ABSTRACT
LINDSEY, ERICK ALLEN. Development of Novel Small Molecules for Combating Infectious Disease. (Under the direction of Dr. Christian Melander.)
Acquired immunodeficiency syndrome (AIDS) and its causative agent human immunodeficiency virus (HIV) are responsible for nearly 30 million deaths since 1981. Current antiretroviral therapies such as Raltegravir have greatly increased both the quality and life expectancy of HIV infected patients, however the persistence of HIV associated dementia is problematic. The inability of most antiretroviral therapies to cross the blood brain barrier makes the treatment of HIV associated dementia a unique challenge. It has been posited that the use of gold nanoparticle based therapeutics may allow for the targeting of HIV located within traditionally inaccessible tissues. To this end, a thiol containing Raltegravir derivative has been successfully synthesized and conjugated to a 2.0 nm gold nanoparticle. Initial results indicate that these conjugates are capable of inhibiting viral replication by approximately 80% when used at 50 nM.
Development of Novel Small Molecules for Combating Infectious Disease
by
Erick Allen Lindsey
A dissertation submitted to the Graduate Faculty of North Carolina State University
in partial fulfillment of the requirements for the degree of
Doctor of Philosophy
Chemistry
Raleigh, North Carolina 2013
APPROVED BY:
______________________________ ______________________________
Dr. Christian Melander Dr. Daniel L. Comins
Committee Chair
______________________________ ______________________________
ii DEDICATION
iii BIOGRAPHY
The author, Erick Allen Lindsey was born in McBain, MI on November 21st 1984 to Charles and Deborah Lindsey. He graduated McBain Public School in 2004, Erick then attended Grand Valley State University (GVSU). In 2008, he graduated from GVSU receiving a Bachelor of Science in Chemistry as well as a minor in mathematics. While at GVSU, Erick also had the opportunity to perform undergraduate research under the direction of Professor Felix Ngassa. He received many awards and honors while at GVSU including the 2008 Student Summer Scholars fellowship and the Outstanding Senior Student Award.
iv ACKNOWLEDGMENTS
I would like to first and foremost thank my wife, Meegan. You have supported me in every way throughout this process. Without you this would not be possible. I cannot show enough appreciation for all you have done for me over our many years together. Mom and Dad, you have undoubtedly played a crucial role in my success both here at NCSU and in life. Thank you for pushing me in the past and for your continued encouragement on future endeavors. Keven and Audrey, thanks for your support over the many years. You have listened countless times to my research ideas, talks, and philosophies, and still accepted me as family. A special thanks to all of my family members for everything that you have done and continue to do for me by way of encouragement throughout this process.
Dr. Christian Melander, you have been a great mentor and boss over the last five years. Your guidance has been crucial during this time and has been instrumental in my development as a scientist. I will always be thankful for your hard work. I would like to thank my committee members: Dr. Daniel Comins, Dr. Reza Ghiladi, and Dr. Gavin Williams for their guidance and input during the course of my studies.
v and science. Christopher Brackett, it has been a rewarding experience to work with someone who cares so deeply about the work they are carrying out, thank you. Thanks also for the many rides home. Rob Furlani, our conversations over the past years have been at the very least both interesting and insightful. I truly appreciate everything you and the Melander lab have done for me. I would also like to thank the members of the Comins, Deiters, and Ghiladi labs for their countless hours of scientific discussion.
While at NCSU I have had the opportunity to work with three impressive undergraduate students. Cristina Alcaraz, Trey Mullikin, and Stew Harsant, you three have taught me so much over the years. Thank you for everything and I hope all of you enjoyed your experience.
vi TABLE OF CONTENTS
LIST OF TABLES ...x
LIST OF FIGURES ... xi
LIST OF SCHEMES... xiii
CHAPTER 1: THE HUMAN IMMUNODEFICIENCY VIRUS: STRUCTURE, LIFECYCLE, AND COMMONLY USED THERAPEUTIC STRATEGIES ...1
1.1 VIRAL INTRODUCTION AND HIV STRUCTURE ...1
1.2 MECHANISM OF HIV INFECTION AND REPLICATION ...3
1.3 CURRENT ANTIRETROVIRAL TREATMENT METHODS ...8
1.4 CONCLUSIONS...12
REFERENCES ...13
CHAPTER 2: THE DEVELOPMENT OF NOVEL ANTI-HIV GOLD NANOPARTICLE THERAPEUTICS ...18
2.1 INTRODUCTION ...18
2.2 INHIBITION OF HIV FUSION WITH MULTIVALENT GOLD NANOPARTICLES 19 2.3 INHIBITION OF VIRAL DNA INTEGRATION ...21
2.4 CONCLUSIONS...27
2.5 EXPERIMENTAL SECTION ...28
vii
CHAPTER 3: INTRODUCTION TO MICROBIAL BIOFILMS ...39
3.1 BACTERIAL BIOFILM OVERVIEW ...39
3.2 SIGNIFICANCE OF BACTERIAL BIOFILMS...40
3.3 RELEVANT BACTERIAL STRAINS ...41
3.4 KNOWN BIOFILM MODULATORS ...43
3.5 CONCLUSIONS...51
REFERENCES ...52
CHAPTER 4: SYNTHESIS AND BIOLOGICAL EVALUATION OF 2-AMINOIMIDAZOLE – CARBAMATE HYBRID BIOFILM AND ANTI-MICROBIAL AGENTS ...57
4.1 INTRODUCTION ...57
4.2 RESULTS AND DISCUSSION ...59
4.3 CONCLUSIONS...68
4.4 EXPERIMENTAL SECTION ...68
APPENDIX ...76
REFERENCES ...110
CHAPTER 5: THE DISCOVERY OF N-1 SUBSTITUTED 2-AMINOBENZIMIDAZOLES AS ZINC-DEPENDENT S. AUREUS BIOFILM INHBITORS ...113
viii
5.2 RESULTS AND DISCUSSION ...115
5.3 CONCLUSIONS...122
5.4 EXPERIMENTAL SECTION ...124
APPENDIX ...154
REFERENCES ...261
CHAPTER 6: INVESTIGATION OF 2-AMINOPYRIMIDINE AS A NOVEL SCAFFOLD FOR BIOFILM MODULATION ...264
6.1 INTRODUCTION ...264
6.2 RESULTS AND DISCUSSION ...266
6.3 CONCLUSIONS...276
6.4 EXPERIMENTAL SECTION ...276
APPENDIX ...304
REFERENCES ...378
CHAPTER 7: ATTEMPTING TO REGULATE PORPHYROMONAS GINGIVALIS BIOIFLMS WITH 2-AMINOBENZIMIDAZOLE DERIVATIVES ...382
7.1 INTRODUCTION ...382
7.2 RESULTS AND DISCUSSION ...383
7.3 CONCLUSIONS...386
7.4 EXPERIMENTAL SECTION ...388
ix CHAPTER 8: COMBATING MYCOBACTERIUM TUBERCULOSIS WITH THE
USE OF ANTI-BIOFILM ADJUVANTS ...434
8.1 INTRODUCTION ...434
8.2 FIRST GENERATION MYCOBACTERIUM TUBERCULOSIS ADJUVANTS...437
8.3 SECOND GENERATION OF MYCOBACTERIUM BIOFILM MODULATORS ...441
8.4 CONCLUSIONS AND FUTURE WORK ...444
8.5 EXPERIMENTAL SECTION ...445
APPENDIX ...457
x LIST OF TABLES
Table 2.1: Survey of reaction conditions for the deprotection of 2.18 ...26
Table 4.1: Biofilm inhibition against various bacterial strains ...63
Table 4.2: MIC values against various bacterial strains ...65
Table 4.3: Biofilm dispersal against various bacterial strains ...67
Table 4.4: Blood lysis assay of the 2-AI/(-)-menthyl carbamates ...68
Table 5.1: MIC values against various bacterial strains ...120
Table 5.2: Biofilm inhibition values against two MRSA strains ...121
Table 6.1: Percent inhibition of biofilm formation at 200 µM ...269
Table 6.2: Summary of IC50 values for the 2-aminopyrimidine amide scaffolds ...270
Table 6.3: Percent inhibition of biofilm formation at 200 µM for the 2nd generation 2-aminopyrimidines ...273
Table 6.4: Summary of IC50 values for the second generation 2-aminopyrimidine scaffolds ...274
Table 7.1: Biofilm inhibition against P. gingivalis ...385
Table 8.1: Summary of IC50 values for the first generation mycobacterium biofilm modulators...439
Table 8.2: Isoniazid resistance suppression against Mtb ...441
xi LIST OF FIGURES
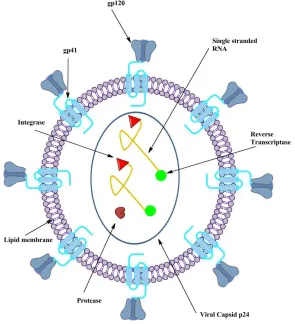
Figure 1.1 Structure of an HIV virion ...2
Figure 1.2: Binding and Fusion of HIV to a CD4+ host cell ...4
Figure 1.3: The process of reverse transcription ...6
Figure 1.4 Integration of viral DNA ...7
Figure 1.5: Examples of ARV entry inhibitors ...9
Figure 1.6: NRTI, NNRTI, and RNase H inhibitors ...10
Figure 1.7: Integrase inhibitors Raltegravir and Elvitegravir ...11
Figure 1.8: Examples of protease and virion maturation inhibitors...12
Figure 2.1: TAK-779 (2.1) and SDC-1721 (2.2) ...20
Figure 2.2: Inhibition of peripheral blood mononuclear cells HIV infection ...21
Figure 2.3: Mechanism of viral DNA integration ...22
Figure 2.4: Raltegravir and modified Raltegravir target for conjugation to gold nano-particles ...23
Figure 2.5: Inhibition of viral replication by 2.21 and 2.21 conjugated gold nano-particles .27 Figure 3.1: The five stages of biofilm development ...40
Figure 3.2: Halogenated furanones and N-acyl homoserine lactones ...44
Figure 3.3: Gram-negative bacteria quorum sensing pathway ...45
Figure 3.4: Gram-positive bacteria quorum sensing pathway ...46
Figure 3.5: Lead biofilm modulators found using high throughput screens ...47
Figure 3.6: Inspirations and evolution of novel anti-biofilm agents ...49
xii
Figure 3.8: Schematic of two-component systems ...51
Figure 4.1: 2-Aminoimidazole anti-biofilm agents based on bromoageliferin ...58
Figure 4.2: (-)-Menthyl carbamate anti-biofilm agent ...59
Figure 4.3: Hybrid 2-AI/(-)-menthyl carbamate anti-biofilm targets ...61
Figure 5.1: Various anti-biofilm agents based on bromoageliferin ...114
Figure 5.2: Remaining N-1-substituted 2-ABI’s examined for biological activity ...117
Figure 5.3: Mitigating effects of divalent cations on two respective N-1-subsituted 2ABIs ...123
Figure 6.1: Known anti-biofilm small molecule scaffolds ...265
Figure 6.2: Example of the anti-biofilm imidazo[1,2-a]pyrimidinium salt and its corresponding 2-AI ...266
Figure 6.3: Evolution of the 2-aminopyrimidine class of biofilm modulators ...267
Figure 6.4: Known 2-aminoimidazoles that suppress antibiotic resistance ...275
Figure 7.1: Lead 2-ABI for the inhibition of P. gingivalis biofilm formation and second generation targets ...383
Figure 7.2: Biotinylated 2-ABI target ...386
Figure 8.1: Examples of mycolic acids used in Mycobacteria ...434
Figure 8.2: Front line Mycobacterium tuberculosis antibiotics ...435
Figure 8.3: Mtb display of biofilm-like mediated isoniazid resistance ...436
Figure 8.4: Mtb display of biofilm-like mediated front-line drug resistance ...436
xiii LIST OF SCHEMES
Scheme 2.1: Synthesis of Raltegravir pyrimidone core ...24
Scheme 2.2: Synthesis of Raltegravir’s active pharmacophores ...24
Scheme 2.3: Completion of modified Raltegravir for conjugation to gold nano-particles ...25
Scheme 4.1: Preparation of 2-AI/(-)-menthyl carbamate hybrids ...62
Scheme 5.1: Synthesis of N-1-substiuted 2-ABI nitro-precursors...115
Scheme 5.2: Synthesis of N-1-substituted 2-ABIs via a one pot reduction-cyclization strategy ...116
Scheme 6.1: Preparation of 2-aminopyrimidines via Sonogashira coupling and hydrogenation ...268
Scheme 6.2: Preparation of 2nd generation 2-aminopyrimidines ...272
Scheme 7.1: Synthesis of second generation heterocyclic-2-ABIs ...384
Scheme 7.2: Synthesis of biotinylated 2-ABI-quinoline derivative ...387
Scheme 8.1: Synthesis of second generation 5-substiuted 2-AP ...442
1 CHAPTER 1
THE HUMAN IMMUNODEFICIENCY VIRUS: STRUCTURE, LIFECYCLE, AND COMMONLY USED THERAPEUTIC STRATEGIES
1.1: Viral introduction and HIV structure
Viruses are the most prevalent biological entity on this planet.1 They have been found to infect living organisms from every branch of the phylogenic tree including archaea, bacteria, and eurkaryota.2 Virus particles, also known as virions, consist of three main components. The first and most crucial of these three components are the nucleic acids, which are responsible for the genetic information of the virion. The genomic information is encapsulated and protected by a protein coat. These core structures are in turn protected by the glycoprotein viral envelope. With only these few fundamental molecules virions lack the operational machinery to reproduce without a host cell.
Human immunodeficiency virus (HIV) is the primary causative agent of acquired immunodeficiency syndrome3 (AIDS) and is therefore responsible for nearly 30 million deaths since 1981.3 There are currently 34.2 million people living with HIV globally, making it one of the largest pandemics to ever affect the human population.4 In 2011 alone, more than 2.5 million people became infected with HIV, while AIDS resulted in 1.7 million deaths.4 HIV is a member of the Retroviridae family of viruses. Retroviruses, such as HIV, use ribonucleic acid rather than deoxyribonucleic acid (DNA) to encode their genetic information. 5
2 Figure 1.1. The virion’s lipid membrane is littered with multimeric glycopeptides (gp) known as gp-120 and gp-41.7 Glycopeptide 41 is an α-helical transmembrane protein that is anchored in the lipid bilayer.8 Glycoprotein 120 is non-covalently bound to gp41, and is located on the surface of the virion.9 As seen in Figure 1.1, the key genetic and biochemical components of the virion are contained within the viral capsid. This capsid consists of a virally encoded protein known as p-24.10 Every HIV particle also contains two identical single stranded RNA molecules, as well as three virally encoded enzymes: reverse transcriptase (RT), integrase, and protease. 1112
3 1.2: Mechanism of HIV infection and replication
In order for HIV replication to occur, the virus must come into contact with a host cell that possesses the cellular machinery for both transcription and translation. The HIV life cycle can be divided into two phases, early and late. The early phase of the HIV life cycle concludes with the integration of viral DNA into the host cell’s genome.13
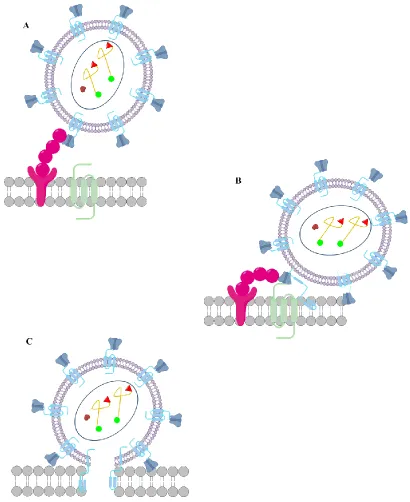
The late phase includes the expression of viral genes and continues until the release of progeny virions.14 The early phase of HIV replication is initiated by the adsorption of the virion to the host cell. HIV displays a remarkable preference for the infection of T-lymphocytes and macrophages,15 due to the high density of the glycoprotein known as cluster of differentiation 4, (CD4) on these cells. 16 HIV’s gp-120 binds to CD4 and induces a conformational change (Figure 1.2 A), which is enhanced by the co-receptor C-C chemokine receptor type 5 (CCR5).17 The binding of gp-120 to both CD4 and CCR5 results in the disassociation of gp-120 and gp-41 (Figure 1.2 B).18 The disassociation of gp-120 and gp-41 induces a conformational shift in gp-41 and places it in a conformation that is suitable for viral fusion (Figure 1.2 C).19
4 Figure 1.2: Binding and Fusion of HIV to a CD4+ host cell A. Binding of gp-120 to CD4 B. Binding of gp-120 to CD4 and CCR5 followed by the disassociation of gp-120 and gp-41 C. Membrane fusion
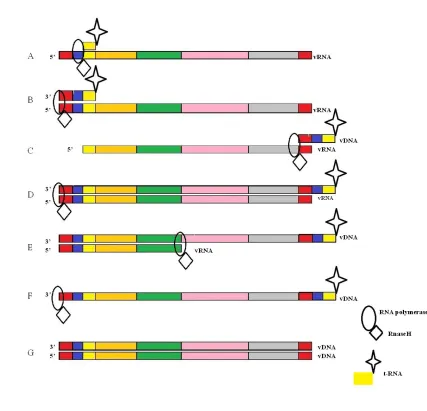
5 RNA (Figure 1.3B) producing a DNA oligo of the primer binding, U5 and R regions of the genome. The RNase H domain is then responsible for the cleavage of the of the P-O-3’ bond of the RNA strand at the U5 and primer binding boundary.25 This Mg (II) catalyzed cleavage results in the disassociation of the viral RNA-DNA hybrid, releasing the viral DNA primer.26 The viral DNA primer then pairs to the R region of the viral RNA at the 3’ terminus (Figure 1.3 C).27 The p-66 polymerase domain of RT then reverse transcribes the viral RNA in a 3’ to 5’ fashion, while this is occurring the p-51 RNase H domain cleaves the viral RNA until only a single strand of complementary viral DNA exists (Figure 1.3 D – F).28 The single stranded viral DNA is then replicated by RT resulting in double stranded DNA that contains the entirety of the HIV genome.23
6 Figure 1.3: The process of reverse transcription A. t-RNA primer binding B: Construction of DNA primer C. Completion of reverse transcription D – F. RNase H mediated RNA cleavage G. DNA replication resulting in double stranded viral DNA, the final product of reverse transcription
7 The remaining structural deficiencies are then repaired, most likely by a cellular DNA repair enzyme, and the incorporation of the now proviral DNA is complete (Figure 1.4 C).12
Figure 1.4 Integration of viral DNA A. Revealing of the Cytosine-Adenosine dinucleotide sequence B. Integration of the viral DNA C. Fully incorporated viral DNA
8 enzymes ensure that HIV proteins such as gp-41, gp-120, and p-24 are operational and can therefore be used in viable future progeny. During normal cellular processes, the proviral DNA is transcribed back into viral RNA. At this point the virus has used the host cell’s machinery to replicate its entire genome; all that remains for the completion of the replication cycle is the assembly and budding process. In order for assembly to occur, the virally encoded gp-160 must be cleaved to its corresponding gp-120 and gp-41 counterparts.36 The assembly of these corresponding viral components occurs spontaneously.36 The gp-120/gp-41 complex then anchors itself to the host’s cellular membrane and the remaining viral components are encapsulated.37 The budding process is thought to be analogous to the binding and fusion events discussed earlier, however it is still poorly understood.38
1.3: Current antiretroviral treatment methods:
Due to the virulence and induced disease states presented by HIV, much effort has been placed on the development of therapeutics known as antiretrovirals 39. Antiretroviral therapies are generally classified by the phase of the retrovirus life cycle that the drug acts upon. These ARV therapies can be broken down into entry inhibitors, RT inhibitors, integrase inhibitors, protease inhibitors and maturation inhibitors.
1.3.1: Entry inhibitors:
9 This binding alters the conformation of CCR5 in a way that hampers HIV’s gp-120 recognition and therefore limits viral binding.42 ARVs that inhibit the fusion process (Figure 1.2 C) are also classified as entry inhibitors. Roche has developed the biomimetic peptide enfuvirtide (1.2, Fuzeon®).43 The competitive binding of 1.2 to gp-41 results in a conformation that is unable to initiate fusion between the host cell and virion.44
Figure 1.5: Examples of ARV entry inhibitors
1.3.2: Reverse transcriptase inhibitors:
10 to be non-competitive inhibitors of RT.48 NNRTIs such as efavirenz (1.4) (Sustiva®) elicit their activity by allosterically inhibiting the enzyme’s ability to continue the chain polymerization process.49 It is suspected that these allosteric interactions limit enzyme flexibility and thereby inhibit polymerization.48
The inhibition of the RNase H domain has recently garnered much attention because of its crucial role in the replication cycle. Derivatives such as 1.5 and 1.6 have been shown to inhibit RNase H activity in HIV.50,51 These derivatives are thought to bind Mg (II) and thereby inhibit the disassociation of the viral RNA-DNA hybrid.52,53
Figure 1.6: NTRI, NNTRI, and RNase H inhibitors
1.3.3: Integrase inhibitors
11 synthesizing the new phosphate linkages that are of necessity for the incorporation of proviral DNA into the host’s cellular DNA.55
Recently, another integrase inhibitor known as elvitegravir (1.8) has been approved by the FDA. Elvitegravir has been shown to display the same mechanism of action as 1.7.56
Figure 1.7: Integrase inhibitors Raltegravir and Elvitegravir 1.3.4: Protease and Maturation inhibitors:
Protease inhibitors prevent HIV replication by binding to the viral proteases. As discussed earlier, the proteases function to generate viable viral proteins. ARVs such as tipranavir (1.9) and indinavir (1.10) competitively inhibit the processing of the Gag poly-proteins, which are responsible for the p-24 viral capsid as well as the cleavage of gp-160.
57-59
12 Figure 1.8: Examples of protease and virion maturation inhibitors
1.4: Conclusions
13 References
(1) Edwards, R. A.; Rohwer, F. Nat. Rev. Microbiol. 2005, 3, 504.
(2) Koonin, E. V.; Senkevich, T. G.; Dolja, V. V. Biol. Direct 2006, 1, 29. (3) UNAIDS 2010.
(4) Organization, W. H. 2013.
(5) Zhang, H.; Dornadula, G.; Orenstein, J.; Pomerantz, R. J. J. Hum. Virol. 2000, 3, 165.
(6) Sougrat, R.; Bartesaghi, A.; Lifson, J. D.; Bennett, A. E.; Bess, J. W.; Zabransky, D. J.; Subramaniam, S. PLoS pathogens 2007, 3, e63.
(7) Chan, D. C.; Kim, P. S. Cell 1998, 93, 681.
(8) Buzon, V.; Natrajan, G.; Schibli, D.; Campelo, F.; Kozlov, M. M.; Weissenhorn, W. PLoS pathogens 2010, 6, e1000880.
(9) Pantophlet, R.; Burton, D. R. Annu. Rev. Immunol. 2006, 24, 739. (10) Montagnier, L. In Encyclopedia of Virology 1999, p 763.
(11) Oliver, R. C.; Brown, L. J. Periodontology 2000 1993, 2, 117.
(12) Lever, M. L. The Molecular Biology of HIV/AIDS; John Wiley & Sons, 1996. (13) Citterio, P.; Rusconi, S. Expert Opin. Invest. Drugs 2007, 16, 11.
(14) Bukrinskaya, A. G. Arch. Virol. 2004, 149, 1067.
14 (18) Moore, J. P.; McKeating, J. A.; Weiss, R. A.; Sattentau, Q. J. Science 1990, 250, 1139.
(19) Blumenthal, R.; Durell, S.; Viard, M. J. Biol. Chem. 2012, 287, 40841. (20) Arhel, N. Retrovirology 2010, 7, 96.
(21) Jacobo-Molina, A.; Ding, J.; Nanni, R. G.; Clark, A. D., Jr.; Lu, X.; Tantillo, C.; Williams, R. L.; Kamer, G.; Ferris, A. L.; Clark, P.; et al. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 6320.
(22) Kohlstaedt, L. A.; Wang, J.; Friedman, J. M.; Rice, P. A.; Steitz, T. A. Science 1992, 256, 1783.
(23) Jacobo-Molina, A.; Arnold, E. Biochemistry 1991, 30, 6351.
(24) Le Grice, S. F.; Naas, T.; Wohlgensinger, B.; Schatz, O. The EMBO journal 1991, 10, 3905.
(25) Yang, W.; Lee, J. Y.; Nowotny, M. Molecular cell 2006, 22, 5.
(26) De Vivo, M.; Dal Peraro, M.; Klein, M. L. J Am Chem Soc 2008, 130, 10955. (27) Le Grice, S. F. J. Biol. Chem 2012, 287, 40850.
(28) Herschhorn, A.; Hizi, A. CMLS 2010, 67, 2717.
(29) Lodi, P. J.; Ernst, J. A.; Kuszewski, J.; Hickman, A. B.; Engelman, A.; Craigie, R.; Clore, G. M.; Gronenborn, A. M. Biochemistry 1995, 34, 9826.
(30) Cai, M.; Zheng, R.; Caffrey, M.; Craigie, R.; Clore, G. M.; Gronenborn, A. M. Nat. Struct. Biol. 1997, 4, 567.
15 (33) Roth, M. J.; Schwartzberg, P. L.; Goff, S. P. Cell 1989, 58, 47.
(34) Engelman, A.; Mizuuchi, K.; Craigie, R. Cell 1991, 67, 1211.
(35) Okumura, Y.; Yano, M.; Murakami, M.; Mori, S.; Towatari, T.; Kido, H. FEBS letters 1999, 442, 39.
(36) Ganser-Pornillos, B. K.; Yeager, M.; Pornillos, O. Adv. Exp. Med. Biol. 2012, 726, 441.
(37) Sundquist, W. I.; Krausslich, H. G. Cold Spring Harbor perspectives in medicine 2012, 2, a006924.
(38) Gan, X.; Gould, S. J. PloS one 2012, 7, e29421.
(39) Yoon, S. S.; Hennigan, R. F.; Hilliard, G. M.; Ochsner, U. A.; Parvatiyar, K.; Kamani, M. C.; Allen, H. L.; DeKievit, T. R.; Gardner, P. R.; Schwab, U.; Rowe, J. J.; Iglewski, B. H.; McDermott, T. R.; Mason, R. P.; Wozniak, D. J.; Hancock, R. E.; Parsek, M. R.; Noah, T. L.; Boucher, R. C.; Hassett, D. J. Developmental cell 2002, 3, 593.
(40) Manoussos Perros, D. A. P., Blanda Luzia Christa Stammen, Anthony Wood; Pfizer: United States, 2008; Vol. 7368460.
(41) Duncan Robert Armour, D. A. P., Blanda Luzia Christa Stammen, Manoussos Perros, Martin Paul Edwards; Pfizer: United States, 2003; Vol. 6586430.
(42) Dorr, P.; Westby, M.; Dobbs, S.; Griffin, P.; Irvine, B.; Macartney, M.; Mori, J.; Rickett, G.; Smith-Burchnell, C.; Napier, C.; Webster, R.; Armour, D.; Price, D.; Stammen, B.; Wood, A.; Perros, M. Antimicrob Agents Chemother 2005, 49, 4721.
(43) Wild, D. P. B. T. J. M. C. T.; Duke University: 1995; Vol. 5464933.
16 (45) Fischl, M. A.; Richman, D. D.; Grieco, M. H.; Gottlieb, M. S.; Volberding, P. A.; Laskin, O. L.; Leedom, J. M.; Groopman, J. E.; Mildvan, D.; Schooley, R. T.; et al. The New England journal of medicine 1987, 317, 185.
(46) Furman, P. A.; Fyfe, J. A.; St Clair, M. H.; Weinhold, K.; Rideout, J. L.; Freeman, G. A.; Lehrman, S. N.; Bolognesi, D. P.; Broder, S.; Mitsuya, H.; et al. Proc. Natl. Acad. Sci. U. S. A. 1986, 83, 8333.
(47) Whitley, R. J. J. Antimicrob. Chemother. 1996, 37 Suppl B, 151. (48) de Bethune, M. P. Antiviral research 2010, 85, 75.
(49) Maga, G.; Ubiali, D.; Salvetti, R.; Pregnolato, M.; Spadari, S. Antimicrob Agents Chemother 2000, 44, 1186.
(50) Dayam, R.; Sanchez, T.; Neamati, N. J Med Chem 2005, 48, 8009.
(51) Dayam, R.; Sanchez, T.; Clement, O.; Shoemaker, R.; Sei, S.; Neamati, N. J Med Chem 2005, 48, 111.
(52) Kawasuji, T.; Yoshinaga, T.; Sato, A.; Yodo, M.; Fujiwara, T.; Kiyama, R. Bioorg Med Chem 2006, 14, 8430.
(53) Kawasuji, T.; Fuji, M.; Yoshinaga, T.; Sato, A.; Fujiwara, T.; Kiyama, R. Bioorg Med Chem 2006, 14, 8420.
17 (55) Bacchi, A.; Carcelli, M.; Compari, C.; Fisicaro, E.; Pala, N.; Rispoli, G.; Rogolino, D.; Sanchez, T. W.; Sechi, M.; Sinisi, V.; Neamati, N. J Med Chem 2011, 54, 8407.
(56) Serrao, E.; Odde, S.; Ramkumar, K.; Neamati, N. Retrovirology 2009, 6, 25. (57) Romines, K. R.; Bundy, G. L.; Schwarts, T. M.; Tommasi, R. A.; Strohbach, J. W.; Turner, S. R.; Thaisrivongs, S.; Aristoff, P. A.; Johnosn, P. D.; Skulnick, H. I.; Skaletzky, L. L.; Anderson, D. J.; Morris, J.; Gammill, R. B.; Luke, G. P. Compounds Useful to Treat Retroviral Infections. U. S. Patent 6,169,181 Jan 2, 2001.
(58) Ferry, J. J.; Baldwin, J. R.; Borin, M. T. Method for Improving the Pharmacokinetics of Tipranavir. U. S. Patent 6,147,095 Nov 14, 2000.
(59) Chodakewita, J. A.; Emini, E. A. Combination Therapy for HIV Infection. U. S. Patent 6,689,761 Feb 10, 2004.
18 CHAPTER 2
THE DEVELOPMENT OF NOVEL ANTI-HIV GOLD NANOPARTICLE THERAPEUTICS
2.1 Introduction
Antiretroviral (ARV) therapies have made great advances in the last 10 years, increasing the quality of life for HIV/AIDS patients; however additional advances in the field are still required. One of the main remaining obstacles in the treatment of HIV is the occurrence of HIV encephalopathy.1 The infection of the central nervous system (CNS) presents many obstacles for the treatment of HIV, including the necessity for therapeutics to cross the blood brain barrier (BBB). The BBB prevents many ARV therapies from entering the CNS and therefore creates a safe haven for HIV.2 Once in the brain, microgilia and macrophages sustain the infection, resulting in the dysregulation of macrophages and the overproduction of proinflammatory cytokines and chemkines ultimately resulting in neuronal damage and the development of HIV associated dementia (HAD).3-5 HAD is clinically characterized by a decline in the infected patients cognitive, behavioral and motor skills, and affects nearly 40% of HIV/AIDS infected patients.6,7
19 cross the BBB and deliver highly effective ARV therapeutics to the CNS. One such novel technology that may allow for the delivery of ARV therapies to the CNS is the use of conjugated gold nanoparticles.
Gold nanoparticle based therapeutics have multiple benefits over small molecule therapeutics. It is possible to covalently conjugate several thiolated therapeutics to a single particle, thereby creating a multivalent drug. The use of multivalency has been exploited by biological systems in many cellular processes resulting in a dramatically enhanced affinity. For example, multivalent binding of hemagglutinin ligands of an influenza virus and the sialic acid surface receptors of erythrocytes occurs with a binding constant (Kd) of 1013 M-1,
while the Kd is estimated to be only 103 M-1 for a single hemagglutinin ligand binding to
sialic acid.9 Such an increase in affinity could greatly increase the efficacy of the ARV therapies. Gold nanoparticles can be coated with tens to hundreds of organic ligands; therefore one could use a combination of ARV therapies, as well as beneficial co-ligands that could easily aid in the therapeutic targeting of desired tissues.
2.2 Inhibition of HIV fusion with multivalent gold nanoparticles10
20 attempt to prove that the multivalency of the nanoparticle can compensate for a low affinity ligand, Dr. Eric Ballard replaced the quaternary ammonium salt with a pegylated thiol tail thereby synthesizing SDC-1721 (2.2). Once synthesized, 2.2 was conjugated to a 2.0 nm diameter gold nanoparticle.
Figure 2.1: TAK-779 (2.1) and SDC-1721 (2.2)
21 both TAK-779 (2.1) and SDC-1721 conjugated gold nanoparticles equally inhibit luciferase activity.10,12,13
Figure 2.2: Inhibition of peripheral blood mononuclear cells HIV infection A. TAK-779 (2.1) HIV inhibition B. SDC-1721 (2.2) HIV inhibition C. Conjugated gold nano-particle HIV inhibition D. Gold nano-nano-particle inhibition E. Inhibition of viral entry as measured by luminescence decrease
2.3: Inhibition of viral DNA integration
22 HIV-integrase please see Chapter 1. Integrase inhibitors are attractive options as ARV therapies as they are unique to retroviridae.14 The HIV-integrase active site contains three catalytically crucial residues: D116, D64 and E152.15 These residues bind Mg2+ cofactors that act as lewis acid catalysts in the strand transfer process (Figure 2.3A).16 Raltegravir (2.3) (Figure 2.4), the first FDA approved inhibitor of HIV-integrase, has been shown to inhibit the viral DNA strand transfer reaction of HIV integrase.17,18 Raltegravir inhibits the strand transfer event by binding these cofactors, (Figure 2.3B) thereby blocking the host cell’s DNA from interacting with HIV-integrase and ceasing viral DNA integration.
23 Figure 2.4: Raltegravir and modified Raltegravir target for conjugation to gold nano-particles
24 Scheme 2.1: Synthesis of Raltegravir pyrimidone core
Selective benzoylation of the phenol moiety in 2.9 was then accomplished using benzoic anhydride in pyridine (Scheme 2.2). N-methylation of the pyrmidone core provided 2.11-N in 41% yield through the use of LiH and dimethylsulfate in dioxane while the O-methyl adduct, 2.11-O was isolated in 44% overall yield. Deprotection of 2.11-N was carried out under standard hydrogenation conditions yielding the amine handle (2.12) for the instillation of the 1,3,4-oxadiazole moiety. Coupling of 2.12 and 2.15 provided the active Raltegravir core 2.16.
25 Phenol deprotection and amide formation was accomplished by treatment of 2.16 with the mono-protected diamine 2.17 (Scheme 2.3). Deprotection of 2.18 proved problematic due to the decomposition of the 1,3,4-oxadiazole moiety (See Table 2.1), however with careful monitoring 2.19 can be prepared using 15% TFA in chloroform. The coupling of 2.19 and commercially available 2.20 also proved to be challenging, due to purification issues. The use of polymer-supported EDC alleviated this issue and provided 2.20, which was taken directly onto the next step. Finally acetate deprotection provided the desired thiolated-Raltegravir derivative 2.21.
26 The modified Raltegravir derivative, 2.21, was then conjugated to 2 nm diameter gold nano-particles by the Feldheim laboratory in ratios of 4:1, 7:1, and 22:1. The ability of 2.21 and the conjugated gold nano-particles to inhibit viral replication was then examined; this preliminary data is summarized in Figure 2.5. Unfortunately, 2.21 was unable to inhibit viral replication at the highest concentration tested (100 nM). Of the conjugated nano-particles, the 4:1 ratio of 2.21:gold appears to be the most active resulting in approximately 70% inhibition of viral replication at 50 nM. In general the conjugated gold nano-particles were more effective at 50 nM than at higher concentrations, this could be due to an aggregation effect.
Table 2.1: Survey of reaction conditions for the deprotection of 2.18
Reaction Conditions Reaction Outcome
Dichloromethane / 50% TFA decomposition
Dichloromethane / 25% TFA decomposition
Dichloromethane / 10 % TFA decomposition
Chloroform / 50% TFA decomposition
Chloroform / 25% TFA 49%*
Chloroform / 15% TFA 88%$
* Determined by 1H NMR with significant amounts of decomposition observed
$
27 Figure 2.5: Inhibition of viral replication by 2.21 and 2.21 conjugated gold nano-particles
2.4 Conclusions:
28 2.5: Experimental Section
Synthesis Experimental:
All reagents used for chemical synthesis were purchased from commercially available sources and used without further purification. Chromatography was performed using 60 Å
mesh standard grade silica gel from Sorbtech. NMR solvents were obtained from Cambridge Isotope Labs and used as is. All 1H NMR (300 MHz or 400 MHz) and 13C NMR (75 MHz or 100 MHz) spectra were recorded at 25 ºC on Varian Mercury spectrometers. Chemical shifts (δ) are given in ppm relative to tetramethylsilane or the respective NMR solvent; coupling constants (J) are in hertz (Hz). Abbreviations used are s = singlet, bs = broad singlet, d = doublet, dd = doublet of doublets, t = triplet, dt = doublet of triplets, bt = broad triplet, qt = quartet, m = multiplet, bm = broad multiplet, p = pentet, and br = broad. Mass spectra were obtained at the NCSU Department of Chemistry Mass Spectrometry Facility. Funding was obtained from the North Carolina Biotechnology Center and the NCSU Department of Chemistry. Infrared spectra were obtained on a FT/IR-4100 spectrophotometer (νmax in
29 tert-butyl-4-((5-hydroxy-1-methyl-2-(2-(5-methyl-1,3,4-oxadiazole-2-carboxamido) propan-2-yl)-6-oxo-1,6-dihydropyrimidine-4-carboxamido)methyl)benzylcarbamate: Methanol (3mL) was added to a 10 mL round bottom flask containing a stir bar and 133 mg (0.292 mmol) of 2.16. To this solution, 166 mg (0.701 mmol) of 1-(N-Boc-aminomethyl)-4-(aminomethyl)benzene (2.17) was added. The reaction flask was then fitted with a reflux condenser and heated to reflux for 16 hours. Upon completion, the reaction mixture was cooled to room temperature and concentrated. The residue was then dissolved in 11 mL of 0.5 N NaOH and transferred to a seperatory funnel. The aqueous layer was then washed three times with 10 mL of diethyl ether. The aqueous layer was then carefully neutralized (pH ~ 6) with 6 N HCl and extracted five times with methylene chloride. The combined organic layers were then dried using anhydrous MgSO4, filtered and concentrated. The
product was obtained a white solid (140 mg, 86%, m.p. 226-228 OC). 1H NMR (400 MHz, CDCl3) δ 11.99 (s, 1H), 8.62 (s, 1H), 7.91 (t, J = 6 Hz, 1H), 7.26 (d, J = 6 Hz, 2H), 7.21 (d, J
= 7.6 Hz, 2H), 5.10 (bs, 1H), 4.54 (d, J = 6 Hz, 2H), 4.23(d, J = 6 Hz, 2H), 3.61 (s, 3H), 2.52 (s, 3H), 1.80 (s, 6H), 1.38 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 168.2, 166.2, 15.5, 158.6,
156.0, 152.3, 150.6, 146.7, 138.7, 136.2, 128.0, 127.8, 124.3, 79.4, 58.0, 44.2, 42.8, 33.3, 28.4, 26.9, 11.1; IR νmax 3404, 1689, 1680, 1650, 1612, 1609, 1590, 1510, 1301, 1251 cm-1;
30 N-(2-(5-hydroxy-4-((4-((6-mercaptohexanamido)methyl)benzyl)carbamoyl)-1-methyl-6-oxo-1,6-dihydropyrimidin-2-yl)propan-2-yl)-5-methyl-1,3,4-oxadiazole-2-carboxamide: A solution of tert-butyl-4-((5-hydroxy-1-methyl-2-(2-(5-methyl-1,3,4-oxadiazole-2-
carboxamido)-propan-2-yl)-6-oxo-1,6-dihydropyrimidine-4-carboxamido)-methyl)-benzylcarbamate (100mg) in 1 mL of anhydrous chloroform was cooled to 0 oC. A 1 mL solution of 30% TFA in chloroform was then added dropwise, the reaction was allowed to warm to room temp and stirred for two hours. At this time the mixture was diluted with 15 mL of chloroform and concentrated. The crude reaction material was then dissolved in 1mL of DMF and then 1.1 equivalent of polymer-supported EDC was added. Triethylamine (2.0 equiv) was added dropwise and the reaction mixture was stirred for 20 hours. The polymer supported EDC was then filtered off and the filtrate was concentrated. The crude material was dissolved in 2 mL of methanol and then 2 mL of a 2% K2CO3 solution was added
dropwise. The residue was then dissolved in 11 mL of 0.5 N NaOH and transferred to a seperatory funnel. The aqueous layer was washed three times with 10 mL of diethyl ether. The aqueous layer was then carefully neutralized (pH ~ 6) with 6 N HCl and extracted five times with methylene chloride. The combined organic layers were dried using anhydrous MgSO4, filtered and concentrated. The crude residue was then purified using Discovery
31 acetonitrile/water gradient. The product was obtained as a white solid (23%, m.p. 172 – 181
o
C). 1H NMR (400 MHz, CDCl3) δ 12.02 (s, 1H), 8.44 (s, 1H), 7.93 (t, J = 6 Hz, 1H), 7.33
(d, J = 6 Hz, 2H), 7.21 (d, J = 7.6 Hz, 2H), 6.07 (t, J = 5.2 Hz, 1H), 4.59 (d, J = 6 Hz, 2H), 4.40(d, J = 6 Hz, 2H), 3.64 (s, 3H), 2.63 (s, 3H), 2.52 (p, J = 7.6 Hz, 2H), 2.36 (t, J = 7.6 Hz, 2H), 2.22 (t, J = 8 Hz, 2H) 1.86 (s, 6H), 1.69-1.59 (m, 4H), 1.47-1.40 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 177.7, 173.1, 168.3, 166.7, 159.6, 158.8, 152.1, 150.5, 147.0, 138.2,
136.7, 128.4, 128.3, 124.3, 58.1, 43.4, 43.0, 36.6, 33.9, 33.7, 33.4, 29.9, 28.0, 27.0, 25.3, 24.6, 11.4; IR νmax 3404, 2075, 1689, 1680, 1650, 1612, 1609, 1590, 1575, 1301, 1251 cm-1;
36 References
(1) Xia, C.; Luo, D.; Yu, X.; Jiang, S.; Liu, S. Microbes infect. 2011, 13, 419. (2) Andersson, L. M.; Hagberg, L.; Rosengren, L.; Fuchs, D.; Blennow, K.; Gisslen, M. BMC infectious diseases 2006, 6, 141.
(3) Zhou, L.; Rua, R.; Ng, T.; Vongrad, V.; Ho, Y. S.; Geczy, C.; Hsu, K.; Brew, B. J.; Saksena, N. K. BMC infectious diseases 2009, 9, 192.
(4) Clifford, D. B. Topics in HIV medicine : a publication of the International AIDS Society, USA 2008, 16, 94.
(5) Gras, G.; Kaul, M. Retrovirology 2010, 7, 30.
(6) Anitinori, A.; Arendt, G.; Becker, J. T.; Brew, B. J.; Byrd, D. A.; Cherner, M.; Clifford, D. B.; Cinque, P.; Epstein, L. G.; Goodkin, K.; Gisslen, M.; Grant, I.; Heaton, R. K.; Joseph, F.; Marder, K.; Marra, C. M.; McArthur, J. C.; Nunn, M.; Price, R. W.; Pulliam, L.; Robertson, K. R.; Sacktor, N.; Valcour, V.; Wojna, V. E. Neurology 2007, 69, 1789.
(7) Antinori, A.; Arendt, G.; Becker, J. T.; Brew, B. J.; Byrd, D. A.; Cherner, M.; Clifford, D. B.; Cinque, P.; Epstein, L. G.; Goodkin, K.; Gisslen, M.; Grant, I.; Heaton, R. K.; Joseph, J.; Marder, K.; Marra, C. M.; McArthur, J. C.; Nunn, M.; Price, R. W.; Pulliam, L.; Robertson, K. R.; Sacktor, N.; Valcour, V.; Wojna, V. E. Neurology 2007, 69, 1789.
(8) McArthur, J. H.; Palenicek, J. G.; Bowersox, L. L. The Nursing clinics of North America 1988, 23, 823.
37 (10) Bowman, M. C.; Ballard, T. E.; Ackerson, C. J.; Feldheim, D. L.; Margolis, D. M.; Melander, C. J Am Chem Soc 2008, 130, 6896.
(11) Barbaro, G.; Scozzafava, A.; Mastrolorenzo, A.; Supuran, C. T. Curr. Pharm. Des. 2005, 11, 1805.
(12) Wei, X.; Decker, J. M.; Liu, H.; Zhang, Z.; Arani, R. B.; Kilby, J. M.; Saag, M. S.; Wu, X.; Shaw, G. M.; Kappes, J. C. Antimicrob Agents Chemother 2002, 46, 1896.
(13) Wei, X.; Decker, J. M.; Wang, S.; Hui, H.; Kappes, J. C.; Wu, X.; Salazar-Gonzalez, J. F.; Salazar, M. G.; Kilby, J. M.; Saag, M. S.; Komarova, N. L.; Nowak, M. A.; Hahn, B. H.; Kwong, P. D.; Shaw, G. M. Nature 2003, 422, 307.
(14) Krishnan, L.; Engelman, A. J. Biol. Chem. 2012, 287, 40858.
(15) Lodi, P. J.; Ernst, J. A.; Kuszewski, J.; Hickman, A. B.; Engelman, A.; Craigie, R.; Clore, G. M.; Gronenborn, A. M. Biochemistry 1995, 34, 9826.
(16) Hazuda, D. J. Current opinion in HIV and AIDS 2012, 7, 383.
(17) Hare, S.; Vos, A. M.; Clayton, R. F.; Thuring, J. W.; Cummings, M. D.; Cherepanov, P. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 20057.
(18) Hare, S.; Gupta, S. S.; Valkov, E.; Engelman, A.; Cherepanov, P. Nature 2010, 464, 232.
(19) Bacchi, A.; Carcelli, M.; Compari, C.; Fisicaro, E.; Pala, N.; Rispoli, G.; Rogolino, D.; Sanchez, T. W.; Sechi, M.; Sinisi, V.; Neamati, N. J Med Chem 2011, 54, 8407.
38 Monteagudo, E.; Muraglia, E.; Nizi, E.; Orvieto, F.; Pace, P.; Pescatore, G.; Scarpelli, R.; Stillmock, K.; Witmer, M. V.; Rowley, M. J Med Chem 2008, 51, 5843.
(21) Savarino, A. Retrovirology 2007, 4, 21.
39 CHAPTER 3
INTRODUCTION TO MICROBIAL BIOFILMS
3.1: Bacterial Biofilm Overview
Microbial biofilms, first discovered in the 17th century, are ubiquitous in nature.1 Biofilm formation is a developmental process in which bacteria undergo a regulated lifestyle switch from a nomadic unicellular state to a sedentary multicellular state.2,3 These microbial communities also possess distinct gene transcription profiles and growth rates as compared to their free floating, planktonic, counterparts.4 This sessile community is encompassed by a self-produced extracellular polymeric substance (EPS).
40 biofilm, releasing viable planktonic cells. The dispersal of planktonic bacteria is thought to occur upon nutrient depletion and waste accumulation. These events slow the production of the EPS and biofilm degradation is initiated.9
Figure 3.1: The five stages of biofilm development
3.2: Significance of Bacterial Biofilms
41 Biofilm mediated bacterial infections have been shown to be at least 1000-fold more resistant to current antibiotic treatments.14 This antibiotic resistance is the cause of chronic bacterial infections and is thought to occur through three potential mechanisms.12 The first of these resistance mechanisms is thought to decrease antibiotic penetration.15 The complex three dimensional architecture of the EPS is thought to be responsible for the inhibition of microbicidal penetration. The second mechanism of antibiotic resistance is due to the fact that biofilms display altered physiological environments. The environmental changes, such as oxygen depletion and pH differences, could potentially decrease antibiotic efficacy.16 The third mechanism of antibiotic resistance is the existence of persister cells.10 Most antibiotics, such as penicillin, require metabolically active bacteria. This allows for these dormant cells to persist in the presence of the therapeutic agent. Persister cells are then able to repopulate the bacterial biofilm once antibiotic therapy is halted.
3.3: Relevant Bacterial Strains Escherichia coli:
E. coli is a rod shaped Gram-negative bacterium. E. coli has been found to be the cause of 90% of all urinary tract infections, and is regarded as the primary cause of food-borne diarrheal disease.17,18 E. coli is the fourth most common organism in surgical site infections,19 and has also been shown to cause neonatal meningitis, as well as sepsis.20
Acinetobacter baumannii:
42 many infections such as pneumonia, urinary tract infections, septicemia, and wound infections. Unfortunately, it has been shown that A. baumannii has evolved into a multi-drug resistant (MDR) organism.22 MDR strains of A. baumannii (MDRAB) have been responsible for the infection of soldiers in both Iraq and Afghanistan. Soldiers are prone to opportunistic pathogens such as MDRAB due to the harsh living conditions and large number of wounds sustained during combat. These soldiers are most often diagnosed with osteomyelitis, an infection of the bone marrow, which may result in the removal of the infected bone tissue.23
Pseudomonas aeruginosa
P. aeruginosa is a Gram-negative highly opportunistic bacterium that is the cause of both mammalian and plant infections. P. aeruginosa rarely causes infection in healthy subjects but is the main cause of morbidity and mortality in cystic fibrosis patients. P. aeruginosa is known to infect victims that have AIDS, severe burns, and/or cancer.24 Burn victims diagnosed with P. aeruginosa infection show a 77% mortality rate.25 P. aeruginosa is also the third most common organism found in nosocomial infections that occur in intensive care units.
Staphylococcus aureus:
43 Methicillin resistant S. aureus (MRSA) has become especially alarming; in the 1980s only 1-5% of S. aureus isolates were methicillin resistant. Today it is estimated that 60-80% of S. aureus strains isolated from hospitals are methicillin-resistant.28 MRSA was found to be the leading cause of nosocomial bacteremia.29 MRSA is quickly becoming a community-acquired infection, being transmitted through the contact of children in day-care facilities and athletics.28
3.4: Known Biofilm Modulators
3.4.1: Quorum Sensing Disruptors
44 Figure 3.2: Halogenated furanones and N-acyl homoserine lactones
45 Figure 3.3: Gram-negative bacteria quorum sensing pathway
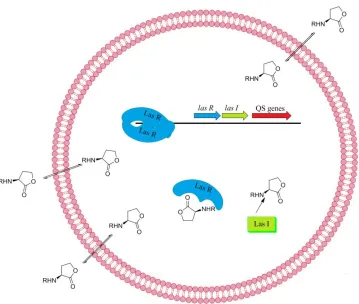
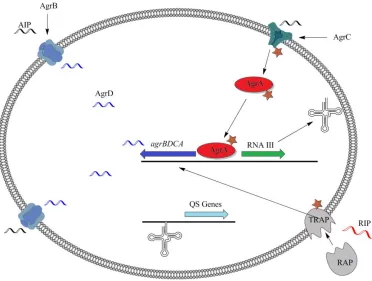
46 AIP-AgrC binding the phosphorylation of ArgA occurs, initiating transcription of RNAIII. It is RNAIII that is the actual regulator of many quorum sensing genes in S. aureus. To date few inhibitors of Gram-positive quorum sensing pathways are known. The most effective of these is the RNAIII-inhibiting peptide, RIP, which has been shown to compete in the RAP/TRAP binding event (Figure 3.4).43,44
Figure 3.4: Gram-positive bacteria quorum sensing pathway
3.4.2: High Throughput Chemical Screens
47 determined that ursolic acid (3.9) was able to inhibit E. coli biofilms at concentrations as low as 22 µM (Figure 3.5).45 The Hergenrother lab has shown that the simple ferric ammonium citrate (3.10) was able to inhibit P. aeruginosa biofilms at moderately low concentrations (IC50 = 60 µM).46 Clardy’s group has screened over 66,000 compounds, and has identified
30 compounds with IC50 values less than 20 µM. Their lead compound (3.11) was found to
have an IC50 value of 530 nM against P. aeruginosa.47 Of the thousands of compounds
screened, these high throughput screens have only turned up a handful of active compounds creating the need for another approach to biofilm modulators.
Figure 3.5: Lead biofilm modulators found using high throughput screens
3.4.3: Marine Natural Product Inspired Small Molecules
48 through a non-microbicidal mechanism while 3.16 was shown to have antimicrobial activity at high concentrations.49
49 Figure 3.6: Inspirations and evolution of novel anti-biofilm agents
50 expression. Pathogenic bacteria use two-component systems as regulators of virulence and antibiotic resistance. For example, BfmR has been shown to play a crucial role in the regulation of chaperone-pili formation, which are required for A. baumannii biofilm formation.56 BfmR has also been shown to be an active regulator of P. aeruginosa biofilm maturation.57
Figure 3.7: Affinity reagent used for the identification of BfmR as the 2-AI target
The simple carbamate 3.14 was found to have mediocre anti-biofilm activity against R. salexigens.48 Compound 3.14 was also found to possess anti-biofilm activity against methicillin-resistant S. aureus and MDRAB (albeit at high concentrations). This promising result prompted an extensive structure activity relationship study that provided compound 3.19 as a simple molecule that was able to inhibit MRSA biofilms with an IC50 value of 4.87
51 Figure 3.8: Schematic of two-component systems
3.5 Conclusions:
52 References
(1) Richards, J. J.; Melander, C. Chembiochem 2009, 10, 2287.
(2) Musk, D. J., Jr.; Hergenrother, P. J. Curr. Med. Chem. 2006, 13, 2163.
(3) Lemon, K. P.; Earl, A. M.; Vlamakis, H. C.; Aguilar, C.; Kolter, R. Curr. Top. Microbiol. Immunol. 2008, 322, 1.
(4) Donlan, R. M.; Costerton, J. W. Clin Microbiol Rev 2002, 15, 167.
(5) Hall-Stoodley, L.; Costerton, J. W.; Stoodley, P. Nat. Rev. Microbiol. 2004, 2, 95.
(6) Stoodley, P.; Sauer, K.; Davies, D. G.; Costerton, J. W. Annu. Rev. Microbiol. 2002, 56, 187.
(7) Characklis, W. G.; McFeters, G. A.; Marshal, K. C. Physiological ecology in biofilm systems; In: Biofilms Characklis, W. G.; Marshal K. C. EDS John Wiley and Sons; 1990, p 195.
(8) Wingernder, J., Neu, TR, Flemming, H. ; Springer, Ed.; Springer: Berlin, 1999, p 93.
(9) O'Toole, G.; Kaplan, H. B.; Kolter, R. Annu. Rev. Microbiol. 2000, 54, 49. (10) Davies, D. Nat. Rev. Drug discovery 2003, 2, 114.
(11) Mah, T. F.; O'Toole, G. A. Trends Microbiol. 2001, 9, 34.
53 Iglewski, B. H.; McDermott, T. R.; Mason, R. P.; Wozniak, D. J.; Hancock, R. E.; Parsek, M. R.; Noah, T. L.; Boucher, R. C.; Hassett, D. J. Dev. Cell 2002, 3, 593.
(14) Rasmussen, T. B.; Givskov, M. Int J Med Microbiol 2006, 296, 149.
(15) Suci, P. A.; Mittelman, M. W.; Yu, F. P.; Geesey, G. G. Antimicrob Agents Chemother 1994, 38, 2125.
(16) Stewart, P. S.; Costerton, J. W. Lancet 2001, 358, 135. (17) Nataro, J. P.; Kaper, J. B. Clin Microbiol Rev 1998, 11, 142. (18) Russo, T. A.; Johnson, J. R. Microbes Infect. 2003, 5, 449.
(19) Favero, M. S.; Gaynes, R. P.; Horan, T. C.; Emori, T. G.; Stroud, L. A.; Archibald, L. K.; Keita-Perse, O.; Wright, G. C.; Culver, D. H.; Edwards, J. R.; Henderson, T. S.; Tolson, J. S. Peavy, G. E. American journal of infection control 1996, 24, 380.
(20) de Louvois, J. J. Antimicrob. Chemother. 1994, 34 Suppl A, 61.
(21) Dijkshoorn, L.; Nemec, A.; Seifert, H. Nat. Rev. Microbiol. 2007, 5, 939. (22) Forster, D. H.; Daschner, F. D. European journal of clinical microbiology & infectious diseases : official publication of the European Society of Clinical Microbiology 1998, 17, 73.
(23) Davis, K. A.; Moran, K. A.; McAllister, C. K.; Gray, P. J. Emerg. Infect. Dis. 2005, 11, 1218.
54 (25) McManus, A. T.; Mason, A. D., Jr.; McManus, W. F.; Pruitt, B. A., Jr. Eur. J. Clin. Microbiol. 1985, 4, 219.
(26) National Institutes of Health Press Release:
http://www.niaid.nih.gov/news/newreleases/1999/pages/staph.aspx 1999.
(27) Cenci-Goga, B. T.; Karama, M.; Rossitto, P. V.; Morgante, R. A.; Cullor, J. S. J. Food Prot. 2003, 66, 1693.
(28) Taubes, G. Science 2008, 321, 356.
(29) Smith, K.; Hunter, I. S. J. Med. Microbiol. 2008, 57, 966.
(30) Fuqua, C.; Parsek, M. R.; Greenberg, E. P. Annu. Rev. Genet. 2001, 35, 439. (31) Hentzer, M.; Riedel, K.; Rasmussen, T. B.; Heydorn, A.; Andersen, J. B.; Parsek, M. R.; Rice, S. A.; Eberl, L.; Molin, S.; Hoiby, N.; Kjelleberg, S.; Givskov, M. Microbiology (Reading, England) 2002, 148, 87.
(32) Manefield, M.; Welch, M.; Givskov, M.; Salmond, G. P.; Kjelleberg, S. FEMS Microbiol. Lett. 2001, 205, 131.
(33) Ren, D.; Sims, J. J.; Wood, T. K. Appl. Environ. Microbiol. 2001, 3, 731. (34) Ren, D.; Sims, J. J.; Wood, T. K. Lett. Appl. Microbiol. 2002, 34, 293. (35) Suga, H.; Smith, K. M. Curr. Opin. Chem. Biol. 2003, 7, 586.
(36) Bruhn, J. B.; Dalsgaard, I.; Nielsen, K. F.; Buchholtz, C.; Larsen, J. L.; Gram, L. Dis. Aquat. Organ. 2005, 65, 43.
55 (39) Geske, G. D.; Wezeman, R. J.; Siegel, A. P.; Blackwell, H. E. J Am Chem Soc 2005, 127, 12762.
(40) Glansdorp, F. G.; Thomas, G. L.; Lee, J. K.; Dutton, J. M.; Salmond, G. P.; Welch, M.; Spring, D. R. Org Biomol Chem 2004, 2, 3329.
(41) Waters, C. M.; Bassler, B. L. Annu. Rev. Cell Dev. Biol. 2005, 21, 319. (42) Lyon, G. J.; Novick, R. P. Peptides 2004, 25, 1389.
(43) Balaban, N.; Novick, R. P. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 1619. (44) Gov, Y.; Bitler, A.; Dell'Acqua, G.; Torres, J. V.; Balaban, N. Peptides 2001, 22, 1609.
(45) Ren, D.; Zuo, R.; Gonzalez Barrios, A. F.; Bedzyk, L. A.; Eldridge, G. R.; Pasmore, M. E.; Wood, T. K. Appl.Environ. Microbiol. 2005, 71, 4022.
(46) Musk, D. J.; Banko, D. A.; Hergenrother, P. J. Chem. Biol. 2005, 12, 789. (47) Junker, L. M.; Clardy, J. Antimicrob Agents Chemother 2007, 51, 3582. (48) Yamada, A.; Kitamura, H.; Yamaguchi, K.; Fukuzawa, S.; Kamijima, C.; Yazawa, K.; Kuramoto, M.; Wang, G. Y. S.; Fujitani, Y.; Uemura, D. Bull. Chem. Soc. Jpn 1997, 70, 3061.
(49) Huigens, R. W., 3rd; Richards, J. J.; Parise, G.; Ballard, T. E.; Zeng, W.; Deora, R.; Melander, C. J Am Chem Soc 2007, 129, 6966.
(50) Richards, J. J.; Ballard, T. E.; Huigens, R. W., 3rd; Melander, C. Chembiochem 2008, 9, 1267.
56 (52) Richards, J. J.; Ballard, T. E.; Melander, C. Org Biomol Chem 2008, 6, 1356. (53) Richards, J. J.; Reed, C. S.; Melander, C. Bioorg Med Chem Lett 2008, 18, 4325.
(54) Rogers, S. A.; Melander, C. Angew Chem Int Ed Engl 2008, 47, 5229.
(55) Thompson, R. J.; Bobay, B. G.; Stowe, S. D.; Olson, A. L.; Peng, L.; Su, Z.; Actis, L. A.; Melander, C.; Cavanagh, J. Biochemistry 2012, 51, 9776.
(56) Tomaras, A. P.; Flagler, M. J.; Dorsey, C. W.; Gaddy, J. A.; Actis, L. A. Microbiology (Reading, England) 2008, 154, 3398.
(57) Petrova, O. E.; Schurr, J. R.; Schurr, M. J.; Sauer, K. Mol Microbiol 2011, 81, 767.
57 CHAPTER 4
SYNTHESIS AND BIOLOGICAL EVALUATION OF 2-AMINOIMIDAZOLE – CARBAMATE HYBRID ANTI-BIOFILM AND ANTI-MICROBIAL AGENTS
4.1 Introduction
Biofilms represent a particularly hardy phenotype of bacterial growth.1 Owing to their encasement in a robust extracellular matrix of biomolecules, bacteria in these surface-adhered communities are uniquely resilient, often displaying resistance toward conventional antibiotics, antiseptics, and host defense mechanisms.2 Indeed, more than 80% of all bacterial infections are the direct result of biofilms comprising medically relevant pathogens.3 Biofilms have been implicated in persistent infections of medical implants4,5, and are responsible for the mortality and morbidity of cystic fibrosis patients.6
Despite the involvement of bacterial biofilms in a host of medical maladies, the development of small, drug-like compound classes that influence their formation and maintenance has lagged significantly.7 Currently, relatively few scaffolds are known to possess anti-biofilm activity, and these include homoserine lactones8,9, brominated furanones10,11, and ursine triterpenes.12,13 Additionally, computer aided drug design protocols14 and high throughput screening methods15,16 have also led to the discovery of a few novel scaffolds that possess anti-biofilm activity. Despite these advances, potent biofilm modulators are still sorely underdeveloped.
58 Our inspiration for the design of these molecules was to extract and systematically optimize structural motifs embedded within the marine natural product bromoageliferin (4.1, Figure 4.1).17-23 The 2-aminoimidazole (2-AI) heterocycle has proven crucial for the observed biological activity of compounds 4.2 – 4.4. These studies have culminated in the discovery of lead compound 4.5 that is active against both Gram-positive and Gram-negative bacteria.
Figure 4.1: 2-Aminoimidazole anti-biofilm agents based on bromoageliferin
59 the systematic optimization of the SCRC3P79 (Cytophaga sp.) bacterial metabolite 4.6. Unlike our 2-AI scaffold, the (-)-menthyl carbamate series lacked the ability to disperse pre-formed biofilms and demonstrated poor anti-biofilm properties against non-staphylococcal strains. Nonetheless, these shortcomings were somewhat offset by the trivial synthesis of compounds such as 4.7.
Figure 4.2: (-)-Menthyl carbamate anti-biofilm agent
We next sought to investigate hybrid scaffolds that included structural motifs from both classes of molecules. Namely, we proposed a series of targets that blended the 2-AI head group from our bromoageliferin analogues (4.2 – 4.5) with the (-)-menthyl carbamate moiety found in 4.7. Presented herein is an account of the successful marriage of these two classes of biofilm inhibitors and the evaluation of these hybrid structures as anti-biofilm and anti-microbial agents.
4.2: Results and Discussion:
60 directly tethered to the 2-AI head group with an intervening triazole similar to compound 4.5. Compounds 4.11 – 4.13 represent (-)-menthyl carbamate analogues of 2-AI amide derivatives 4.3 and 4.4. Finally, 4.14 and 4.15 were designed to closely resemble our lead compound 4.5 by replacing the aryl olefin in 4.5 with a (-)-menthyl carbamate linkage. Additionally, we elected to prepare the acetamido analogue of 2-AI/(-)-menthyl carbamate hybrid 4.8 as a control compound featuring the 2-AI head group capped with an acetamido group in lieu of the (-)-menthyl carbamate.
61 Figure 4.3: Hybrid 2-AI/(-)-menthyl carbamate anti-biofilm targets
62 inhibit 50% biofilm formation) as judged by a crystal violet reporter assay.29 The data for these experiments is collected in Table 4.1. During the course of these experiments, most of the compounds displayed a precipitous drop in biofilm inhibition activity over a narrow
Scheme 4.1: Preparation of 2-AI/(-)-menthyl carbamate hybrids
concentration range. For example, the compounds would exhibit > 90% inhibition at 10 µM concentrations, but a dismal < 10% inhibition at 5 µM. Such behavior is commonly diagnostic of an underlying microbicidal mechanism for biofilm inhibition. Such dose response behavior precludes the ability to calculate a reliable IC50 value and compounds
exhibiting this behavior are indicated with a (-) in Table 4.1. It should be noted that all of the compounds exhibited this type of behavior for MSSA.
Some compounds, however, returned dose-response data suitable for the determination of IC50 values. Against MRSA, compounds 4.14 and 4.15 returned IC50 values
63 4.14 and 4.15 gave IC50 values of 58.8 and 40.3 µM, respectively, against PA14. For A.
baumannii, IC50 values of 19.2, 18.4, 16.7, 19.2, 16.7, and 94.9 µM were obtained for 4.8 –
Table 4.1: Biofilm inhibition (IC50 values in µM) against various bacterial strains
Compound MRSA PA14 A. baumannii
4.8 - >200 19.2 ± 2.0
4.9 - >200 18.4 ± 1.0
4.10 - >200 16.7 ± 1.5
4.11 - 18.0 ± 5.1 19.2 ± 2.0
4.12 - 18.0 ± 5.3 -
4.13 - - 16.7 ± 1.2
4.14 29.9 ± 5.8 58.7 ± 1.5 94.9 ± 0.2
4.15 20.5 ± 4.9 40.3 ± 5.2 -
4.16 >200 >200 >200
4.11, 4.13, and 4.14, respectively. Control acetamido compound 4.16 exhibited no anti-biofilm activity for any of the four bacterial strains at the highest concentration tested (200 µM), confirming the necessity of the carbamate moiety for anti-biofilm activity.
We next conducted growth curves at the IC50 concentrations for each of the
64 activity was due to the inhibition of bacterial growth. The same was true for compounds 4.8, 4.9, 4.11, 4.13, and 4.14 against A. baumannii. Importantly, however, compounds 4.11, 4.12, 4.13, and 4.15 inhibited PA14 biofilm formation via a non-microbicidal mechanism. Additionally, compound 4.10 was found to inhibit A. baumannii biofilms in a non-toxic fashion.
Given that most of the 2-AI/(-)-menthyl carbamate hybrids exhibited anti-biofilm activity by toxic means, we elected to evaluate their potential as anti-microbial agents. To determine the extent of microbicidal activity, we measured the minimum inhibitory concentration30 (MIC) of each compound against MRSA, MSSA, PA14, and A. baumannii. Additionally, we evaluated the compounds against multi-drug resistant A. baumannii (MDRAB). The data for this study is summarized in Table 4.2.
65 Table 4.2: MIC values (µg mL-1) against various bacterial strains
Compound MRSA MSSA PA14 A. baumannii MDRAB
4.8 8 4 >128 16 16
4.9 4 4 >128 32 >128
4.10 4 4 >128 >128 >128
4.11 32 32 32 32 32
4.12 8 8 64 16 16
4.13 8 8 64 8 8
4.14 64 16 >128 64 128
4.15 16 16 64 64 64
4.16 >128 >128 >128 >128 >128
anti-66 microbial behavior up to the highest concentration tested (128 µg mL-1). This result serves to highlight the importance of both the 2-AI head and the (-)-menthyl carbamate functionality in eliciting the observed anti-microbial properties.
Given the observed anti-biofilm activity via both toxic and non-toxic means, we were eager to evaluate whether or not several of the 2-AI/(-)-menthyl carbamate hybrids could disperse pre-formed bacterial biofilms. This study was of particular interest given the dichotomy between the two parent scaffolds. While our 2-AI leads (Figure 4.1) effectively disperse pre-formed biofilms across order, class and phylum, our (-)-menthyl carbamate lead 4.7 failed to exhibit any biofilm dispersal capability.
In order to assess the potential for biofilm dispersal, we chose compounds 4.8 and 4.13, based on the fact that they exhibited the lowest MIC values against the S. aureus and A. baumannii strains. Additionally, we elected to screen 4.10 for non-toxic dispersal activity given that it inhibited the formation of A. baumannii biofilms in a non-microbicidal fashion. Pre-formed biofilms from MRSA, MSSA, and A. baumannii were treated with lead compounds 4.8, 4.10 and 4.13. Dose-response curves were generated to determine the EC50
values for biofilm dispersal (the concentration required to disperse 50% of a preformed biofilm); this data is summarized in Table 4.3.
While compound 4.8 failed to disperse pre-formed staphylococcal biofilms, it exhibited an EC50 value of 37.5 µM against A. baumannii. Compound 4.10 gave EC50 values
67 against A. baumannii with an EC50 value of 20.6 µM (13.1 µg mL-1). Finally, compound
4.13 had EC50 values of 53.5, 38.8, and 68.3 µM against MRSA, MSSA, and A. baumannii,
respectively. In addition to the data collected in Table 4.3, we also screened the entire library against pre-formed P. aeruginosa biofilms owing to their relatively low toxicity in the MIC studies against that strain. Unfortunately, none of the compounds in this study effectively dispersed pre-formed PA14 biofilms.
Table 4.3: Biofilm dispersal (EC50 values (µM)) against various bacterial strains
Compound MRSA MSSA A. baumannii
4.8 >200 >200 37.5 ± 2.3
4.10 38.1 ± 5.7 32.5 ± 4.6 20.6 ± 1.7
4.13 53.5 ± 5.1 38.8 ± 4.5 68.3 ± 2.7
In a final set of experiments, all of the compounds in the study were preliminarily assessed for mammalian cytotoxicity, through the use of a red blood cell hemolysis assay using difibrinated sheep’s blood.30
HD50 values (the concentration at which 50% hemolysis
68 Table 4.4: Blood lysis (HD50 values in µM) assay of the 2-AI/(-)-menthyl carbamates
Compound HD50 Compound HD50
4.8 93.8 ± 3.5 4.13 473.7 ± 15.0
4.9 46.0 ± 4.9 4.14 474.0 ± 7.2
4.10 42.7 ± 1.7 4.15 317.6 ± 14.6
4.11 463.7 ± 0.7 4.16 >800
4.12 228.8 ± 29.9
4.3: Conclusion:
In conclusion, we have introduced a new class of hybrid 2-aminoimidazole/(-)-menthyl carbamate anti-biofilm agents that exhibit the ability to inhibit biofilm formation and to disperse mature, pre-formed biofilms. While most of the 2-AI/(-)-menthyl carbamate hybrids elicited their anti-biofilm activity via underlying microbicidal means, this study also resulted in a panel of non-toxic inhibitors for the P. aeruginosa strain PA14 (i.e. compounds 4.11, 4.12, 4.14, and 4.15). Additionally, compound 4.10 both inhibited and dispersed biofilms of A. baumannii in a non-toxic fashion.
4.4: Experimental Section Synthesis Experimental