THIONYL CHLORIDE INDUCED CONVENIENT SYNTHESIS
OF BENZAMIDES FROM 3-BROMO-5-NITROBENZOIC ACID
AND AMINES UNDER SOLVENT FREE CONDITIONS
Shritesh D. Jagtap,
[a]Aniket P. Sarkate,
[a]*Arjun L. Khandare,
[b]Ishudeep K.
Narula,
[a]Kshipra S. Karnik,
[a]Dattatraya N. Pansare,
[c]Rohini N. Shelke
[c]Keywords:thionyl chloride;coupling reaction;benzamides;benzoic acid; solvent free.
A new method of thionyl chloride induced convenient synthesis of novel benzamides under solvent free conditions has been developed using benzoic acid and amines. The benzamides were synthesized through a coupling reaction of benzoic acid and different amines using thionyl chloride at room temperature. The abovementioned technique assists in the preparation of substituted benzamides which were obtained in good yields within 2-4 h using conventional heating. The developed method is flexible, economic, environment friendly; also is catalyst, ligand and solvent free and has major importance in industry and laboratory.
* Corresponding Authors
E-Mail: [email protected]
[a] Department of Chemical Technology, Dr. Babasaheb Ambedkar Marathwada University, Aurangabad, 431004 (MS), India
[b] Food Toxicology Division, Food and Drug Toxicology Research Centre, National Institute of Nutrition, Tarnaka, Hyderabad-500007, Telangana, India.
[c] Department of Chemistry, Deogiri College, Station road, Aurangabad- 431005 (MS), India
INTRODUCTION
Preparation of bromo and nitro-substituted benzoic acids as precursors for preparation of pharmaceutically active benzamides is an important challenge of organic chemistry. The nitration of aromatic compounds may be achieved with many nitrating reagents but the conditions are incompatible with a range of compounds which are sensitive to oxidizing or strongly acidic conditions.1,2 Nitration of benzoic acid 62% nitric acid is successfully carried out under solvent-free condition in a biphasic mode in the presence of the Bronsted acidic ionic liquids; the only by-product is water and ionic liquids are capable of being reused without any separation.3In this paper we give information about optimization of the initial reported method.
The classical bromination reactions involve the use of hazardous elemental bromine.4 That’s why bromination reaction has been still attracting attention to develop the more practical method without the use of hazardous and highly toxic elemental bromine.5 Several acid-catalyzed N-bromosuccinimide (NBS) ring brominations have been reported and continue to be of interest, mainly in connection with the bromination of polyalkyl benzenes.5,6 In terms of ease of handling and availability, N-bromosuccinimide (NBS) is a superior brominating reagent.7 In this paper we give information about optimization the previously reported method to brominate 3-nitrobenzoic acid with NBS and sulfuric acid.
Benzamides are an important class of chemicals that have been widely used as chemical intermediates in organic synthesis, raw materials for engineering plastics, detergents, and lubricants.8 The conversion of carboxylic acids to amides is one of the most important transformations in organic synthesis. In general, the conversion of carboxylic acids to carboxamides requires an activation of the carboxyl group. The solvents in most organic reactions increase the reaction yield, stereoselectivity and chemoselectivity.9 The violent exothermic reactions are also controlled by the solvent which involves quick electron transfer from metal and that with a highly reactive species.10 On the other hand, the use of a solvent may decrease the reaction rate, produces waste solvent, enhances the production cost, and make a chemical operation risky when a flammable solvent is used.11-12 Solvent-free reactions are therefore becoming important from the viewpoint of green chemistry.13
We have been engaged in the development of aromatic compounds as anti-cancer agents and have shown structure– activity relationships with several substituted benzoic acids (containing NO2 and Br group) to which a heterocyclic aromatic amide group containing ring bound,14 therefore the amidation reactions of 3-bromo-5-nitro benzoic acid was performed in the presence of SOCl2 as promoter in a solvent and catalyst free conditions.15
In continuation of our work,16-33 we have developed the new protocol synthesis of benzamides from 3-bromo-5-nitrobenzoic acid and amines.
MATERIALS AND METHOD
spectra were recorded on a 400 MHz Varian NMR spectrometer. The 13C were recorded on a 100 MHz Varian NMR spectrometer. Mass spectra were taken with Micromass-QUATTRO-II of WATER mass spectrometer.
COOH
NO2
Br
HN R2
SOCl2
C
NO2
Br O
R1 N
R2
1 2 3
R1
Scheme 1. Synthesis of 3-bromo-5-nitrobenzamides
General procedure for the synthesis of 3-bromo-5-nitro-benzamides
Nitration of benzoic acid
Benzoic acid (10 g, 0.0406 mmol) and cc. sulfuric acid (25ml) were added to a reaction vessel and nitrating mixture (H2SO4+HNO3) (H2SO4:HNO3= 1:1.5, 16.5 ml) was added drop wise at 0-5°C temperature for 45 minutes and further the reaction was stirred for 1 h. Then the reaction mixture was quenched with the addition of cracked ice (precipitate formation was observed) and the product was obtained by vacuum filtration. Further the product was recrystallized with methanol. Melting point observed was 139-141°C.
Bromination of 3-nitrobenzoic acid
3-Nitrobenzoic acid (10 g, 0.0598 mmol), NBS (N-bromo succinamide, (11.66 g, 0.05979 mmol), and conc. H2SO4 (20ml) were added to a reaction vessel. The mixture was then heated to 80°C temperature for 2 h. Then the reaction was quenched with the addition of cracked ice (precipitate formation was observed) and the product was obtained by vacuum filtration. The reaction product was recrystallized from methanol. Melting point was found to be 159-161°C.
Coupling reaction
3-Bromo-5-nitro benzoic acid 1 (1 g, 0.0406 mmol), amines (primary or secondary) 2 (1eq.) and thionyl chloride (5 ml) were put into a reaction vessel. The mixture was then stirred at room temperature for 2-4 h. It was then extracted with ethyl acetate; the solvents were removed under vacuum and the obtained product was recrystallized from ethanol to yield the corresponding substituted benzamides. The 1H and 13C NMR spectra are given in the Electronic Supplementary Information (ESI).
The following are the spectral analysis of the synthesized compounds:
Synthesis of 3-bromo-N-methyl-5-nitrobenzamide (3a)
1H NMR (400 MHz, DMSO-d
6) &8.67 (m, 1H), 8.48 (s, 1H), 8.26 (s, 1H), 7.96 (s, 1H), 2.89 (d. 3H, J=5 Hz). 13C NMR 167.9, 148.3, 136.0, 134.7, 129.2, 124.5, 121.57, 26.67
Synthesis of 3-bromo-N-(2-hydroxyethyl)-5-nitrobenzamide (3b)
1H NMR (400 MHz, DMSO-d
6) & 8.70(s, 1H), 8.42(s, 1H), 8.24 (s, 1H), 8.16(t, 1H), 4.50 (t, 1H), 3.55 (d, 1H) (d. 3H, J=5 Hz). 13C NMR 166.2, 148.7, 135.2, 134.4, 128.3, 124.5, 121.5, 60.12, 42.43
Synthesis of 3-bromo-N-butyl-5-nitrobenzamide (3c)
1H NMR (400 MHz, DMSO-d6) 8.69 (s, 1H), 8.49 (s, 1H), 8.27 (s, 1H), 8.05 (td, 1H), 3.30 (t, 2H), 1.54 (t, 2H), 1.34 (t, 1H), 0.91 (d. 3H, J=5 Hz). 13C NMR 166.2, 148.7, 135.3 , 134.2, 127.9, 121.5, 39.6, 31.2, 20.4, 13.7
3-bromo-N,N-dimethyl-5-nitrobenzamide (3d)
1H NMR (400 MHz, DMSO-d
6) 8.59 (s, 1H), 8.44 (s, 1H), 8.13 (s, 1H), 3.01 (s, 6H). 13C NMR 170.2, 148.1, 136.5, 134.5, 120.9, 36.47
(3-Bromo-5-nitrophenyl)(piperazin-1-yl)methanone (3e)
1H NMR (400 MHz, DMSO-d
6) 8.60 (s, 1H), 8.44(s, 1H), 8.14 (s, 1H), 3.56 (t, 3H), 3.48 (d, 2H), 2.81 (t, 3H), 2.16 (d, 1H). 13C NMR 169.0, 148.2, 135.9, 135.1, 127.6, 123.4, 121.3, 47.3, 45.9
3-Bromo-N-(4-methoxybenzyl)-5-nitrobenzamide (3f)
1H NMR (400 MHz, DMSO-d
6) 8.70 (d, 1H), 8.48 (s, 1H), 8.19 (s, 1H), 7.25(d, 2H), 6.86 (d, 3H), 4.44 (s, 1H), 3.79 (s, 6H). 13C NMR 166.2, 158.9, 149.2, 135.7, 135.1, 133.2, 129.1, 128.4, 124.9, 121.7, 113.8, 55.3, 44.4
(3-Bromo-5-nitrophenyl)(2-methylpiperazin-1-yl)methanone (3g)
1H NMR (400 MHz, DMSO-d
6) 8.63 (s, 3H), 8.46 (s, 3H), 8.17 (s, 3H), 4.46 (t, 1H), 3.69 (d, 3H), 3.66 (d, 3H),3.06 (t, 2H), 2.99(d, 4H), 2.93(d, 2H), 2.10(t, 2H), 1.25(t, 8H). 13C NMR 168.9, 148.5, 136.9, 135.0, 127.9, 123.3, 121.4, 51.4, 45.5, 44.4, 16.12
N-Benzyl-3-bromo-5-nitrobenzamide (3h)
1H NMR (400 MHz, DMSO-d
6) 8.69(t, 5H), 8.67(s), 8.65 (s, 2H), 8.45(s, 1H), 7.34(d, 3H), 7.24 (t, 1H), 4.54 (d, 3H) 13C NMR 166.1, 149.1, 138.6, 135.7, 134.4, 128.3, 128.1, 124.9, 121.7, 43.2
3-Bromo-5-nitro-N-(o-tolyl)benzamide (3i)
1H NMR (400 MHz, DMSO-d
6) 9.68(s, 1H), 8.73(s, 1H), 8.45 (s, 1H), 8.20(s, 1H), 7.18 (t, 2H), 2.31(s, 3H). 13C NMR 165.3, 149.1, 138.9, 135.6, 134.9, 133.2, 129.1, 128.5, 127.2, 125.5, 124.2, 121.6, 119.0, 17.7
3-Bromo-N-(furan-2-ylmethyl)-5-nitrobenzamide (3j)
1H NMR (400 MHz, DMSO-d
RESULTS AND DISCUSSION
The substituted benzamides are synthesized as per the established route of synthesis. The thionyl chloride was used as promoter in the synthesis (Scheme 1). We have optimized the condition for the preparation of our substituted products. Some solvents and solvent free conditions were also tested. We have presented the optimization conditions in Table 1.
Table 1. Effect of solvents in the preparation of 3a
aIsolated yield
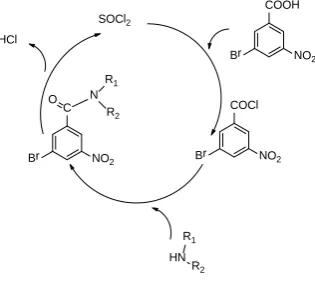
As can be seen from the above data, the reaction goes well in the absence of any solvent and the reaction time as well as the percentage yield of the synthesized compounds better than in the presence of solvents. Using toluene as the solvent the time required for completion was 20 h with only 58% yield (Table 1, Entry 1). Using N,N-dimethylformamide (DMF) the yield drastically decreased to 31% with 18 h of reaction time (Table 1, Entry 2). Methanol was also tried but the obtained yield was again very low (Table 1, Entry 3). Dichloromethane (DCM) was used which to our surprise gave 85% yield in 9 h but the product obtained was somewhat sticky which required further purification (Table 1, Entry 4). Lastly the reaction was carried out in the solvent free condition which not only increased the yield to 94% but also decreased the reaction time to 2 h (Table 1, Entry 5). Hence further derivatization was carried out in a solvent free condition to obtain the required products. The possible reaction mechanism is explained in Figure 1.
The first step is that 3-bromo-5-nitro benzoic acid reacts with the thionyl chloride species to form a 3-bromo-5-nitrobenzoyl chloride complex which acts as an intermediate for the coupling reaction. Due to high electronegativity of benzoyl chloride group, coordination is orientated by the partial charges on NH+ and on the electronegative 3-bromo-5-nitrobenzoyl chloride group which assists the substitution by the amines to get desired product.
SOCl2
HN R2 R1
COOH
NO2 Br
COCl
NO2 Br
C
NO2 Br
O R1 N
R2 HCl
Figure 2.The plausible reaction mechanism
CONCLUSION
We have prepared novel benzamides using thionyl chloride to perform thecoupling reaction of 3-bromo-5-nitrobenzic acid and amines. Our method has minimum environmental impact, is flexible and economic. The reaction is done without using any transition metal catalyst, ligand, base, toxic or hazardous reagent, additives, promoters and organic solvent (i.e. solvent free). We believe that this substituted benzamide compound protocol has major importance in industry, laboratory as well as a drug intermediate due to all its advantages.
Table 2. Synthetic details of the prepared 3-bromo-5-nitrobenzamides
Entry Amines Prepared compound Time, h %Yield Melting or boiling
point, oC
3a
H3C
NH2
Br NO2
H N
CH3
O 2 94 143
3b
H2N OH
Br NO2
H N O
OH 2 90 136.8
3c
NH2
Br NO2
H N
O 2 91.5 141
3d
NH
Br NO2
N
O 3 85 298 (BP)
Entry Solvents Time, h Yield,a %
1 Toluene 20 58
2 DMF 18 31
3 Methanol 12 32
4 Dichloromethane 9 85
3e
HN NH
Br NO2
N O
NH 3 80 142.9
3f
OCH3
H2N
Br NO2 H N O
OCH3 4 91 210 (BP)
3g
HN NH
Br NO2 N O
NH 3 85 260 (BP)
3h
H2N
Br NO2 H N O
4 90 240 (BP)
3i
NH2
Br NO2
H N
O 2 90 250 (BP)
3j
O NH2
Br NO2 H N O
O 3 80 400 (BP)
ACKNOWLEDGMENTS
The authors are thankful to The Head, Department of Chemical Technology, Dr. Babasaheb Ambedkar Marathwada university, Aurangabad 431004 (MS) providing the laboratory facilities.
REFERENCES
1Zolfigol, M., Ghaemi, E., Madrakian, E, 2001, 6, 614-620.
https://dx.doi.org/10.3390/60700614
2Bak, R., Smallridge, A., Tetrahedron Lett., 2001,42, 6767-6769.
https://doi.org/10.1016/S0040-4039(01)01378-8
3Qiao, K., Chiaki, Y., Chem Lett., 2004 33,
808-809.https://doi.org/10.1246/cl.2004.808
4Moriuchi, T., Yamaguchi, M., Kikushima, K., Hirao, T.,
Tetrahedron Lett., 2007, 48, 2667-2670.
https://doi.org/10.1016/j.tetlet.2007.02.074
5 Moriuchi, T., Yamaguchi, M., Kikushima, K., Hirao, T.,
Tetrahedron Lett., 2005, 51, 340-342.
https://doi.org/10.1016/j.tetlet.2009.11.016
6Hoffmann, K., Carlsen, P., Syn. Comm., 1999, 29, 1607-1610.
https://doi.org/10.1080/00397919908086142
7Carreño, M., Ruano, G., Miguel, G., Toledo, A., Urbano, A., J.
Org. Chem., 1995, 60, 5328-5331.
https://doi.org/10.1021/jo00121a064
8Tanemura, K., Suzuki, T., Nishidi, Y., Satsumabayashi, K.,
Horaguchi, T., Chem Comm., 2004, 470-471.
https://doi.org/10.1039/B315340A
9Fujwara, H., Ogasawara, Y., Kotani, M., Yamaguchi, K., Mizuno,
N., Chem. Asian J., 2008, 3, 1715–1721.
https://doi.org/10.1002/asia.200800067
10Ema, T., Nanjo, Y., Shiratori, S., Terao, Y., Kimura, R., Org.
Lett., 2016, 18, 5764−5767.
https://doi.org/10.1021/acs.orglett.6b03115
11Ema, T., Miyazaki, Y., Koyama, S., Yano, Y., Sakai, T., Chem
Comm., 2012, 48, 4489−4491. https://doi.org/10.1039/C2CC30591G
12Ema,T., Miyazaki, Y., Shimonishi, J., Maeda, C., Hasegawa, J., J.
Am. Chem. Soc., 2014, 136, 15270−15279.
https://doi.org/10.1021/ja507665a
13Maeda, C., Taniguchi, T., Ogawa, K., Ema, T., Angew, Chem.
Int., 2015, 54, 134−138.
https://doi.org/10.1002/anie.201409729
. 14Maeda, C., Shimonishi, J., Miyazaki, R., Hasegawa, J., Ema, T.,
Chem. Eur. J., 2016, 22, 6556−6563. https://doi.org/10.1002/chem.201600164
15Susanta, S.,Becker, F., Banik, K., Tetrahedron Lett., 2000, 41,
8017-8020. https://doi.org/10.1016/S0040-4039(00)01397-6
16Desmukh, S.V., Pawar, C.D., Pansare, D. N., Chavan, S.L.,
Pawar, R.P., Ubale, M.B. Eur. Chem. Bull. 2019, 8(4), 115-122. DOI: http://dx.doi.org/10.17628/ecb.2019.8.115-122
17Walle, M. R., Pansare, D. N.,Kamble, S. S., Pawar, R. P., Ingale,
R.D., Eur. Chem. Bull. 2019, 8(3), 101-104. DOI: 10.17628/ecb.2019.8.101-104
18Gaikwad, D. D., Pawar, C. D., Pansare, D.N.,Chavan, S. L.,
19Pravin N. Chavan, Dattatraya N. Pansare, Rohini N. Shelke. J.
Chin. Chem. Soc. 2019, in press https://doi.org/10.1002/jccs.201800411
20Shelke, R.N., Pansare, D. N., Pawar R. P., Shinde, D. B., Thopate,
S. R., Eur. Chem. Bull. 2019, 8(2), 63-70. DOI: 10.17628/ecb.2019.8.63-70
21Pansare, D. N., Shelke, R. N., Khade, M. C., Jadhav, V. N.,
Pawar, C. D., Jadhav, R. A., Bembalkar, S. R., Eur. Chem. Bull. 2019, 8(1), 7-14. DOI: 10.17628/ecb.2019.8.7-14
22Pansare, D. N., Shelke, R. N., Khade, M. C., Jadhav, V. N.,
Pawar, C. D., Deshmukh, S.U., Dhas, A. K., Chavan, P. N., Sarkate, A. P., Pawar, R. P., Shinde, D. B., Thopate, S. R.,
Eur. Chem. Bull. 2019, 8(1), 1-6. DOI:
10.17628/ecb.2019.8.1-6
23Shelke, R. N., Pansare, D. N., Khade, M. C., Jadhav, V. N.,
Pawar, C. D., Deshmukh, S.U., Sarkate, A. P., Gore N. S., Pawar, R. P., Shinde, D. B., Thopate, S. R., Eur. Chem. Bull.
2019, 8(2), 63. DOI: 10.17628/ecb.2019.8.63-70
24Pansare, D. N., Shelke, R. N., Pawar, C. D., Lett. Org. Chem.,
2017, 14(7), 517.
https://doi.org/10.2174/1570178614666170524142722
25Pansare, D. N., Shelke, R. N., Shinde, D. B., J. Het. Chem., 2017,
54(6), 3077. https://doi.org/10.1002/jhet.2919
26Pansare, D. N., Shinde, D. B., J. Saudi. Chem. Soc., 2017, 21,
434. https://doi.org/10.1016/j.jscs.2015.10.005
27Tiwari, S. V., Siddiqui, S., Seijas, J. A., Vazquez-Tato, M. P.,
Sarkate, A. P., Lokwani, D. K., Nikalje, A. P. G., Molecules,
2017, 22 (6), 995.
https://doi.org/10.3390/molecules22060995
28Tiwari, S. V., Seijas, J. A., Vazquez-Tato, M. P., Sarkate, A. P.,
Lokwani, D. K., Nikalje, A. P. G., Molecules 2016, 21 (8), 894. https://doi.org/10.3390/molecules21080894
29Pawar, C. D., Sarkate, A. P., Karnik, K. S., Bahekar, S. S.,
Pansare, D. N., Shelke R. N., Jawale, C. S., Shinde, D. B.,
Bioorg. Med. Chem. Lett., 2016, 26 (15), 3525.
https://doi.org/10.1016/j.bmcl.2016.06.030
30Dofe, V. S., Sarkate, A. P., Lokwani, D. K., Kathwate, S. H., Gill,
C. H., Res. Chem. Int., 2017, 43 (1), 15.
https://doi.org/10.1007/s11164-016-2602-z
31Sarkate, A. P., Bahekar, S. S., Wadhai, V. M., Ghandge, G. N.,
Wakte, P. S., Shinde, D. B., Synlett 2013, 24, 1513. DOI: 10.1055/s-0033-1338869
32Doherty, W., Adler, N., Knox, A., Nolan, D., McGouran, J.,
Nikalje, A. P. G., Lokwani, D., Sarkate, A., Evans, P., Eur. J.
Org. Chem., 2017, 1, 175.
https://doi.org/10.1002/ejoc.201601221
33Lokwani, D., Azad, R., Sarkate, A., Reddanna, P., Shinde, D.,
Bioorg. Med. Chem., 2015, 23 (15), 4533.
https://doi.org/10.1016/j.bmc.2015.06.008
This paper was presented at the “International Symposium on Exploring New Horizons in Chemical Sciences”, January 10– 12, 2019, Aurangabad, India (ENHCS–2019).