown that, in the case where the pattern of migration is symmetrical such as the finite island model, the expected number of segregating sites is independent of the migration rate when two or three DNA sequences are randomly sampled from the same subpopulation, but depends on the migration rate when more than three DNA sequences are sampled. It is also shown that the population subdivision can increase the amount of DNA polymorphism even in a subpopulation in some cases.
T
HE amount of DNA polymorphism in a popula-tion can be measured by the average number of
(pairwise) nucleotide differences or the number of
segregating sites in a sample. Although their statistical
properties are already known, they have been ob-
tained under the assumption of random mating pop-
ulation (WATTERSON 1975; TAJIMA 1983). Natural
populations, however, are often subdivided into a
number of subpopulations. LI (1976) has studied the
distribution of nucleotide differences between two
randomly chosen DNA sequences in a subdivided
population by using the finite island model (MARU-
YAMA 1970; LATTER 1973; NEI 1975). Although his results give useful information on DNA polymor-
phism in a subdivided population, it is not clear
whether or not they can be applied to the case where more than two DNA sequences are sampled from the population.
In this paper I shall examine the expected number
of segregating sites in a number of DNA sequences
randomly sampled from a subdivided population by
using the two subpopulation model (NEI and FELDMAN
1972; LI and NEI 1975, 1977). Although one of the main reasons, why the two subpopulation model is
used instead of the other models such as the infinite
island model (WRIGHT 1931), the finite island model
and the stepping stone model (KIMURA and WEISS
1964; WEISS and KIMURA 1965; MARUYAMA 1969,
1977), is its simplicity, this model has the advantage of being flexible, as will be shown later.
MODEL
In this paper we assume that a population is subdi-
vided into two subpopulations, say, subpopulations 1
and 2. We denote the effective sizes of subpopulations
Genetics 123: 229-240 (September, 1989)
1 and 2 by N1 and N2, respectively. We also denote
the probability that a DNA sequence in subpopulation
1 comes from subpopulation 2 in the immediately
previous generation by m l , and the probability that a
DNA sequence in subpopulation 2 comes from sub-
population 1 in the immediately previous generation
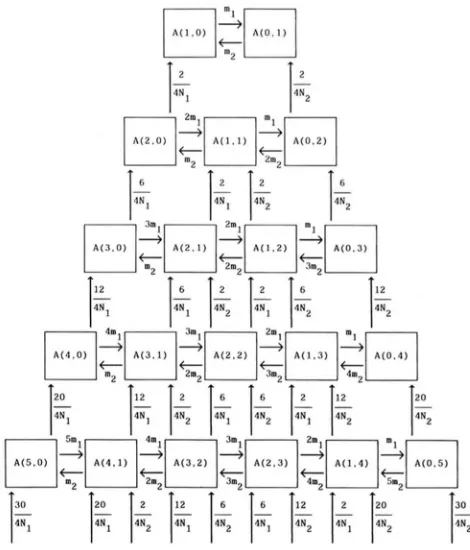
by m2. T h e scheme of this model is shown in Figure 1.
We denote by tr the mutation rate per DNA se-
quence per generation, and assume that the number
of nucleotide sites on a DNA sequence is so large that
a newly arisen mutation takes place at a site different
from the sites where the previous mutations have
occurred [the infinite site model (KIMURA 1969)]. We
also assume that the mutants are selectively neutral
[the neutral mutation model (KIMURA 1968, 1983)l.
Finally, we assume that m l , m2, l/N1 and 1/N2 are so
small that their higher order terms can be ignored.
THEORY
In this paper we use the genealogical relationship
of DNA sequences (GRIFFITHS 1980; KINGMAN 1982;
HUDSON 1983; TAJIMA 1983). Let A(i, j ) be the state
that i a n d j DNA sequences are randomly chosen from
subpopulations 1 and 2, respectively.
When one DNA sequence is randomly chosen from the population, there are two states, namely the state
that the DNA sequence is chosen from subpopulation
1 and the state that the DNA sequence is randomly
chosen from subpopulation 2. According to the defi-
nition, the former is denoted by A( 1 , 0) and the latter by A(0, 1). T h e probability that a DNA sequence in
subpopulation 1 comes from subpopulation 2 in the
immediately previous generation is ml. In terms of
S a h p o p u I a t i o t 8 I
r d
. - T I
S u b p o p u l a t i o n 2-
N 2
E f f e c t i v e s i r e o f s u b p o p u l a t i o n
FIGURE 1 ."Scheme of the two subpopulation model used.
probability that A ( l , 0 ) changes to A(0, 1) in one generation is m l . In the same way, the probability that
A(0, 1) changes to A ( l , 0 ) in one generation is m2.
These events are shown in Figure
2.
I t should be keptin mind that we are going back to the past.
When two DNA sequences are randomly chosen
from the population, there are three states, namely
A(2, 0), A( 1 , 1) and A(0, 2). T h e probability that one
of two DNA sequences in subpopulation 1 comes from
subpopulation 2 in the immediately previous genera-
tion is 2 m l ( l
-
m l )=
2 m l . Therefore, the probability that A(2, 0 ) changes to A ( l , 1 ) in one generation is2 m l . In the same way, the probability that A(0, 2)
changes to A( 1 , 1) in one generation is 2m2 approxi- mately, and the probability that A ( l , 1 ) changes to
A(2, 0 ) or A ( 0 , 2 ) in one generation is approximately
m2 or m l , respectively.
Let us now consider the effect of random genetic drift on the change of state. T h e probability that two
DNA sequences in subpopulation 1 are derived from
a common ancestral DNA sequence in the immedi-
ately previous generation is 2/(4N1), so that the prob- ability that A(2,O) changes to A ( 1 , 0 ) in one generation is 2/(4N1). Similarly, the probability that A ( 0 , 2 )
changes to A(0, 1) in one generation is 2/(4N2). These
events are also shown in Figure 2.
In the same way, we can obtain the necessary prob-
abilities when the number of DNA sequences is larger
than two, which approximately become as follows:
Prob(A(i, j ) to A(i
-
1 , j+
1)in one generation) = iml (1)
Prob(A(i, j ) to A(i
+
1 , j-
1)in one generation) = jm2 (2)
Prob{A(i, j ) to A(i
-
1 , j )in one generation) =
i(i
-
1 ) / 4 N I (3)Prob(A(i, j ) to A(i, j
-
1)in one generation) = j ( j
-
1)/4N2. (4)For the derivations of (3) and (4), see HUDSON (1983)
or TAJIMA (1983). In the formulations of ( 1 ) and ( 2 )
FIGURE Z.-Flowchart of the state, A ( i j ) , that i and j DNA
sequences are randomly chosen from subpopulations 1 and 2, respectively.
the higher order terms of m1 and m2 were ignored.
This also means that each state can change only to the
adjacent states. Figure 2 shows these probabilities
when the number of DNA sequences is smaller than
six. We should again keep in mind that we are going back to the past.
Let us now obtain the expected number of segre- gating sites in a sample. Let S ( i , j ) be the expected
number of segregating sites in a sample of n DNA
sequences among which i DNA sequences are sampled
from subpopulation 1 and j ( = n
-
i) DNA sequencesfrom subpopulation
2.
Under the infinite site model,this number is equal to the expected number of mu- tations which take place while A(i, j ) changes to A( 1,
0 ) or A(0, 1). First, we notice that the expected num-
ber of generations, during which A(i, j ) remains the same, is equal to the reciprocal of the probability that
A(i, j ) changes to the other states in one generation. Using (1)-(4), this probability is given by
i(i
-
1 ) + jo'-
1)B
= iml+
jm2+
-
-
4N1 4N2 9 ( 5 )
so that the expected number of generations is 1/B.
Since on the average nv mutations take place in each
generation, the expected number of mutations oc-
curred while A(i, j ) remains the same is given by
C = nv/B
(6) . .
-
-
nM1M2DNA Polymorphism in Subpopulations
where MI = 4Nlv, M2 = 4NzV, Q1 = 4Nlml and Q 2 = S(1, 1)
23 1
4N2m2.
When A(i, j) changes, it changes to one of four
possible states, A(i
-
1, j+
l), A(i+
1, j-
l), A(i-
1 ,j) and A(i, j
-
1). These probabilities can be obtainedfrom (1)-(4). That is, the probability that A(i, j)
changes to A(i
-
1, j+
1) when it changes is iml/B, that of A(i+
1, j-
1) is jm2/B, that of A(i-
1, j ) isi(i
-
1)/(4NlB) and that of A(i, j-
1) is j ( j-
l)/ (4N2B). Therefore, S ( i , j ) can be given byS ( i , j ) = C + - S ( i - im 1 l , j + 1) B
j m 2
+-S(i+ 1 , j - l)+-S(i-
i(i-
1) 1 , j )B 4N1B
( 7 )
+
JU
q i , j-
1) 4N2B-
-
nMIMz+
avM1+
bvM2j ( j
-
1+
@)M1+
i (i-
1+
Q1)MzQ2(1
+
@)M:+
2( 1+
Q 1 )Q4
1+
Q2)MI+
Ql(1+
Q1)Mz '-
-
d
(9)
S(0, 2)
-
- @M:
+
@(3+
~ Q I ) M I M Z+
Ql(1+
Q ) M ;Qz(1
+
@)M1+
QI(1+
Q1)MzWhen n = 3, from
( 7 )
we haveS(3, 0)
-
-
MI+
2S(2, 0)+
QlS(2, 1)2
+
Q1S(2, 1)
~ M I M z
+
@M1S(3, 0)-
-
+
2MZ[S(I, 1)+
QlS(1, 2)] Q2M1+
2(1+
Q1)Mz 'where S(1, 2)
ad = j [ ( j
-
l)S(i, j-
1)+
@S(i+
1, j-
l)],~ M I M z
+
2M1[S(1, 1)+
@S(2, l)]and
-
+
QIMZS(0, 3)-
2(1
+
@)M1+
QlM2, (10)
bq =
i[(i
-
1)S(i-
1, j)+
@S(i-
1, j+
l)].Incidentally,
(7)
can be also obtained from S ( 0 , 3)S ( i , j) = (1
-
B)S(i, j)+
imlS(i-
1, j+
1) +jmzS(i+
1 , j-
1)-
M2+
2S(O, 2)+
QZS(1, 2)-
2
+
Q2+
[i(i-
1)/(4N])]S(i-
1, j)+
[ j ( j-
1)/(4N~)]S(i, j-
1)+
nv, which can be derived by using the theory of Markov chains with transient states. Solving this equation, we have( 7 ) .
We notice S(1, 0) = S ( 0 , 1) = 0 from the definition
of S ( i , j ) . T h e above formulae look complicated. T h e
computations of S ( i , j ) , however, are straightforward although they are not always easy.
For example, when n = 2, from
(7)
we haveM1
+
QlS(1, 1) 1+
Q1S(2, 0) =
S(1, 1) =
S(0, 2) =
9
2M1M2
+
@M1S(2, 0)+
QIMZS(O~ 2), (8)QZMI
+
Q1M2M2
+
@S(1, 1)1
+
Q2 ,which are equivalent to equations (9) in STROBECK
(1 987) and equations (1 3) in SLATKIN (1 987). Solving (8), we have
S(2, 0)
-
-
@U
+ @)M:+
Ql(3+
2QZ)MIMZ+
@M;@(l
+
Q2)MI+
Ql(1+
Q1)MZSince S(2, 0), S(1, 1) and S(0, 2) are already known, we can solve (1 0). Practically, however, it is not easy to obtain the solution of (1 0). If we use a computer, we can avoid such a difficulty. Especially, we can easily obtain the solution of (1 0) if the software for solving simultaneous linear equations is available. In the case where such a program package is not available, we can solve (10) by using (10) repeatedly. That is, we first substitute arbitrary values of S ( 3 , 0), S(2, l), S(1,
2) and S(0, 3) into (10). Then we obtain new values
of S ( 3 , 0), S(2, l), S(1, 2) and S(0, 3). We again
substitute these values into (lo), then obtain new
values. Repeating this process many times, we finally obtain the true values of S ( 3 , 0), S(2, l ) , S(1, 2) and S(0, 3). T h e number of iterations required depends
on the values of Q1 and Q, and the accuracy we seek.
In general, many iterations are required for large
values of Q1 and Q2.
In the case where n is larger than 3, we can obtain
the values of S(n, 0), S(n
-
1 , l),. . .
and S(0, n) from S(n-
1, 0), S(n-
2, l),.
. .
and S(0, n-
1 ) by using(7).
In this case we need the software for solvingsimultaneous linear equations, or we can use the same
iteration method as mentioned in the case of n = 3.
At any rate we can obtain those values for n = 4 from
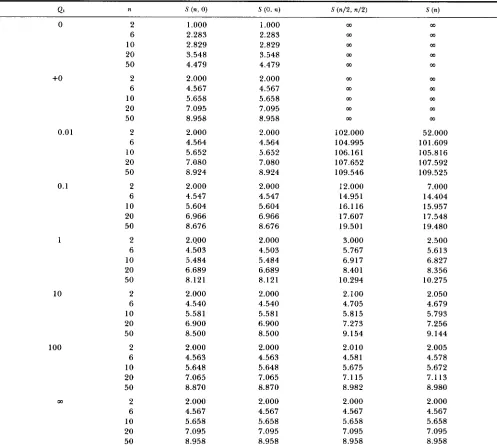
TABLE 1
Expected number of segregating sites in the case of M , = = 1 and Q =
+O 0.01 0.1 1 10 100 m
0 2
6 10 20 50 2 6 10 20 50 2 6 10 20 50 2 6 10 20 50 2 6 10 20 50 2 6 10 20 50 2 6 10 20 50 2 6 10 20 5 0
1
.ooo
2.283 2.829 3.548 4.479 2.000 4.567 5.658 7.095 8.958 2.000 4.564 5.652 7.080 8.924 2.000 4.547 5.604 6.966 8.676 2.400 4.503 5.484 6.689 8.121 2.000 4.540 5.581 6.900 8.500 2.000 4.563 5.648 7.065 8.870 2.000 4.567 5.658 7.095 8.958
1
.ooo
2.283 2.829 3.548 4.479 2.000 4.567 5.658 7.095 8.958 2.000 4.564 5.652 7.080 8.924 2.000 4.547 5.604 6.966 8.676 2.000 4.503 5.484 6.689 8.121 2.000 4.540 5.581 6.900 8.500 2.000 4.563 5.648 7.065 8.870 2.000 4.567 5.658 7.095 8.958 m m m m m m m m m m 102.000 104.995 106.161 107.652 109.546 12.000 14.951 16.1 16 17.607 19.501 3.000 5.767 6.91 7 8.401 10.294 2.100 4.705 5.815 7.273 9.154 2.010 4.581 5.675 7.115 8.982 2.000 4.567 5.658 7.095 8.958 m m m m m m m m m m 52.000 101.609 105.816 107.592 109.525 7.000 14.404 15.957 17.548 19.480 2.500 5.613 6.827 8.356 10.275 2.050 4.679 5.793 7.256 9.144 2.005 4.578 5.672 7.113 8.980 2.000 4.567 5.658 7.095 8.958+O means that Q1 is close to 0, but not 0.
from those of n = 4. In this way we can obtain the values of S(n, 0), S(n
-
1, l),. . .
and S(0, n ) for anyn.
Let us next consider the case where n DNA se-
quences are randomly sampled from the entire pop- ulation. Let S ( n ) be the expected number of segregat-
ing sites in n DNA sequences randomly sampled from
the entire population. T h e probability that i and
n
-
i DNA sequences are randomly chosen fromsubpopulations 1 and 2, respectively, given that n
DNA sequences are randomly chosen from the entire
population, is given by
n ! N’;N;-:
P(i, n
-
;‘
-
“’’
-
i !(n-
i) ! ( N I+
N2)n-
n! M\M;”-
i !(n
-
i) ! ( M I+
M2)nso that S(n) can be obtained from
n
S(n) = P(i, n
-
i ) S ( i , n-
i). (12)DNA Polymorphism in Subpopulations 233
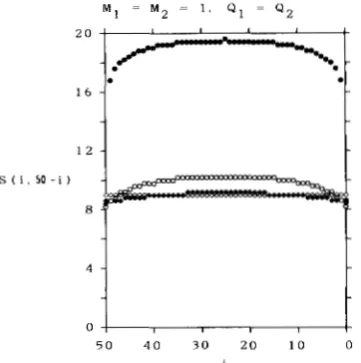
M 1 = M 2 = 1 , Q1 = Q 2
2 o
.
j
-.
[
l 6 1 2
1.
S ( i . 5 0 - i )
8
l 5
m
M I = 0 . 1 . M 2 = 1 . 9, Q , = Q 2
1 2
1
S ( i . 5 0 - i )
1
t
0 1 I
c
5 0 4 0 3 0 2 0 1 0 0
1
FIGURE 3.-Expected number, S ( i , 50
-
i ) , of segregating sites in a sample of 50 DNA sequences among which i DNA sequences are sampled from subpopulation 1 and 50-
i DNA sequences from subpopulation 2. M I = M Z = 1 and QI = Qz are assumed. 0, QI =0.1; 0, QI = 1 ; +, Q1 = 10; 0, Q1 = m.
NUMERICAL COMPUTATIONS
Let us now examine the effect of population sub- division or migration on the number of segregating sites in a sample by using the method developed in
the foregoing section. Here we will consider the six
cases.
(a) N1 = N z , m l = m2.
First we consider the case where the sizes of two subpopulations are the same and the migration rate
from subpopulation
2
to subpopulation 1 is the sameas that from subpopulation 1 to subpopulation
2.
Inthis case M1 = M z and Q1 = Q2. This is a special case of the finite island model. Table 1 gives the values of
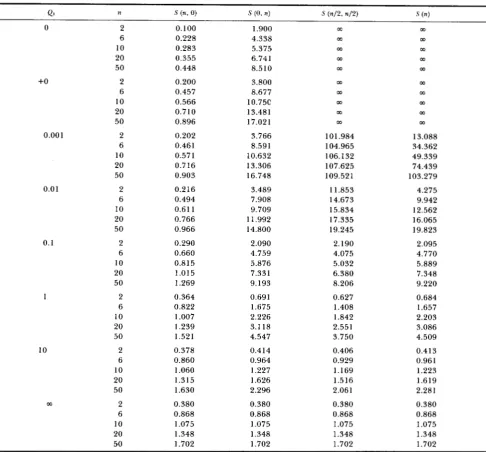
S(n, 0), S ( 0 , n), S ( n / 2 , n / 2 ) and S(n) in this case. In this table +O means that Ql or ml is close to 0, but not
0. From this table, we can see that, in the case where
two DNA sequences are sampled from the same sub- population, the expected number of segregating sites
[S(2, 0) or S ( 0 ,
2)]
is the same as long as the migrationrate is not 0. This is consistent with the results ob- tained by LI (1 976) using the finite island model. That
is, the expected number of nucleotide differences
between the two DNA sequences randomly sampled
from the same subpopulation is independent of the migration rate unless the migration rate is 0. This is also true in the case where three DNA sequences are sampled, namely S(3, 0) = S ( 0 , 3) = 3 for Q1 =
Q2
>
0. When the sample size is more than three, however, the expected number of segregating sites in a sample depends on the migration rate. Table 1 shows that
S(n, 0) or S ( 0 , n) takes the smallest value when m l is 1, and that the value of S(n, 0) or S ( 0 , n) obtained by approaching m l to 0 is the same as that for the infi-
5 0 4 0 3 0 2 0 I O o
1
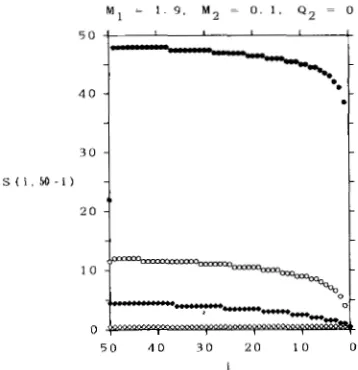
FIGURE 4.-Expected number, S ( i , 50 - i ) , of segregating sites in a sample of 50 DNA sequences among which i DNA sequences are sampled from subpopulation 1 and 50
-
i DNA sequences from subpopulation 2. M I = 0.1, M2 = 1.9 and QI = Q 2 are assumed. 0 ,Q, = 0.1; 0, Q1 = 1; +, Ql = 10; 0, Q, = m.
nitely large value of ml. T h e values of S ( n / 2 , n/2) and
S(n) decrease as the migration rate increases, as ex-
pected. Figure 3 shows S ( i , j ) when n = 50. From this
figure, we can see the effect of migration on the value
of S(i, j ) .
(b) N 1
<
N z , QI = Q2.Next, we consider the case where the sizes of two subpopulations are different and the number of indi-
viduals which migrate from subpopulation 1 to sub-
population 2 is the same as that from subpopulation
2 to subpopulation 1. Table
2
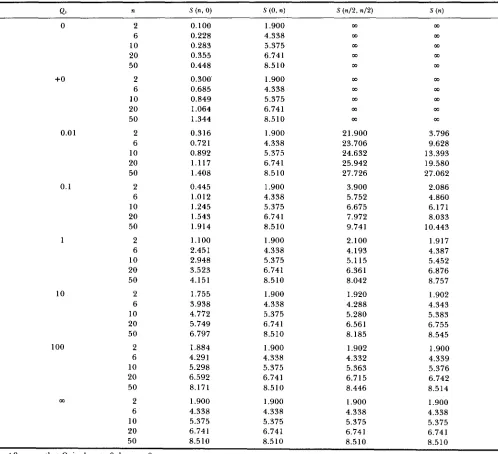
shows the case whereM1 = 0.1 and M2 = 1.9. First, we notice that, as the
migration rate increases, the expected number of
segregating sites approaches the number of segregat- ing sites expected under the random mating popula- tion with size N 1
+
N2. Next, we notice that, as themigration rate decreases, S(n, 0) decreases but S ( 0 , n)
increases. T h e latter property is important, since the expected number of segregating sites [ S ( O , n)] in n
DNA sequences sampled from the large subpopula-
tion is larger than that of random mating population with size N1
+
N2. Figure 4 also shows the effect ofmigration on the expected number of segregating sites
in a sample.
(c) N 1
<
N 2 , ml = m2.Here, we examine the case where the sizes of two
subpopulations are different and the proportion of
immigrants in each subpopulation is the same in every
generation. Table 3 shows the case where MI = 0.1,
M I - 0 . 1. M 2 - 1. 9 , Q 2 = 1 9 Q ,
1 5
S ( i . 5 0 - i )
1 0
5
0 -I I
5 0 4 0 3 0 2 0 1 0 0
1
FIGURE 5.-Expected number, S ( i , 50
-
i), of segregating sites in a sample of 50 DNA sequences among which i DNA sequences are sampled from subpopulation 1 and 50-
i DNA sequences from subpopulation 2. MI = 0.1, MP = 1.9 and Q:! = 19Q are assumed.A , Q 1 = O . O l ; O , Q I = O . l ; O , Q 1 = l ; O , Q l = ~ .
MI ~ 0 . 1. M 2 = 1. 9 . Q 2 ~ 0
l 5
-
l 2
i
t
5 0 4 0 3 0 2 0 1 0 0
I
FIGURE 6.-Expected number, S ( i , 50 - i), of segregating sites in a sample of 50 DNA sequences among which i DNA sequences are sampled from subpopulation 1 and 50
-
i DNA sequences from subpopulation 2. M I = 0.1, MP = 1.9 and Q2 = 0 are assumed. 0 ,41 = 0.1; 0,
el
= 1 ; +, Q1 = 10; 0, = m.sites in n DNA sequences randomly sampled from the
same subpopulation approaches that of random mat- ing population with size ~ N I N ~ / ( N ~
+
N2).
Large val- ues of S ( 0 , n) are also observed in the case of smallis also shown in Figure 5 .
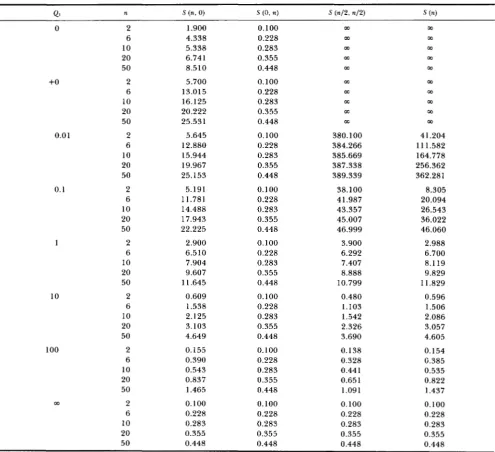
(d) N 1
<
N P , m2 = 0.In this and the following cases we assume that the migration takes place only in one direction, namely
from subpopulation 2 to subpopulation 1. Therefore,
the expected number of segregating sites in DNA
sequences randomly sampled from subpopulation 2 is
1 8
S ( i . 5 0 - i )
1 2
6
0
5 0 4 0 3 0 2 0 1 0 0
1
FIGURE 7,"Expected number, S ( i , 50 - i), of segregating sites in a sample of 50 DNA sequences among which i DNA sequences are sampled from subpopulation 1 and 50
-
i DNA sequences from subpopulation 2. M I = M2 = 1 and 42 = 0 are assumed. 0 , Q1 =0.1; 0, Q1 = 1 ; +, QI = 10; 0, QI = m.
M I - 1. 9 . M 2 = 0 . 1 . Q 2 = 0
5 0 4 4
I
-.
-.
i
0
5 0 4 0 3 0 2 0 1 0 0
1
FIGURE 8.-Expected number, S(i, 50 - i), of segregating sites
in a sample of 50 DNA sequences among which i DNA sequences are sampled from subpopulation 1 and 50
-
i DNA sequences from subpopulation 2. MI = 1.9, M:! = 0.1 and42
= 0 are assumed. 0,Ql = 0.1; 0, Q1 = 1; +, Q I = 10; 0 , Q , = m.
independent of the rate of migration from subpopu-
lation
2
to subpopulation 1. Table4
shows the casewhere M , = 0.1 and MP = 1.9. As expected, the
expected number of segregating sites in DNA se-
quences randomly sampled from subpopulation 1 in-
creases as the migration rate increases, and reaches
the same value as that of subpopulation 2. Figure 6 also shows the effect of migration on the expected number of segregating sites.
(e) N 1 = N P , m2 = 0.
Table 5 shows the case where MI = M P = 1 and
4
2
DNA Polymorphism in Subpopulations 235
TABLE 2
Expected number of segregating sites in the case of M , = 0.1, M 2 = 1.9 and =
41 n S (n, 0) S (0, n) S ( 4 2 , n I 2 ) s ( 4
0 2 0.100 1.900 m
6 0.228 4.338 m
10 0.283 5.375 m m
20 0.355 6.741 m
50 0.448 8.510 m
m
03
m
m
+O
0.01
0.1
1
10
100
m
2 0.290 2.090
6 0.662 4.772
10 0.820 5.913
20 1.029 7.415
50 1.299 9.362
m m m m m
m
m
m
m
m
2 0.307 2.089 2 1
.ooo
3.8816 0.700 4.770 22.926 9.747
10 0.867 5.909 23.878 13.421
20 1.086 7.409 25.208 19.361
2 0.445 2.082 3.900 2.250
10 1.245 5.881 6.831 6.557
20 1.544 7.366 8.163 8.409
2 1.145 2.045 2.190 2.057
6 2.552 4.666 4.401 4.697
10 3.070 5.775 5.366 5.821
20 3.670 7.228 6.663 7.300
50 4.325 9.096 8.397 9.208
2 1.845 2.008 2.019 2.009
6 4.137 4.585 4.509 4.587
10 5.008 5.680 5.547 5.683
20 6.024 7.121 6.880 7.126
50 7.108 8.984 8.561 8.996
2 1.983 2.001 2.002 2.001
6 4.516 4.569 4.560 4.569
10 5.574 5.660 5.644 5.661
20 6.931 7.098 7.063 7.099
50 8.583 8.961 8.876 8.962
50 1.368 9.351 27.005 26.472
6 1.012 4.751 5.873 5.208
50 1.917 9.282 9.960 10.738
2 2.000 2.000 2.000 2.000
6 4.567 4.567 4.567 4.567
10 5.658 5.658 5.658 5.658
20 7.095 7.095 7.095 7.095
50 8.958 8.958 8.958 8.958
+O means that Ql is close to 0, but not 0.
segregating sites in DNA sequences randomly sampled
from subpopulation 1 decreases as the migration rate
increases. Large values of S ( n , 0) are observed when
the migration rate is small. T h e effect of migration
on S ( i , j ) is also shown in Figure
7.
( f ) N I
>
NP, m2 = 0.Here we examine the case where the migration takes place only in the direction from a small subpop- ulation to a large subpopulation. This is an unusual case, so that this type of migration is very rare in nature, if any. Table 6 shows the case where M I =
1.9, M2 = 0.1 and Q 2 = 0. Extremely large values of
S(n, 0) are observed when the migration rate is small.
Figure 8 also shows the relationship between the
migration rate and the expected number of segregat- ing sites in a sample.
CONCLUSION AND DISCUSSION
In this paper we have examined the effect of pop-
ulation subdivision or migration on the expected num-
ber of segregating sites in a sample by using the two subpopulation model, and obtained the following re- sults: (1) T h e expected number of segregating sites in
+O
0.001
0.0 1
0.1
1
10
m
TABLE 3
Expected number of segregating sites in the case of M , = 0.1, M 2 = 1.9 and = 19@
QI n S (n, 0) S (0, n) S (n/2,n/2) S (n)
0 2 0.100 1.900 m m
6 10
m
20
m
50 0.448 8.510 m m
m
2 0.200 3.800 m
6 0.457 8.677 m
m
10
m
0.566 10.750 m
20 0.710 m m
m
13.48 1
50 0.896 17.021 m m
0.228 4.338 0.283 5.375
0.355
m m
6.741 m
2 0.202 3.766 101.984 13.088
6 0.461 8.591 104.965 34.362
10 0.571 10.632 106.132 49.339
20 0.716 13.306 107.625 74.439
50 0.903 16.748 109.521 103.279
2 0.2 16 3.489 11.853 4.275
6 0.494 7.908 14.673 9.942
10 0.61 1 9.709 15.834 12.562
20 0.766 1 1.992 17.335 16.065
50 0.966 14.800 19.245 19.823
2 0.290 2.090 2.190 2.095
6 0.660 4.759 4.075 4.770
10 0.815 5.876 5.032 5.889
20 1.015 7.331 6.380 7.348
50 1.269 9.193 8.206 9.220
2 0.364 0.691 0.627 0.684
6 0.822 1.675 1.408 1.657
10 1.007 2.226 1.842 2.203
20 1.239 3.1 18 2.551 3.086
50 1.521 4.547 3.750 4.509
2 0.378 0.414 0.406 0.41 3
6 0.860 0.964 0.929 0.961
10 1.060 1.227 1.169 1.223
20 1.315 1.626 1.516 1.619
50 1.630 2.296 2.061 2.281
2 0.380 0.380 0.380 0.380
6 0.868 0.868 0.868 0.868
10 1.075 1.075 1.075 1.075
20 1.348 1.348 1.348 1.348
50 1.702 1.702 1.702 1.702
+O means that Q1 is close to 0, but not 0.
subpopulation generally depends on the migration
rate. T h e expected number of segregating sites in the two DNA sequences randomly sampled from the same subpopulation, however, is independent of the migra- tion rate when the pattern of migration is symmetrical such as the finite island model or case (a). This is also
true when the three DNA sequences are sampled.
When the sample size is larger than three, this is not the case.
(2)
In some cases [in cases (b), (c), (e) and(f)], the expected number of segregating sites in DNA
sequences randomly sampled from the same subpop- ulation can be larger than that expected from the
random mating population whose effective size is the
same as that of the entire population or the sum of
the two effective sizes of subpopulations. In other
words, the population subdivision can increase the
amount of DNA polymorphism even in a subpopula-
tion in some cases.
Recently Tajima (1989) has developed a statistical
method for testing the neutral mutation hypothesis
by using the relationship between the number of
segregating sites and the average number of (pairwise)
nucleotide differences among a sample of DNA se-
quences. This method assumes that the population
DNA Polymorphism in Subpopulations 237
TABLE 4
Expected number of segregating sites in the case of M, = 0.1, M p = 1.9 and @ = 0
0.01
0.1
1
10
100
0 2 0.100 1 .goo m m
6 0.228 4.338 m m
10 0.283 5.375 m m
20 0.355 6.741 m m
50 0.448 8.510 m m
+O 2 0.300 1 .goo m m
6 0.685 4.338 m m
10 0.849 5.375 m m
20 1.064 6.741 m m
50 1.344 8.510 m m
2 0.316 1.900 2 1.900 3.796
6 0.72 1 4.338 23.706 9.628
10 0.892 5.375 24.632 13.393
20 1.117 6.741 25.942 19.580
50 1.408 8.5 10 27.726 27.062
2 0.445 1 .goo 3.900 2.086
6 1.012 4.338 5.752 4.860
10 1.245 5.375 6.675 6.171
20 1.543 6.741 7.972 8.033
50 1.914 8.510 9.741 10.443
2 1.100 1.900 2.100 1.917
6 2.451 4.338 4.193 4.387
10 2.948 5.375 5.115 5.452
20 3.523 6.741 6.361 6.876
50 4.151 8.510 8.042 8.757
2 1.755 1.900 1.920 1.902
10 4.772 5.375 5.280 5.383
20 5.749 6.741 6.56 1 6.755
2 1.884 1 .goo 1.902 1.900
6 4.291 4.338 4.332 4.339
5.298 5.375 5.363 5.376
50 8.171 8.510 8.446 8.514
1.900 4.338
6 3.938 4.338 4.288 4.343
50 6.797 8.510 8.185 8.545
10 20 6.592 6.741 6.715 6.742
m 2 1 .goo 1 .goo 1.900
4.338 4.338 4.338
6
10 20 6.741 6.741 6.741 50
5.375 5.375 5.375 5.375
6.741 8.510 8.510 8.510 8.510
0.01
0.1
1
10
100
0 2 1.000 1
.ooo
m6 2.283 2.283 m
10 2.829 2.829 m
20 3.548 3.548
50 4.479
+O 2 3.000 1.000 m
m
m
m
m m
4.479 m m
6
10 20
50 13.438 4.479 m m
m
6.850 2.283 m m
8.487 2.829 m
10.643
m m
3.548 m
2 2.980 1.000 201
.ooo
101.49510 8.418 2.829 205.153 204.614
20 10.542 3.548 206.644 206.583
6 6.800 2.283 203.987 197.507
50 13.280 4.479 208.538 208.5 17
1 1.455
6.396 2.283 23.883 23.040
10 7.867 2.829 25.040 24.858
20 9.743 3.548 26.528 26.468
50 12.070 4.479 28.421 28.399
2 2.000 1
.ooo
3.000 2.2506 4.488 2.283 5.300 5.099
10 5.446 2.829 6.375 6.264
20 6.610 3.548 7.817 7.765
50 7.980 4.479 9.693 9.673
2 1.182 1
.ooo
1.200 1.1456 2.757 2.283 2.719 2.687
10 3.500 2.829 3.470 3.443
20 4.541 3.548 4.584 4.562
50 5.934 4.479 6.224 6.210
2 1.020 1.000 1.020 1.015
6 2.342 2.283 2.331 2.328
10 2.924 2.829 2.905 2.902
20 3.726 3.548 3.692 3.690
50 4.862 4.479 4.809 4.806
m 2 1.000 1.000 1.000 1.000
6 2.283 2.283 2.283 2.283
10 2.829 2.829 2.829 2.829
20 3.548 3.548 3.548 3.548
50 4.479 4.479 4.479 4.479
2 2.818 1.000 2 1.000
6
DNA Polymorphism in Subpopulations 239
TABLE 6
Expected number of segregating sites in the case of M, = 1.9, M n = 0.1 and = 0
+O
0.01
0.1
1
10
100
0 2 1 .goo 0.100 m m
6 4.338 0.228 m m
10 5.338 0.283 m m
20 6.741 0.355 m m
50 8.510 0.448 m m
2 5.700 0.100 m m
6 13.015 0.228 cc m
10 16.125 0.283 cc
20 20.222 0.355 m m
50 25.531 0.448 cc m
m
2 5.645 0.100 380.100 4 1.204
6 12.880 0.228 384.266 11 1.582
10 15.944 0.283 385.669 164.778
20 19.967 0.355 387.338 256.362
50 25.153 0.448 389.339 362.281
2 5.191 0.100 38.100 8.305
6 11.781 0.228 41.987 20.094
10 14.488 0.283 43.357 26.543
20 17.943 0.355 45.007 36.022
50 22.225 0.448 46.999 46.060
2 2.900 0.100 3.900 2.988
6 6.510 0.228 6.292 6.700
10 7.904 0.283 7.407 8.1 19
20 9.607 0.355 8.888 9.829
50 1 1.645 0.448 10.799 1 1.829
2 0.609 0.100 0.480 0.596
6 1.538 0.228 1.103 1.506
10 2.125 0.283 1.542 2.086
20 3.103 0.355 2.326 3.057
50 4.649 0.448 3.690 4.605
2 0.155 0.100 0.138 0.154
10 0.543 0.283 0.441 0.535
20 0.837 0.355 0.65 1 0.822
m 2 0.100 0.100 0.100 0.100
6 0.390 0.228 0.328 0.385
50 1.465 0.448 1.091 1.437
6 0.228 0.228 0.228 0.228
10 0.283 0.283 0.283
20
0.283 0.355 0.355 0.355 0.355
50 0.448 0.448 0.448 0.448
rium unless the migration rate is very small. That is, the expected number of segregating sites in a sample is close to that expected in the random mating popu-
lation, when
Q1
is larger than 10. This means that themigration of only a few number of individuals in every
generation is sufficient for his method to be applicable
as long as the effective population sizes stay constant. In this paper we have used the two subpopulation
model, and obtained interesting results of which some
could not be obtained from the other models. Al-
though the actual pattern of migration in natural
populations seems to be more complicated than the two subpopulation model, this model might be useful for obtaining a rough idea about the effect of popu- lation subdivision or migration on DNA polymor- phisms.
I thank two anonymous referees for useful comments on this paper.
LITERATURE CITED
GRIFFITHS, R. C., 1980 Lines of descent in the diffusion approx- imation of neutral Wright-Fisher models. Theor. Popul. Biol. 17: 37-50.
HUDSON, R. R., 1983 Testing the constant-rate neutral allele model with protein sequence data. Evolution 37: 203-217.
KIMURA, M., 1968 Evolutionary rate at the molecular level. Na- ture 217: 624-626.
KIMURA, M., 1969 The number of heterozygous nucleotide sites maintained in a finite population due to steady flux of muta- tions. Genetics 61: 893-903.
KIMURA, M., 1983 The Neutral Theory of Molecular Evolution.
Cambridge University Press, London.
KIMURA, M., and G. H. WEISS, 1964 The stepping stone model of population structure and the decrease of genetic correlation with distance. Genetics 4 9 561-576.
KINGMAN, J. F. C., 1982 On the genealogy of large populations. J. Appl. Probab. 19A: 27-43.
LATTER, B. D. H., 1973 T h e island model of population differ- entiation: A general solution. Genetics 73: 147-157.
LI, W.-H., 1976 Distribution of nucleotide differences between two randomly chosen cistrons in a subdivided population: the finite island model. Theor. Popul. Biol. 10: 303-308.
LI, W.-H., A N D M. NEI, 1975 Drift variances of heterozygosity and genetic distance in transient states. Genet. Res. 25: 229- 248.
LI, W.-H., and M. NEI, 1977 Persistence of common alleles in two related populations or species. Genetics 86: 901-914.
MARUYAMA, T., 1969 Genetic correlation in the stepping stone model with non-symmetrical migration rate. J. Appl. Probab. 6: 463-477.
MARUYAMA, T., 1970 Effective number of alleles in a subdivided population. Theor. Popul. Biol. 1: 273-306.
MARUYAMA, T., 1977 Stochastic Problems in Population Genetics.
Springer-Verlag, Berlin.
NEI, M., 1975 Molecular Population Genetics and Evolution. North- Holland, Amsterdam.
NEI, M., and M. W. FELDMAN, 1972 Identity of genes by descent within and between populations under mutation and migration pressures. Theor. Popul. Biol. 3: 460-465.
SLATKIN, M., 1987 The average number of sites separating DNA sequences drawn from a subdivided population. Theor. Popul. Biol. 32: 42-49.
STROBECK, C., 1987 Average number of nucleotide differences in a sample from a single subpopulation: a test for population subdivision. Genetics 117: 149-153.
TAJIMA, F., 1983 Evolutionary relationship of DNA sequences in finite populations. Genetics 105: 437-460.
TAJIMA, F., 1989 Statistical method for testing the neutral muta- tion hypothesis by DNA polymorphism. Genetics (in press). WATTERSON, G. A , , 1975 On the number of segregating sites in
genetic models without recombination. Theor. POPU~. Biol. 7:
256-276.
WEISS, G. H., and M. KIMURA, 1965 A mathematical analysis of
the stepping stone model of genetic correlation. J. Appl. Probab. 2: 129-149.
WRIGHT, S . , 1931 Evolution in Mendelian populations. Genetics 16: 97-159.