ROLES AND REGULATION OF STAT3 IN INNATE IMMUNITY
Hung-Ching Hsia
A dissertation submitted to the faculty of the University of North Carolina at Chapel Hill in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the department
of Cell Biology and Physiology in the School of Medicine
Chapel Hill 2017
Approved by: Albert Baldwin Douglas Cyr Blossom Damania Ben Major
ABSTRACT
Hung-Ching Hsia: Roles and Regulation of STAT3 in Innate Immunity (Under the direction of Albert Baldwin)
Innate immunity is the first line of host defense to microbial infections. The rapid
induction of the innate immune response is achieved through recognition of pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs). Upon
recognition of PAMPs and DAMPs, cells initiate immune responses by expressing type I interferons (IFN) and interferon-stimulated genes (ISGs) to establish an antiviral and
antimicrobial state, and by secreting cytokines and chemokines to recruit and activate immune cells such as macrophages and monocytes. These processes are precisely regulated to initiate a swift and effective defense against pathogens.
My dissertation focuses on the roles and regulation of signal transducer and activator of transcription 3 (STAT3) in these responses. In the first project, I investigated the role of STAT3 in myeloid cells with regards to innate immune responses during viral infection. We discovered that STAT3 knockout mice are more susceptible to herpes simplex virus-1 (HSV-1) due to an attenuated IFN response and to defects in dendritic cell and natural killer cell activation,
ACKNOWLEDGEMENTS
This dissertation would not have been possible without the help from a lot of wonderful people. First and foremost, I would like to express my gratitude to my advisor, Dr. Al Baldwin, for providing a supportive environment that allowed me to pursue interesting scientific
questions. He guided me through the process of dissertation research and cultivated my critical thinking skills to become a good scientist. The trainings I received from him are invaluable and will be forever useful. I am also grateful to my committee members, Dr. Doug Cyr, Dr. Blossom Damania, Dr. Ben Major, and Dr. Yue Xiong, for offering their time and knowledgeable insights. Their constructive feedbacks helped shape my research tremendously. Special thanks are owed to Dr. Damania, who provided expert knowledge and supports in immunology and virology as I wandered into unfamiliar territories. Thanks are also due to Dr. Jessica Hutti for her numerous thoughtful inputs, Dr. Zhe Ma and Dr. Zhigang Zhang for their suggestions and assistance in viral infection experiments, and Mr. Aaron Ebbs, Mr. Jose Roques, and Mr. Charles Stopford for their superb support in animal studies. I wish to thank the past and current members of the Baldwin Lab for constant feedbacks, daily support, and awesome happy hour that made science even more exciting. I am also fortunate to have a group of amazing friends, who are my best emotional support system no matter where in the world they are. Finally, I would like to thank my grandma and parents, who have no idea what I do but still believe in me and love me
TABLE OF CONTENTS
LIST OF TABLES ... x
LIST OF FIGURES ... xi
LIST OF ABBREVIATIONS ... xiii
CHAPTER 1: INTRODUCTION ...1
1.1 STAT3 ... 1
1.1.1 General introduction ... 1
1.1.2 STAT3 in human diseases ... 2
1.2 TBK1 and IKKε ... 4
1.2.1 General introduction ... 4
1.2.2 TBK1 and IKKε in innate immunity ... 5
1.2.3 TBK1 and IKKε in cancers ... 6
1.3 Responses Elicited by Cytosolic DNA ... 7
1.3.1 Induction of the inflammasomes by cytosolic DNA ... 7
1.3.2 Induction of interferons by cytosolic DNA ... 8
1.3.3 Induction of the NF-κB pathway by cytosolic DNA ... 12
1.3.4 Cytosolic DNA and autoimmunity ... 12
1.3.5 Cytosolic DNA and microbial infections ... 13
1.4 Regulation of STATs by Innate Immune Signaling Pathways ... 13
CHAPTER 2: STAT3 REGULATES HOST DEFENSE AND PROTECTS MICE
AGAINST HERPES SIMPLEX VIRUS-1 INFECTION ...29
2.1 Summary ... 29
2.2 Introduction ... 30
2.3 Results ... 33
2.3.1 Characterization of Stat3fl/fl LysM-Cre+/+ mice ... 33
2.3.2 Stat3 knockout BMMs show attenuated type I interferon response to HSV-1 infection ... 34
2.3.3 Myeloid Stat3 knockout mice are more susceptible to HSV-1 infection ... 36
2.3.4 Susceptibility of Stat3 knockout mice is not caused by immunopathology in the brain ... 37
2.3.5 Stat3 knockout mice express reduced levels of antiviral cytokines in the spleen ... 38
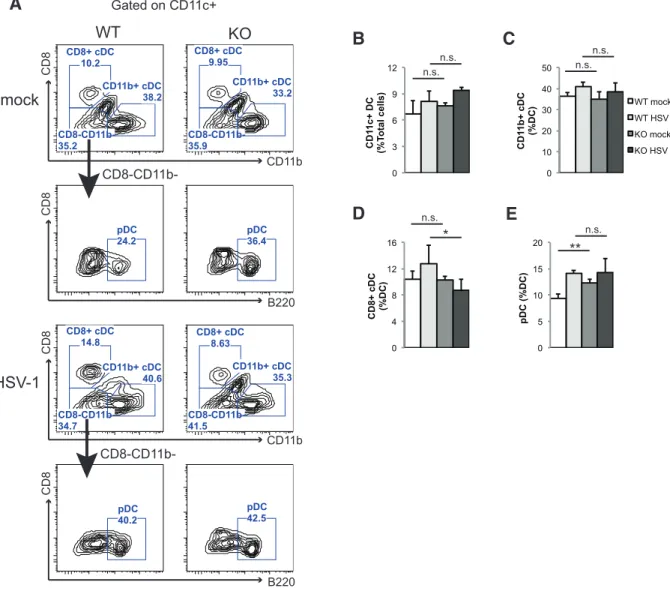
2.3.6 Stat3 knockout mice have decreased CD8+ cDC frequency during HSV-1 infection ... 39
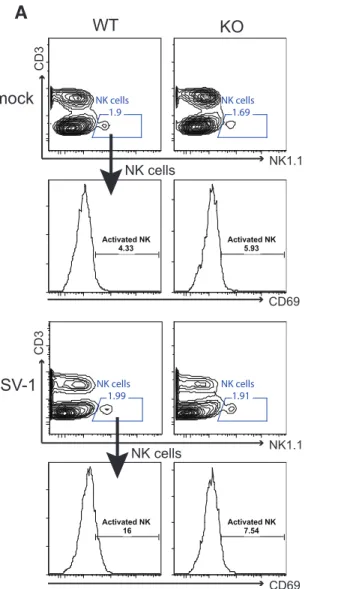
2.3.7 Stat3 knockout mice have impaired NK and T cell activation during HSV-1 infection ... 40
2.4 Discussion ... 41
2.5 Materials and Methods ... 46
REFERENCES ... 60
CHAPTER 3: CYTOSOLIC DNA PROMOTES STAT3 PHOSPHORYLATION BY TBK1 TO RESTRAIN STAT3 ACTIVITY ...66
3.1 Summary ... 66
3.2 Introduction ... 67
3.3 Results ... 69
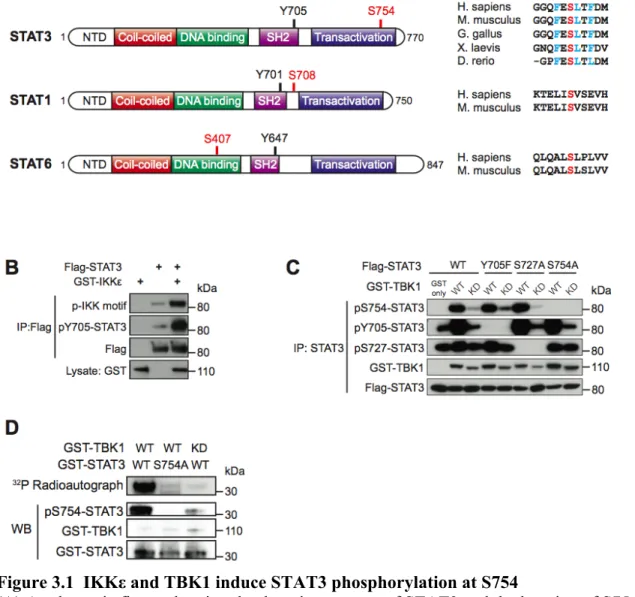
3.3.1 TBK1 directly phosphorylates STAT3 at serine 754 ... 69
3.3.2 STAT3 is phosphorylated at S754 in response to cytosolic dsDNA ... 70
3.3.4 The cGAS-STING-TBK1 pathway induces STAT3 S754 phosphorylation
in response to cytosolic DNA ... 73
3.3.5 Secreted cytokines induce activation of STAT3 in response to cytosolic DNA ... 74
3.3.6 S754 phosphorylation of STAT3 restricts cytosolic DNA-induced STAT3 target gene expression ... 75
3.3.7 S754 phosphorylation suppresses the transcriptional activity of STAT3 ... 76
3.4 Discussion ... 78
3.5 Materials and Methods ... 82
REFERENCES ... 102
CHAPTER 4: FUTURE DIRECTIONS AND CONCLUSIONS ...107
4.1 Roles and Regulation of STAT3 Activity in Diseases ... 107
4.1.1 Regulation of STAT3 activity: A balancing act ... 107
4.1.2 Regulation of STAT3 activity in viral Infection ... 109
4.1.3 Regulation of STAT3 activity in tumors and the tumor microenvironment ... 109
4.2 The Mechanism of STAT3 Inhibition by S754 Phosphorylation ... 112
4.3 Concluding Remarks ... 114
4.4 Materials and Methods ... 115
LIST OF TABLES
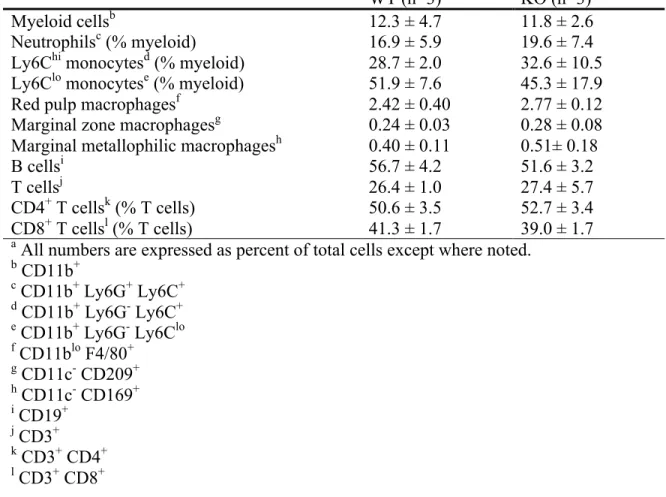
Table 2.1 Cellular composition of the spleena ... 50
LIST OF FIGURES
Figure 1.1 A schematic overview of the STAT3 pathway ... 15
Figure 1.2 Signaling pathways induced by cytosolic DNA ... 16
Figure 2.1 STAT3 knockout BMMs show attenuated type I interferon response to HSV-1 infection ... 52
Figure 2.2 Myeloid STAT3 knockout mice are more susceptible to HSV-1 infection ... 53
Figure 2.3 Susceptibility of STAT3 knockout mice is not caused by immunopathology in the brain ... 54
Figure 2.4 STAT3 knockout mice express reduced levels of antiviral cytokines in the spleen ... 55
Figure 2.5 STAT3 knockout mice have decreased CD8+ cDC frequency during HSV-1 infection ... 56
Figure 2.6 STAT3 knockout mice have impaired NK and T cell activation during HSV-1 infection ... 57
Supplemental Figure 2.1 Myeloid-specific deletion of STAT3 ... 58
Supplemental Figure 2.2 KO mice showed marginally higher viral loads in the brain ... 59
Figure 3.1 IKKε and TBK1 induce STAT3 phosphorylation at S754 ... 88
Figure 3.2 Cytosolic DNA induces STAT3 activation and phosphorylation at S754 ... 89
Figure 3.3 TBK1 activity is required for cytosolic DNA-induced S754 phosphorylation of STAT3 ... 90
Figure 3.4 Cytosolic DNA induces pS754-STAT3 via the cGAS/STING axis ... 91
Figure 3.5 Cytosolic DNA-induced STAT3 activation is mediated by de novo synthesized secreted factors ... 92
Figure 3.6 . S754 phosphorylation restricts STAT3 activation in response to cytosolic DNA ... 94
Figure 3.7 S754 phosphorylation inhibits transcriptional activity of STAT3 ... 96
Supplemental Figure 3.1 Cytosolic DNA induces expression of IFNβ and IL-6 ... 99
Supplemental Figure 3.2 CRISPR-mediated knockout of STAT3 in HEK293T
and THP-1 ... 100
Supplemental Figure 3.3 STAT3 inhibits IFNβ-induced ISRE reporter expression ... 101
Figure 4.1 Cytosolic DNA induces STAT3 S754 phosphorylation and promotes
DC maturation ... 117
LIST OF ABBREVIATIONS
AD-HIES Autosomal dominant hyper IgE syndrome AIM2 Absent in melanoma 2
BMDC Bone marrow-derived dendritic cells BMM Bone marrow-derived macrophages cDC Conventional dendritic cells
cGAS Cyclic GAMP synthase CTL Cytotoxic T lymphocytes
DAMP Danger-associated molecular pattern DBD DNA-binding domain
DDX41 DEAD-box helicase 41 GAS Gamma-activated sequence HSV-1 Herpes simplex virus-1
IFI16 Interferon gamma inducible protein IFN Interferon
IκB Inhibitor of kappaB IKK IκB kinase
IRF Interferon regulatory factor ISG Interferon stimulated genes
ISGF3 Interferon-stimulated gene factor 3 ISRE Interferon-stimulated responsive element JAK Janus kinase
MMMΦ Marginal metallophilic macrophages MOI Multiplicity of infection
MZMΦ Marginal zone macrophages NCOA1 Nuclear receptor coactivator 1 NF-κB Nuclear factor-kappaB
NK Natural killer cells NTD N-terminal domain
pDC plasmacytoid dendritic cells
PAMP Pathogen-associated molecular pattern PTM Post-translational modification
SH2 SRC homology 2
STAT3 Signal transducer and activator of transcription 3 STING Stimulator of interferon genes
CHAPTER 1: INTRODUCTION 1.1 STAT3
1.1.1 General introduction
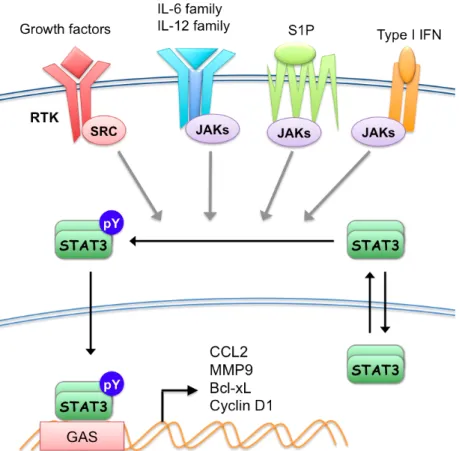
Unphosphorylated STAT3 exists as stable dimers prior to activation because of the homotypic interaction mediated by the NTDs of two STAT3 molecules, but inactive STAT3 dimers are constantly being shuttled in between the nucleus and the cytoplasm (6-9). While activated, Y705 phosphorylation enhances the DNA binding activity of STAT3 and thus the nuclear retention (8,10). Active STAT3 homodimers bind to a 9-bp palindromic sequence with the consensus of TTCNNNGAA (11). However, this gamma-interferon activating sequence (GAS) recognized by STAT3 is very similar to that of STAT1. In fact, STAT3 and STAT1 share identical optimal GAS sequence when analyzed in vitro, but variation in endogenous core and flanking sequences leads to differential recognition by STAT1 and STAT3, resulting in distinct but overlapping target genes controlled by STAT1 and STAT3 (12,13). In the nucleus, STAT3 promotes the transcription of genes involved in inflammation, survival, and cell cycle, such as C-C motif ligand 2 (CCL2), Bcl-xL, metallomatrix protease 2 (MMP2), Survivin, and Cyclin D1 (4) (Figure 1.1). With target genes involved in these essential cellular processes, constitutive activation of STAT3 has been implicated in tumorigenesis and various types of cancers (4,14,15), as discussed in the following paragraphs.
1.1.2 STAT3 in human diseases
STAT3 in myeloid cells results in elevated TNFα and IL-12 in response to bacterial components and susceptibility to bacterial infections (18,19). These mice are also more susceptible to herpes simplex virus-1 (HSV-1) infection, as discussed in Chapter 2 of this dissertation (20).
Conversely, somatic activating mutations of STAT3 have been associated with
lymphoproliferative disorders and hepatocellular adenomas (21-23), whereas germline activating mutations of STAT3 contribute to early-onset autoimmune disorders that affect multiple organs (24), underscoring dysregulated STAT3 activation as a causative factor in enhanced cell
proliferation and aberrant activation of immune cells.
Constitutive activation of STAT3 has been documented in many types of cancers, and the consequences of such dysregulation are multifaceted. Through target genes involved in
inflammation, anti-apoptosis, cell cycle, and invasion and migration, STAT3 supports
tumorigenesis, survival, proliferation, and metastasis of cancer cells (4,25,26). Expression of a constitutively active STAT3 mutant is sufficient to transform immortalized cells (14,27). In K-RAS driven pancreatic cancers, STAT3 is required for the initiation and progression of
inflammatory cytokines (15). STAT3 physically interacts with NF-κB and retains NF-κB in the nucleus for sustained activation and expression of IL-6, which further activates STAT3 and forms an amplification loop (35-39). Finally, STAT3 inhibits the expression of
immunostimulatory genes in cancer cells (40), and induces the expression of IL-23 to activate regulatory T cells (Treg) in the tumor microenvironment, thereby dampening the anti-tumor immune responses through a concerted effort (41). Moreover, IL-10 from tumor cells inhibits activation of immune cells via STAT3 signaling, and genetic deletion of STAT3 in
hematopoietic cells enhances anti-tumor immunity, demonstrating an immunosuppressive role of STAT3 in the tumor microenvironment (42,43).
1.2 TBK1 and IKKε
1.2.1 General introduction
NF-κB is a family of dimeric transcription factors comprising five subunits: p65 (RelA), p100/p52, p105/p50, RelB, and c-Rel. In the canonical NF-κB pathway, the activity of the p65-p50 dimer is tightly regulated by inhibitor of kappaB (IκB) and two IκB kinases (IKKs), IKKα
and IKKβ. Under basal conditions, the p65-p50 NF-κB dimers are inactive and bound by IκBα in the cytosol. Various environmental cues and cytokines such as IL-1 and TNFα can lead to
activation of the IKK complex comprising of IKKα, IKKβ, and IKKγ (NF-κB essential modifier/NEMO). The activated IKK complex phosphorylates IκBα at S32 and S36, which triggers proteasome-dependent degradation of IκBα. The liberated p65-p50 dimers then
inhibitory ankyrin repeats in p100 subunit. Phosphorylation of p100 by IKKα induces
proteasomal processing of p100 into p52, resulting in the active RelB-p52 dimers (Reviewed in (44)). Genetic experiments demonstrated that the IKKs, especially IKKβ, are required for NF-κB activation by lipopolysaccharide (LPS), IL-1, and TNFα (45-47). Following the identification of IKKα and IKKβ, two more related kinases were identified. TANK-binding kinase 1 (TBK1) was originally identified in a screen for proteins interacting with TANK, a TRAF-binding protein that modulates TRAF-mediated NF-κB signaling (48), and IKKε was identified as an IKK-related kinase whose expression can be highly induced by LPS (49). The sequences of TBK1 and IKKε
share approximately 50% identity with each other, and approximately 30% identity with IKKα
and IKKβ (50). The domain structures of TBK1 and IKKε are similar to IKKα and IKKβ, but TBK1 and IKKε lack the NEMO-binding domain in the C terminus.
1.2.2 TBK1 and IKKε in innate immunity
Unlike IKKα and IKKβ that phosphorylate both S32 and S36 of IκBα, TBK1 and IKKε
(TLRs) and TLR-independent receptors (58). In general, the activating signals from these
receptors lead to the assembly of ubiquitin ligase complexes, which bind to and K63-ubiquitinate TBK1 and IKKε. The K63-polyubiquitin chains then cooperate with ubiquitin binding proteins to promote TBK1 and IKKε oligomerization and facilitate their autophosphorylation (59-63). Once activated by autophosphorylation, TBK1 and IKKε mediate IFN expression by directly
phosphorylating interferon regulatory factors IRF3 and IRF7 at a number of serine residues, allowing IRFs to homodimerize and translocate to the nucleus to induce type I IFN transcription (52,64). In addition to its pivotal role in IFN induction, TBK1 also promotes autophagy-mediated clearance of intracellular bacteria by phosphorylating adaptors that target bacteria for the
autophagosome (65,66). With their ability to respond to the presence of PAMPs and DAMPs to induce IFN expression and autophagy, TBK1, and to a lesser extent IKKε, are critical molecules that establish the first line of host defense against pathogens at the cellular level.
1.2.3 TBK1 and IKKε in cancers
In addition to their well-established roles in initiating IFN responses, several studies reported an oncogenic role for TBK1 and IKKε. An RNAi screening showed that KRAS-transformed cancer cell lines require TBK1-mediated activation of NF-κB and transcription of anti-apoptotic genes for survival (67). TBK1 also inhibits oncogenic stress-induced apoptosis via NF-κB independent pathways (68,69). Similarly, IKKε is often overexpressed or amplified in human breast cancers (70), and inhibition of IKKε in related breast cancer cell-lines results in halted growth and cell death (70,71). IKKε facilitates oncogenesis by indirectly promoting
1.3 Responses Elicited by Cytosolic DNA
The subcellular compartmentalization of mammalian cells normally sequesters DNA in the nucleus and mitochondria. DNA in the cytosol is thus a DAMP that signals abnormality and triggers cell-intrinsic immune responses (56,73). Cytosolic DNA can be derived from self-DNA in normal physiological conditions, such as phagocytosed materials, including dead cells and ejected nuclei from erythroid precrusors, or re-activated endogenous retroelements. Additionally, viral or intracellular bacterial infections lead to the presence of foreign DNA in the cytosol (74). Recognition of cytosolic DNA by cellular sensors leads to activation of two major pathways: (i) inflammasome-driven mechanisms that produce IL-1β and IL-18, and (ii) IRF activation that induces IFN expression. The major players in the cytosolic DNA pathway are introduced in the following paragraphs, with emphases on the IFN response (Figure 1.2).
1.3.1 Induction of the inflammasomes by cytosolic DNA
The inflammasome is a molecular complex that regulates the activation of caspase-1 and induces inflammation in response to a variety of PAMPs and DAMPs. Assembly of
inflammasomes is mediated by polymerization of apoptosis-associated speck-like protein containing a CARD (ASC) proteins and the subsequent recruitment of pro-caspase 1. After self-activating proteolytic cleavage, caspase 1 processes the cytokine precursors pro-IL-1β and pro-IL-18 into active IL-1β and IL-18 (75). Cytosolic DNA induces IL-1β and IL-18 secretion through inflammasome formation mediated by the protein absent in melanoma 2 (AIM2). AIM2 is a member of the Pyrin and HIN domain (PYHIN) family (76-78) and binds to dsDNA directly via its HIN domain. The Pyrin domain and HIN domain of AIM2 form an autoinhibitory
the HIN domain liberates the Pyrin domain, allowing it to interact with ASC. Using a DNA molecule as a platform, multiple AIM2 proteins bind to and nucleate on the DNA, thereby facilitating the polymerization of ASC and the formation of inflammasomes in response to cytosolic DNA (77-79).
1.3.2 Induction of interferons by cytosolic DNA
While detection of cytosolic DNA by AIM2 induces inflammasome formation through polymerization of the adaptor ASC, other cytosolic DNA sensors engage different adaptor proteins to promote TBK1 and IRF3 activation, thereby leading to interferon production. The roles of various cytosolic DNA sensors and the quintessential adaptor stimulator of interferon genes (STING) in cytosolic DNA-induced IFN response are discussed as follows.
response mediated by other cytosolic DNA sensors, such as IFI16 and DDX41 (83,89). In addition to cGAMP, STING also recognizes other cyclic di-nucleotides (CDNs) generated by bacteria (90,91). Therefore, STING has an integral role in defending against intracellular bacterial infection by directly sensing bacterial CDN and by indirectly responding to bacterial DNA via cGAS. Several frequent single nucleotide polymorphisms (SNPs) have been identified in the human TMEM173 allele. These SNPs lead to amino acid substitutions that decrease the affinity of STING to bacterial CDNs, and may have implications in the susceptibility to bacterial diseases and autoimmunity of different individuals (92,93).
ZBP1. Z-DNA binding protein 1 (ZBP1) is a DNA-binding protein that was initially identified in macrophages as being encoded by an IFN-inducible gene (97,98). A cytosolic protein, ZBP1 binds to cytosolic dsDNA and contributes to optimal IFN expression in MEFs and L929 cells by promoting TBK1-IRF3 interaction (99). ZBP1 also contributes to IFN responses and resistance to cytomegalovirus, a DNA virus (100). However, it is unlikely that ZBP1 is the principle cytosolic DNA sensor, since genetic deletion of ZBP1 has minimal impact on the ability of cells to respond to cytosolic DNA (83,101).
DDX41. The DEAD-box helicase 41 (DDX41) protein has been demonstrated to participate in the cytosolic DNA-induced IFN response in dendritic cells (89). DDX41 binds to dsDNA through its DEAD-box domain, and interacts and colocalizes with STING upon dsDNA transfection (89). It was later shown that Bruton’s tyrosine kinase (BTK) phosphorylates DDX41 and induces its binding with STING upon cytosolic dsDNA stimulation, leading to activation of the TBK1-IRF3 signaling axis (102). Moreover, DDX41 also binds to cyclic di-GMP and cyclic AMP with even higher affinity than STING, and is required for cyclic GMP and cyclic di-AMP-induced, STING-dependent IFN responses (103). It is worth noting that while expression of IFI16 and ZBP1 remains low unless induced by IFNs, the constitutive expression of DDX41 in cells such as DCs and monocytes suggest a critical role for DDX41 in the first line of cytosolic DNA sensing.
RNA polymerase III. STING has an essential role in IFN expression induced by cytosolic DNA regardless of DNA sequences or sources with one exception (104). Transfection of poly(dA:dT) induces moderate IFN expression even in the absence of STING (104),
transcribed by RNA polymerase III in the cytosol (105,106). The resulting 5’ triphosphate-capped RNA products are recognized by retinoic acid inducible gene-I (RIG-I), thereby inducing IFN expression through a mitochondrial antiviral signaling protein (MAVS)-dependent
mechanism (105,106).
cGAS. While the aforementioned cytosolic DNA sensors are all involved in relaying the presence of cytosolic DNA to IFN induction, knockdown of individual sensors rarely achieves complete abrogation of cytosolic DNA-induced IFN, indicating redundancy between these cytosolic DNA sensors. Moreover, their inducible expression (IFI16, ZBP1), limited distribution pattern in specific cell types (DDX41), and specificity toward AT-rich substrates (RNA Pol III) suggested the existence of additional unidentified cytosolic DNA sensors that are more
1.3.3 Induction of the NF-κB pathway by cytosolic DNA
In addition to type I IFN, cytosolic DNA also activates NF-κB in a STING-depedent manner. STING overexpression is sufficient to activate NF-κB, whereas STING deficiency abolishes cytosolic DNA-induced NF-κB activation (81,104). Further studies demonstrated that both canonical (p65-p50) and non-canonical (RelB-p52) NF-κB are activated downstream of STING, and NF-κB is required for IL-6 expression and contributes to IFNβ expression induced by cytosolic DNA (115). Activation of NF-κB requires IKKα, IKKβ, and the E3 ligases TRAF3 and TRAF6 but, depending on the cell type, TBK1 may or may not be required for activation of IKKβ by cytosolic DNA ((115) and Chapter 3).
1.3.4 Cytosolic DNA and autoimmunity
Clearance of self-DNA in the cytosol under physiological conditions is achieved by DNases in the cytosol and lysosome, and failure to clear self-DNA causes substantial inflammatory and IFN responses as well as autoimmune conditions. For example,
1.3.5 Cytosolic DNA and microbial infections
Cytosolic DNA can also arise from viral or intracellular microbial infections. The DNA genome of herpesvirus and vaccinia virus, as well as the reverse-transcribed HIV DNA, are recognized by intracellular DNA sensors, thereby triggering IFN responses and host antiviral mechanisms (83,100,114,128-133). Not surprisingly, a number of viral proteins have been identified by their ability to target and inhibit the cytosolic DNA pathway to evade host immune surveillance (Reviewed in (134)). Interestingly, an unbiased screening revealed that in addition to its role in controlling DNA virus pathogenesis, cGAS also contributes to limiting RNA virus replication (135). This suggests an unidentified mechanism by which viral RNA activates cGAS. Alternatively, it is possible that low levels of cGAS activation by self-DNA pre-establish a suboptimal antiviral state, such that cGAS deficiency makes cells more susceptible to RNA virus infection. Intracellular bacteria, such as Mycobacterium tuberculosis, induce IFNβ production in a cGAS-STING-dependent manner (136,137). Activation of TBK1 in this scenario also induces ubiquitin-mediated autophagy that targets the M. tuberculosis to lysosome for degradation, thereby serving as an important tool in controlling intracellular mycobacteria replication (138). Similarly, intracellular infection of Legionella, Listeria, and Francisella all engage the cytosolic DNA pathway, while Listeria also produces cyclic-di-AMP that directly activates STING (136,139-141).
1.4 Regulation of STATs by Innate Immune Signaling Pathways
which promotes efficient antiviral responses by inducing CCL2, CCL20, and CCL26 (142). Type I IFN (IFNα and IFNβ) induces STAT1 homodimers to promote GAS-driven gene expression, while type II IFN (IFNγ) induces STAT1-STAT2-IRF9 heterotrimers, which bind to the interferon-stimulated response element (ISRE) and promote a different ISG expression profile. IKKε fine-tunes the balance between GAS-driven and ISRE-driven ISGs by phosphorylating S708 of STAT1. This phosphorylation disrupts STAT1 homodimerization and favors the heterotrimer formation, thereby shifting the IFN-induced gene expression profile toward ISRE-driven ISGs (143,144). It has also been noted that STING deficiency in autoimmune-prone mice leads to hyperactivation of STAT1-mediated IFN responses and exacerbated autoimmunity symptoms, suggesting a negative role of STING in regulation of STAT1 (145). This regulation may be partially mediated by the ability of STING to activate SH2 domain-containing
Figure 1.1 A schematic overview of the STAT3 pathway
Figure 1.2 Signaling pathways induced by cytosolic DNA
The presence of cytosolic DNA is detected by a variety of sensors: cGAS, IFI16, DDX41, AIM2, and RNA polymerase III. While AIM2 induces inflammasome formation and RNA polymerase III activates the dsRNA pathway, the other sensors engage STING to activate TBK1, leading to IRF3 phosphorylation and IFNβ production. IFNβ establishes an antiviral state through
REFERENCES
1. Zhong Z, Wen Z, Darnell JE. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science. 1994 Apr 1;264(5155):95–8.
2. Akira S, Nishio Y, Inoue M, Wang XJ, Wei S, Matsusaka T, et al. Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell. 1994 Apr 8;77(1):63–71.
3. Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer. 2014 Oct 24;14(11):736–46. 4. Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for
STAT3. Nat Rev Cancer. 2009 Nov;9(11):798–809.
5. Lee H, Deng J, Kujawski M, Yang C, Liu Y, Herrmann A, et al. STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nat Med. 2010
Dec;16(12):1421–8.
6. Haan S, Kortylewski M, Behrmann I, Müller-Esterl W, Heinrich PC, Schaper F.
Cytoplasmic STAT proteins associate prior to activation. Biochem J. 2000 Feb 1;345 Pt 3:417–21.
7. Braunstein J, Brutsaert S, Olson R, Schindler C. STATs dimerize in the absence of phosphorylation. J Biol Chem. 2003 Sep 5;278(36):34133–40.
8. Kretzschmar AK, Dinger MC, Henze C, Brocke-Heidrich K, Horn F. Analysis of Stat3 (signal transducer and activator of transcription 3) dimerization by fluorescence resonance energy transfer in living cells. Biochem J. 2004 Jan 15;377(Pt 2):289–97.
9. Reich NC, Liu L. Tracking STAT nuclear traffic. Nat Rev Immunol. 2006 Aug;6(8):602– 12.
10. Sasse J, Hemmann U, Schwartz C, Schniertshauer U, Heesel B, Landgraf C, et al.
Mutational analysis of acute-phase response factor/Stat3 activation and dimerization. Mol Cell Biol. 1997 Aug;17(8):4677–86.
11. Becker S, Groner B, Müller CW. Three-dimensional structure of the Stat3beta homodimer bound to DNA. Nature. 1998 Jul 9;394(6689):145–51.
12. Horvath CM, Wen Z, Darnell JE. A STAT protein domain that determines DNA sequence recognition suggests a novel DNA-binding domain. Genes Dev. 1995 Apr 15;9(8):984–94. 13. Ehret GB, Reichenbach P, Schindler U, Horvath CM, Fritz S, Nabholz M, et al. DNA
14. Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, et al. Stat3 as an oncogene. Cell. 1999 Aug 6;98(3):295–303.
15. Grivennikov SI, Karin M. Dangerous liaisons: STAT3 and NF-kappaB collaboration and crosstalk in cancer. Cytokine and Growth Factor Reviews. 2010 Feb;21(1):11–9.
16. Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, et al. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med. 2007 Oct 18;357(16):1608–19. 17. Siegel AM, Heimall J, Freeman AF, Hsu AP, Brittain E, Brenchley JM, et al. A critical
role for STAT3 transcription factor signaling in the development and maintenance of human T cell memory. Immunity. 2011 Nov 23;35(5):806–18.
18. Takeda K, Clausen BE, Kaisho T, Tsujimura T, Terada N, Förster I, et al. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. 1999 Jan;10(1):39–49.
19. Matsukawa A, Takeda K, Kudo S, Maeda T, Kagayama M, Akira S. Aberrant
inflammation and lethality to septic peritonitis in mice lacking STAT3 in macrophages and neutrophils. J Immunol. 2003 Dec 1;171(11):6198–205.
20. Hsia H-C, Stopford CM, Zhang Z, Damania B, Baldwin AS. Signal transducer and activator of transcription 3 (Stat3) regulates host defense and protects mice against herpes simplex virus-1 (HSV-1) infection. Journal of Leukocyte Biology. 2016 Dec 13.
21. Koskela HLM, Eldfors S, Ellonen P, van Adrichem AJ, Kuusanmäki H, Andersson EI, et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med. 2012 May 17;366(20):1905–13.
22. Jerez A, Clemente MJ, Makishima H, Koskela H, LeBlanc F, Peng Ng K, et al. STAT3 mutations unify the pathogenesis of chronic lymphoproliferative disorders of NK cells and T-cell large granular lymphocyte leukemia. Blood. 2012 Oct 11;120(15):3048–57.
23. Pilati C, Amessou M, Bihl MP, Balabaud C, Van Nhieu JT, Paradis V, et al. Somatic mutations activating STAT3 in human inflammatory hepatocellular adenomas. J Exp Med. 2011 Jul 4;208(7):1359–66.
24. Flanagan SE, Haapaniemi E, Russell MA, Caswell R, Lango Allen H, De Franco E, et al. Activating germline mutations in STAT3 cause early-onset multi-organ autoimmune disease. Nat Genet. 2014 Aug;46(8):812–4.
25. Yuan J, Zhang F, Niu R. Multiple regulation pathways and pivotal biological functions of STAT3 in cancer. Sci Rep. 2015;5:17663.
serve as a hit in the process of malignant transformation of primary cells. Cell Death Differ. 2012 Aug;19(8):1390–7.
28. Corcoran RB, Contino G, Deshpande V, Tzatsos A, Conrad C, Benes CH, et al. STAT3 Plays a Critical Role in KRAS-Induced Pancreatic Tumorigenesis. Cancer Res. 2011 Jul 14;71(14):5020–9.
29. Grabner B, Schramek D, Mueller KM, Moll HP, Svinka J, Hoffmann T, et al. Disruption of STAT3 signalling promotes KRAS-induced lung tumorigenesis. Nat Commun.
2015;6:6285.
30. Zhou J, Qu Z, Yan S, Sun F, Whitsett JA, Shapiro SD, et al. Differential roles of STAT3 in the initiation and growth of lung cancer. Oncogene. 2015 Jul;34(29):3804–14.
31. Kim E, Kim M, Woo D-H, Shin Y, Shin J, Chang N, et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell. 2013 Jun 10;23(6):839–52.
32. Schroeder A, Herrmann A, Cherryholmes G, Kowolik C, Buettner R, Pal S, et al. Loss of Androgen Receptor Expression Promotes a Stem-like Cell Phenotype in Prostate Cancer through STAT3 Signaling. Cancer Res. 2014 Feb 16;74(4):1227–37.
33. Kesanakurti D, Chetty C, Rajasekhar Maddirela D, Gujrati M, Rao JS. Essential role of cooperative NF-κB and Stat3 recruitment to ICAM-1 intronic consensus elements in the regulation of radiation-induced invasion and migration in glioma. Oncogene. 2013 Oct 24;32(43):5144–55.
34. Deng J, Liu Y, Lee H, Herrmann A, Zhang W, Zhang C, et al. S1PR1-STAT3 signaling is crucial for myeloid cell colonization at future metastatic sites. Cancer Cell. 2012 May 15;21(5):642–54.
35. Grivennikov S, Karin E, Terzic J, Mucida D, Yu G-Y, Vallabhapurapu S, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009 Feb 3;15(2):103–13.
36. Yoon S, Woo SU, Kang JH, Kim K, Shin H-J, Gwak H-S, et al. NF-κB and STAT3 cooperatively induce IL6 in starved cancer cells. Oncogene. 2012 Jul 19;31(29):3467–81. 37. Hutti JE, Pfefferle AD, Russell SC, Sircar M, Perou CM, Baldwin AS. Oncogenic PI3K
mutations lead to NF-κB-dependent cytokine expression following growth factor deprivation. Cancer Res. 2012 Jul 1;72(13):3260–9.
38. Lee H, Herrmann A, Deng J-H, Kujawski M, Niu G, LI Z, et al. Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell. 2009 Apr 7;15(4):283–93.
inflammation, and development of colitis-associated cancer. Cancer Cell. 2013 Jan 14;23(1):107–20.
40. Lee H, Deng J, Xin H, Liu Y, Pardoll D, Yu H. A requirement of STAT3 DNA binding precludes Th-1 immunostimulatory gene expression by NF-κB in tumors. Cancer Res. 2011 Jun 1;71(11):3772–80.
41. Kortylewski M, Xin H, Kujawski M, Lee H, Liu Y, Harris T, et al. Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell. 2009 Feb 3;15(2):114–23.
42. Wang T, Niu G, Kortylewski M, Burdelya L, Shain K, Zhang S, et al. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat Med. 2003 Dec 21;10(1):48–54.
43. Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S, et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med. 2005 Nov 20;11(12):1314–21.
44. Rinkenbaugh A, Baldwin A. The NF-κB Pathway and Cancer Stem Cells. Cells. 2016 Jun;5(2):16.
45. Li Q, Lu Q, Hwang JY, Büscher D, Lee KF, Izpisua-Belmonte JC, et al. IKK1-deficient mice exhibit abnormal development of skin and skeleton. Genes Dev. 1999 May
15;13(10):1322–8.
46. Li Q, Van Antwerp D, Mercurio F, Lee KF, Verma IM. Severe liver degeneration in mice lacking the IkappaB kinase 2 gene. Science. 1999 Apr 9;284(5412):321–5.
47. Li Q, Estepa G, Memet S, Israel A, Verma IM. Complete lack of NF-kappaB activity in IKK1 and IKK2 double-deficient mice: additional defect in neurulation. Genes Dev. 2000 Jul 15;14(14):1729–33.
48. Pomerantz JL, Baltimore D. NF-kappaB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO J. 1999 Dec
1;18(23):6694–704.
49. Shimada T, Kawai T, Takeda K, Matsumoto M, Inoue J, Tatsumi Y, et al. IKK-i, a novel lipopolysaccharide-inducible kinase that is related to IkappaB kinases. International Immunology. 1999 Aug;11(8):1357–62.
50. Shen RR, Hahn WC. Emerging roles for the non-canonical IKKs in cancer. Oncogene. 2010 Nov 1;30(6):631–41.
51. Tojima Y, Fujimoto A, Delhase M, Chen Y, Hatakeyama S, Nakayama K, et al. NAK is an IkappaB kinase-activating kinase. Nature. 2000 Apr 13;404(6779):778–82.
interferon antiviral response through an IKK-related pathway. Science. 2003 May 16;300(5622):1148–51.
53. Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, et al. IKKε
and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003 Apr 14;4(5):491–6.
54. Hemmi H, Takeuchi O, Sato S, Yamamoto M, Kaisho T, Sanjo H, et al. The roles of two IkappaB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J Exp Med. 2004 Jun 21;199(12):1641–50.
55. Perry AK, Chow EK, Goodnough JB, Yeh W-C, Cheng G. Differential Requirement for TANK-binding Kinase-1 in Type I Interferon Responses to Toll-like Receptor Activation and Viral Infection. J Exp Med. 2004 Jun 21;199(12):1651–8.
56. Ishii KJ, Coban C, Kato H, Takahashi K, Torii Y, Takeshita F, et al. A Toll-like receptor– independent antiviral response induced by double-stranded B-form DNA. Nat Immunol. 2005 Nov 13;7(1):40–8.
57. Solis M, Romieu-Mourez R, Goubau D, Grandvaux N, Mesplede T, Julkunen I, et al. Involvement of TBK1 and IKKε in lipopolysaccharide-induced activation of the interferon response in primary human macrophages. Eur J Immunol. 2007 Feb;37(2):528–39.
58. Chau T-L, Gioia R, Gatot J-S, Patrascu F, Carpentier I, Chapelle J-P, et al. Are the IKKs and IKK-related kinases TBK1 and IKK-epsilon similarly activated? Trends Biochem Sci. 2008 Apr 1;33(4):171–80.
59. Zhou AY, Shen RR, Kim E, Lock YJ, Xu M, Chen ZJ, et al. IKKε-mediated
tumorigenesis requires K63-linked polyubiquitination by a cIAP1/cIAP2/TRAF2 E3 ubiquitin ligase complex. Cell Rep. 2013 Mar 28;3(3):724–33.
60. Tu D, Zhu Z, Zhou AY, Yun C-H, Lee K-E, Toms AV, et al. Structure and
Ubiquitination-Dependent Activation of TANK-Binding Kinase 1. Cell Rep. The Authors; 2013 Mar 28;3(3):747–58.
61. Larabi A, Devos JM, Ng S-L, Nanao MH, Round A, Maniatis T, et al. Crystal structure and mechanism of activation of TANK-binding kinase 1. Cell Rep. 2013 Mar
28;3(3):734–46.
62. Ma X, Helgason E, Phung QT, Quan CL, Iyer RS, Lee MW, et al. Molecular basis of Tank-binding kinase 1 activation by transautophosphorylation. Proc Natl Acad Sci USA. 2012 Jun 12;109(24):9378–83.
63. Wang L, Li S, Dorf ME. NEMO Binds Ubiquitinated TANK-Binding Kinase 1 (TBK1) to Regulate Innate Immune Responses to RNA Viruses. Meurs EF, editor. PLoS ONE. 2012 Sep 18;7(9):e43756.
IFN-regulatory factor 3-dependent gene expression is defective in Tbk1-deficient mouse embryonic fibroblasts. Proc Natl Acad Sci USA. 2004 Jan 6;101(1):233–8.
65. Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, et al. Phosphorylation of the Autophagy Receptor Optineurin Restricts Salmonella Growth. Science. 2011 Jul 7;333(6039):228–33.
66. Pilli M, Arko-Mensah J, Ponpuak M, Roberts E, Master S, Mandell MA, et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity. 2012 Aug 24;37(2):223–34.
67. Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009 Nov 5;462(7269):108–12.
68. Chien Y, Kim S, Bumeister R, Loo Y-M, Kwon SW, Johnson CL, et al. RalB GTPase-mediated activation of the IkappaB family kinase TBK1 couples innate immune signaling to tumor cell survival. Cell. 2006 Oct 6;127(1):157–70.
69. Ou Y-H, Torres M, Ram R, Formstecher E, Roland C, Cheng T, et al. TBK1 Directly Engages Akt/PKB Survival Signaling to Support Oncogenic Transformation. Mol Cell. 2011 Feb 18;41(4):458–70.
70. Boehm JS, Zhao JJ, Yao J, Kim SY, Firestein R, Dunn IF, et al. Integrative Genomic Approaches Identify IKBKE as a Breast Cancer Oncogene. Cell. 2007 Jun;129(6):1065– 79.
71. Barbie TU, Alexe G, Aref AR, Li S, Zhu Z, Zhang X, et al. Targeting an IKBKE cytokine network impairs triple-negative breast cancer growth. J Clin Invest. 2014 Nov 3.
72. Hutti JE, Shen RR, Abbott DW, Zhou AY, Sprott KM, Asara JM, et al. Phosphorylation of the tumor suppressor CYLD by the breast cancer oncogene IKKepsilon promotes cell transformation. Mol Cell. 2009 May 14;34(4):461–72.
73. Stetson DB, Medzhitov R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity. 2006 Jan;24(1):93–103.
74. Paludan SR, Bowie AG. Immune sensing of DNA. Immunity. 2013 May 23;38(5):870–80. 75. Guo H, Callaway JB, Ting JP-Y. Inflammasomes: mechanism of action, role in disease,
and therapeutics. Nat Med. 2015 Jun 29;21(7):677–87.
76. Muruve DA, Pétrilli V, Zaiss AK, White LR, Clark SA, Ross PJ, et al. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature. 2008 Feb 20;452(7183):103–7.
26;458(7237):509–13.
78. Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009 Mar 26;458(7237):514–8.
79. Jin T, Perry A, Jiang J, Smith P, Curry JA, Unterholzner L, et al. Structures of the HIN domain:DNA complexes reveal ligand binding and activation mechanisms of the AIM2 inflammasome and IFI16 receptor. Immunity. 2012 Apr 20;36(4):561–71.
80. Zhong B, Yang Y, Li S, Wang Y-Y, Li Y, Diao F, et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity. 2008 Oct 17;29(4):538–50.
81. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008 Aug 24;455(7213):674–8.
82. Sun W, Li Y, Chen L, Chen H, You F, Zhou X, et al. ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc Natl Acad Sci USA. 2009 May 26;106(21):8653–8.
83. Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, et al. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol. 2010 Nov;11(11):997–1004. 84. Burdette DL, Vance RE. STING and the innate immune response to nucleic acids in the
cytosol. Nat Immunol. 2012 Dec 14;14(1):19–26.
85. Ouyang S, Song X, Wang Y, Ru H, Shaw N, Jiang Y, et al. Structural analysis of the STING adaptor protein reveals a hydrophobic dimer interface and mode of cyclic di-GMP binding. Immunity. 2012 Jun 29;36(6):1073–86.
86. Tanaka Y, Chen ZJ. STING Specifies IRF3 Phosphorylation by TBK1 in the Cytosolic DNA Signaling Pathway. Sci Signal. 2012 Mar 6;5(214):ra20–0.
87. Zhao B, Shu C, Gao X, Sankaran B, Du F, Shelton CL, et al. Structural basis for concerted recruitment and activation of IRF-3 by innate immune adaptor proteins. Proc Natl Acad Sci USA. 2016 Jun 14;113(24):E3403–12.
88. Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science. 2015 Mar 13;347(6227):aaa2630.
89. Zhang Z, Yuan B, Bao M, Lu N, Kim T, Liu Y-J. The helicase DDX41 senses
intracellular DNA mediated by the adaptor STING in dendritic cells. Nat Immunol. 2011 Sep 4;12(10):959–65.
Function of Sting in the In Vivo Interferon Response to Listeria monocytogenes and Cyclic Dinucleotides. Infection and Immunity. 2011 Jan 21;79(2):688–94.
91. Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, et al. STING is a direct innate immune sensor of cyclic di-GMP. Nature. 2011 Sep 25;478(7370):515–8. 92. Yi G, Brendel VP, Shu C, Li P, Palanathan S, Cheng Kao C. Single nucleotide
polymorphisms of human STING can affect innate immune response to cyclic dinucleotides. PLoS ONE. 2013;8(10):e77846.
93. Jin L, Xu L-G, Yang IV, Davidson EJ, Schwartz DA, Wurfel MM, et al. Identification and characterization of a loss-of-function human MPYS variant. Genes Immun. 2011
Jun;12(4):263–9.
94. Aglipay JA, Lee SW, Okada S, Fujiuchi N, Ohtsuka T, Kwak JC, et al. A member of the Pyrin family, IFI16, is a novel BRCA1-associated protein involved in the p53-mediated apoptosis pathway. Oncogene. 2003 Dec 4;22(55):8931–8.
95. Orzalli MH, Conwell SE, Berrios C, DeCaprio JA, Knipe DM. Nuclear interferon-inducible protein 16 promotes silencing of herpesviral and transfected DNA. Proc Natl Acad Sci USA. 2013 Nov 19;110(47):E4492–501.
96. Morrone SR, Wang T, Constantoulakis LM, Hooy RM, Delannoy MJ, Sohn J.
Cooperative assembly of IFI16 filaments on dsDNA provides insights into host defense strategy. Proc Natl Acad Sci USA. 2014 Jan 7;111(1):E62–71.
97. Fu Y, Comella N, Tognazzi K, Brown LF, Dvorak HF, Kocher O. Cloning of DLM-1, a novel gene that is up-regulated in activated macrophages, using RNA differential display. Nature. 1999 Nov;240(1):157–63.
98. Schwartz T, Behlke J, Lowenhaupt K, Heinemann U, Rich A. Structure of the DLM-1-Z-DNA complex reveals a conserved family of Z-DLM-1-Z-DNA-binding proteins. Nat Struct Biol. 2001 Sep;8(9):761–5.
99. Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. 2007 Jul 26;448(7152):501–5.
100. DeFilippis VR, Alvarado D, Sali T, Rothenburg S, Fruh K. Human Cytomegalovirus Induces the Interferon Response via the DNA Sensor ZBP1. J Virol. 2009 Dec 9;84(1):585–98.
101. Ishii KJ, Kawagoe T, Koyama S, Matsui K, Kumar H, Kawai T, et al. TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature. 2008 Feb 7;451(7179):725–9.
type 1 interferon response. Cell Rep. 2015 Feb 24;10(7):1055–65.
103. Parvatiyar K, Zhang Z, Teles RM, Ouyang S, Jiang Y, Iyer SS, et al. The helicase DDX41 recognizes the bacterial secondary messengers cyclic di-GMP and cyclic di-AMP to activate a type I interferon immune response. Nat Immunol. 2012 Nov 11;13(12):1155– 61.
104. Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009 Oct 8;461(7265):788–92. 105. Chiu Y-H, MacMillan JB, Chen ZJ. RNA polymerase III detects cytosolic DNA and
induces type I interferons through the RIG-I pathway. Cell. 2009 Aug 7;138(3):576–91. 106. Ablasser A, Bauernfeind F, Hartmann G, Latz E, Fitzgerald KA, Hornung V.
RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat Immunol. 2009 Oct;10(10):1065–72.
107. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science. 2013 Feb
14;339(6121):786–91.
108. Kranzusch PJ, Lee AS-Y, Berger JM, Doudna JA. Structure of human cGAS reveals a conserved family of second-messenger enzymes in innate immunity. Cell Rep. 2013 May 30;3(5):1362–8.
109. Civril F, Deimling T, de Oliveira Mann CC, Ablasser A, Moldt M, Witte G, et al. Structural mechanism of cytosolic DNA sensing by cGAS. Nature. 2013 Jun 20;498(7454):332–7.
110. Gao P, Ascano M, Wu Y, Barchet W, Gaffney BL, Zillinger T, et al. Cyclic
[G(2',5')pA(3“,5”)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell. 2013 May 23;153(5):1094–107.
111. Wu J, Sun L, Chen X, Du F, Shi H, Chen C, et al. Cyclic GMP-AMP Is an Endogenous Second Messenger in Innate Immune Signaling by Cytosolic DNA. Science. 2013 Feb 14;339(6121):826–30.
112. Ablasser A, Schmid-Burgk JL, Hemmerling I, Horvath GL, Schmidt T, Latz E, et al. Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature. 2013 Nov 28;503(7477):530–4.
113. Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Röhl I, et al. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature. 2013 May 30.
114. Li XD, Wu J, Gao D, Wang H, Sun L, Chen ZJ. Pivotal Roles of cGAS-cGAMP Signaling in Antiviral Defense and Immune Adjuvant Effects. Science. 2013 Sep
115. Abe T, Barber GN. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-κB activation through TBK1. J Virol. 2014
May;88(10):5328–41.
116. Lehtinen DA, Harvey S, Mulcahy MJ, Hollis T, Perrino FW. The TREX1 double-stranded DNA degradation activity is defective in dominant mutations associated with autoimmune disease. J Biol Chem. 2008 Nov 14;283(46):31649–56.
117. Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, et al. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 cause Aicardi-Goutières syndrome at the AGS1 locus. Nat Genet. 2006 Jul 16;38(8):917–20.
118. Napirei M, Karsunky H, Zevnik B, Stephan H, Mannherz HG, Möröy T. Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat Genet. 2000 Jun;25(2):177– 81.
119. Okabe Y, Kawane K, Akira S, Taniguchi T, Nagata S. Toll-like receptor-independent gene induction program activated by mammalian DNA escaped from apoptotic DNA
degradation. J Exp Med. 2005 Nov 21;202(10):1333–9.
120. Okabe Y, Kawane K, Nagata S. IFN regulatory factor (IRF) 3/7dependent and -independent gene induction by mammalian DNA that escapes degradation. Eur J Immunol. 2008 Nov;38(11):3150–8.
121. Yoshida H, Okabe Y, Kawane K, Fukuyama H, Nagata S. Lethal anemia caused by interferon-beta produced in mouse embryos carrying undigested DNA. Nat Immunol. 2005 Jan;6(1):49–56.
122. Stetson DB, Ko JS, Heidmann T, Medzhitov R. Trex1 Prevents Cell-Intrinsic Initiation of Autoimmunity. Cell. 2008 Aug;134(4):587–98.
123. Ahn J, Gutman D, Saijo S, Barber GN. STING manifests self DNA-dependent inflammatory disease. Proc Natl Acad Sci USA. 2012 Nov 20;109(47):19386–91. 124. Gao D, Li T, Li X-D, Chen X, Li Q-Z, Wight-Carter M, et al. Activation of cyclic
GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc Natl Acad Sci USA. 2015 Oct 20;112(42):E5699–705.
125. Hasan M, Koch J, Rakheja D, Pattnaik AK, Brugarolas J, Dozmorov I, et al. Trex1 regulates lysosomal biogenesis and interferon-independent activation of antiviral genes. Nat Immunol. 2012 Nov 18;14(1):61–71.
126. Ablasser A, Hemmerling I, Schmid-Burgk JL, Behrendt R, Roers A, Hornung V. TREX1 Deficiency Triggers Cell-Autonomous Immunity in a cGAS-Dependent Manner. J Immunol. 2014 Jun 6;192(12):5993–7.
Syndrome. J Immunol. 2015 Sep 1;195(5):1939–43.
128. Horan KA, Hansen K, Jakobsen MR, Holm CK, Soby S, Unterholzner L, et al. Proteasomal Degradation of Herpes Simplex Virus Capsids in Macrophages Releases DNA to the Cytosol for Recognition by DNA Sensors. J Immunol. 2013 Feb
15;190(5):2311–9.
129. Rasmussen SB, Horan KA, Holm CK, Stranks AJ, Mettenleiter TC, Simon AK, et al. Activation of autophagy by α-herpesviruses in myeloid cells is mediated by cytoplasmic viral DNA through a mechanism dependent on stimulator of IFN genes. J Immunol. 2011 Nov 15;187(10):5268–76.
130. Lio C-WJ, McDonald B, Takahashi M, Dhanwani R, Sharma N, Huang J, et al. cGAS-STING Signaling Regulates Initial Innate Control of Cytomegalovirus Infection. J Virol. 2016 Sep 1;90(17):7789–97.
131. Ma Z, Jacobs SR, West JA, Stopford C, Zhang Z, Davis Z, et al. Modulation of the cGAS-STING DNA sensing pathway by gammaherpesviruses. Proc Natl Acad Sci USA. 2015 Aug 4;112(31):E4306–15.
132. Yoh SM, Schneider M, Seifried J, Soonthornvacharin S, Akleh RE, Olivieri KC, et al. PQBP1 Is a Proximal Sensor of the cGAS-Dependent Innate Response to HIV-1. Cell. 2015 Jun 4;161(6):1293–305.
133. Guo H, König R, Deng M, Riess M, Mo J, Zhang L, et al. NLRX1 Sequesters STING to Negatively Regulate the Interferon Response, Thereby Facilitating the Replication of HIV-1 and DNA Viruses. Cell Host and Microbe. 2016 Apr 13;19(4):515–28.
134. Ma Z, Damania B. The cGAS-STING Defense Pathway and Its Counteraction by Viruses. Cell Host and Microbe. 2016 Feb 10;19(2):150–8.
135. Schoggins JW, MacDuff DA, Imanaka N, Gainey MD, Shrestha B, Eitson JL, et al. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature. 2014 Jan 30;505(7485):691–5.
136. Watson RO, Bell SL, MacDuff DA, Kimmey JM, Diner EJ, Olivas J, et al. The Cytosolic Sensor cGAS Detects Mycobacterium tuberculosis DNA to Induce Type I Interferons and Activate Autophagy. Cell Host and Microbe. 2015 Jun 10;17(6):811–9.
137. Collins AC, Cai H, Li T, Franco LH, Li X-D, Nair VR, et al. Cyclic GMP-AMP Synthase Is an Innate Immune DNA Sensor for Mycobacterium tuberculosis. Cell Host and
Microbe. 2015 Jun 10;17(6):820–8.
138. Watson RO, Manzanillo PS, Cox JS. Extracellular M. tuberculosis DNA Targets Bacteria for Autophagy by Activating the Host DNA-Sensing Pathway. Cell. 2012 Aug
17;150(4):803–15.
a type I interferon pathway in controlling bacterial intracellular infection in mice. Cellular Microbiology. 2011 Aug 24;13(11):1668–82.
140. Storek KM, Gertsvolf NA, Ohlson MB, Monack DM. cGAS and Ifi204 cooperate to produce type I IFNs in response to Francisella infection. J Immunol. 2015 Apr 1;194(7):3236–45.
141. Hansen K, Prabakaran T, Laustsen A, Jørgensen SE, Rahbæk SH, Jensen SB, et al. Listeria monocytogenes induces IFNβ expression through an IFI16-, cGAS- and STING-dependent pathway. EMBO J. 2014 Aug 1;33(15):1654–66.
142. Chen H, Sun H, You F, Sun W, Zhou X, Chen L, et al. Activation of STAT6 by STING is critical for antiviral innate immunity. Cell. 2011 Oct 14;147(2):436–46.
143. Tenoever BR, Ng S-L, Chua MA, McWhirter SM, García-Sastre A, Maniatis T. Multiple functions of the IKK-related kinase IKKepsilon in interferon-mediated antiviral immunity. Science. 2007 Mar 2;315(5816):1274–8.
144. Ng S-L, Friedman BA, Schmid S, Gertz J, Myers RM, Tenoever BR, et al. IκB kinase epsilon (IKK(epsilon)) regulates the balance between type I and type II interferon responses. Proc Natl Acad Sci USA. 2011 Dec 27;108(52):21170–5.
145. Sharma S, Campbell AM, Chan J, Schattgen SA, Orlowski GM, Nayar R, et al.
Suppression of systemic autoimmunity by the innate immune adaptor STING. Proc Natl Acad Sci USA. 2015 Feb 2;:201420217.
CHAPTER 2: STAT3 REGULATES HOST DEFENSE AND PROTECTS MICE AGAINST HERPES SIMPLEX VIRUS-1 INFECTION1
2.1 Summary
Signal transducer and activator of transcription 3 (STAT3) mediates cellular responses to multiple cytokines, governs gene expression, and regulates the development and activation of immune cells. STAT3 also modulates reactivation of latent herpes simplex virus-1 (HSV-1) in ganglia. However, it is unclear how STAT3 regulates the innate immune response during the early phase of HSV-1 lytic infection. Many cell types critical for the innate immunity are derived from the myeloid lineage. Therefore, here we used myeloid-specific STAT3 knockout mice to investigate the role of STAT3 in the innate immune response against HSV-1. Our results demonstrated that STAT3 knockout bone marrow-derived macrophages (BMMs) expressed decreased levels of Interferon α (IFNα) and interferon-stimulated genes (ISGs) upon HSV-1 infection. In vivo, knockout mice were more susceptible to HSV-1, as marked by higher viral loads and more significant weight loss. Splenic expression of IFNα and ISGs was reduced in the absence of STAT3, indicating that STAT3 is required for optimal type I interferon response to HSV-1. Expression of TNFα and IL-12, cytokines that have been shown to limit HSV-1 replication and pathogenesis, was also significantly lower in knockout mice. Interestingly, STAT3 knockout mice failed to expand CD8+ conventional DC (cDC) population upon HSV-1
1 This chapter has been accepted by the Journal of Leukocyte Biology and is placed into this
dissertation after additional editing by the author. The original citation is as follows: Hsia H-C, Stopford CM, Zhang Z, Damania B, Baldwin AS. Signal transducer and activator of transcription 3 (Stat3) regulates host defense and protects mice against herpes simplex virus-1 (HSV-1)
infection, and this was accompanied by impaired NK and CD8 T cell activation. Collectively, our data demonstrated that myeloid-specific STAT3 deletion causes defects in multiple aspects of the immune system, and that STAT3 has a protective role at the early stage of systemic HSV-1 infection.
2.2 Introduction
Herpes simplex virus 1 (HSV-1) is estimated to infect more than two thirds of the population worldwide. Common symptoms of HSV-1 infection, including local skin or mucosa membrane lesions, usually resolve in a few days to several weeks (1). Localized HSV-1
infections are also often accompanied by viremia (2,3). Following lytic infection, the virus retreats to neurons where it establishes latency. Latency is lifelong and may be asymptomatic with occasional relapses (1). HSV-1 infection seldom causes severe disease in healthy
individuals. However, newborns, the elderly, and immunocompromised individuals are prone to HSV-1-induced diseases, such as herpes simplex keratitis (HSK) and life-threatening herpes simplex encephalitis (HSE) (1). These susceptible populations also are more likely to exhibit HSV-1 viremia in the blood (2), and it is now becoming more apparent that primary HSV-1 infection and reactivation can occur beyond the mucocutaneous areas in both immunocompetent and immunocompromised individuals (2). It is therefore important to understand the mechanism by which the immune system controls HSV-1 infection.
Both innate and adaptive immunity are critical in controlling HSV-1 infection. Several major cellular mediators of the innate immunity implicated in limiting HSV-1 pathogenesis are of the myeloid lineage, including macrophages, dendritic cells (DCs), and neutrophils.
macrophages in mice leads to higher viral replication and exaggerated pathology (6,7), while adoptive transfer of adult macrophages protects susceptible neonates from lethal HSV-1 infection (8). Macrophages also limit the replication and dissemination of HSV-1 in the peripheral nervous system (9). The protective effects of macrophages are partially mediated through secreted factors, including IL-12, TNFα, and nitric oxide (NO). These factors inhibit viral replication, recruit leukocytes to the site of infection, promote the cellular immune response, and further activate macrophages to protect mice from HSV-1 and other herpesvirus infections (9-13). Specialized macrophages such as marginal zone macrophages (MZMΦs) and marginal metallophilic macrophages (MMMΦs) produce substantial amount of type I interferons to elicit antiviral response during HSV-1 viremia (14). In addition to macrophages, dendritic cells (DCs) also play a pivotal role in the immunity against herpesvirus infections. Mice depleted of DCs have shortened survival and higher viral loads in the nervous system during HSV-1 infection, and the susceptibility is attributed to impaired NK and T cell activation in the absence of DCs (15). Major subsets of DCs include CD11b+ conventional DC (cDC), CD8+ cDC, and plasmacytoid DC (pDC). Both CD8+ cDC and pDC are involved in promoting NK cell activation and anti-HSV cytotoxic T lymphocyte (CTL) immunity (16-18), but only CD8+ cDC is able to cross present and directly activate CD8 T cells (19,20). Importantly, pDCs are capable of
producing large quantities of type I interferons during HSV-1 infection (17). Finally, neutrophils have dual roles in HSV-1 infection. It has been suggested that neutrophils inhibit HSV-1
replication (21), but further examination revealed that neutrophils mediate HSV-1-induced tissue injury and immunopathology (22,23).
implicated in survival, inflammation, proliferation, and migration (24). STAT3 modulates HSV-1 reactivation in the ganglia (25) and governs various aspects of myeloid cell proliferation, differentiation, and responses to infections (26-29). For example, STAT3-null macrophages display heightened immune responses in response to lipopolysaccharide (LPS) and bacterial infections, as marked by the overproduction of pro-inflammatory cytokines (26,30).
Additionally, STAT3-null neutrophils are defective in their bactericidal capacity (30).
Nonetheless, the role of STAT3 in the myeloid lineage during viral infections has not been fully investigated.
Given the critical role of macrophages and DCs in limiting HSV-1 pathogenesis at the early phase of infection, we set out to investigate if STAT3 in the myeloid lineage modulates the innate immune response to HSV-1. Here we show that the type I interferon response was
partially defective in STAT3-null bone marrow-derived macrophages (BMMs) during HSV-1 infection. In vivo, myeloid STAT3 knockout mice exhibited higher viral loads and more significant weight loss during HSV-1 infection compared to wild-type mice. Importantly,
2.3 Results
2.3.1 Characterization of Stat3fl/fl LysM-Cre+/+ mice
To investigate if STAT3 in the myeloid lineage affects the innate immune response to HSV-1, we crossed Stat3fl/fl mice to LysM-Cre mice to generate myeloid-specific STAT3 knockout mice. Cre recombinase driven by the lysozyme (LysM) promoter is specifically expressed in the myeloid lineage, leading to conditional deletion of loxP-flanked target gene in granulocytes, monocytes, and macrophages (31). In agreement with the literature, bone marrow-derived macrophages (BMMs) from Stat3fl/fl LysM-Cre+/+ mice (hereafter referred as knockout mice) showed more than 90% reduction of STAT3 protein comparing to BMMs from Stat3wt/wt
LysM-Cre+/+ mice (hereafter referred as wild-type mice). No noticeable reduction of STAT3 protein levels were observed in the brain, liver, or muscle as determined by immunoblotting (Supplemental Figure 2.1).
The Stat3fl/fl used in this study (see Materials and Methods) differs from the strain used in the previous LysM-Cre/Stat3 mouse study (26). Thus, to determine the effect of myeloid Stat3
deletion with regard to this specific strain, we examined the composition of lymphocytes and myeloid cells in the knockout mice. Lack of STAT3 in the myeloid lineage did not affect the composition of major cell populations in the spleen. Specifically, the percentage of B cells, T cells, neutrophils, monocytes, red pulp macrophages (RpMΦ), marginal metallophilic
accompanied by a mild decrease in B cell and T cell populations in the bone marrow.
Neutrophilia has been reported by other groups using hematopoietic-specific or inducible Stat3
knockout mice (28,29). Our results suggest that myeloid-specific deletion of Stat3 is sufficient to cause the neutrophil expansion phenotype. The number of CD11blo F4/80+ macrophages in the bone marrow did not differ between wild-type and knockout mice (Table 2.2). Similar to the previous study (26), we did not observe a differentiation block of STAT3-null BMMs cultured in vitro (data not shown), suggesting that STAT3 is not crucial in normal macrophage
differentiation. Taken together, our data agreed with the previous studies and independently verified that myeloid Stat3 deletion leads to neutrophil expansion in the bone marrow but has minimal effects on splenic cells or BMM differentiation by using a different Stat3fl/fl strain.
2.3.2 Stat3 knockout BMMs show attenuated type I interferon response to HSV-1 infection
HSV-1 is able to enter and infect BMMs and peritoneal macrophages, but few infectious viral particles are produced following virus entry, indicating that macrophages are
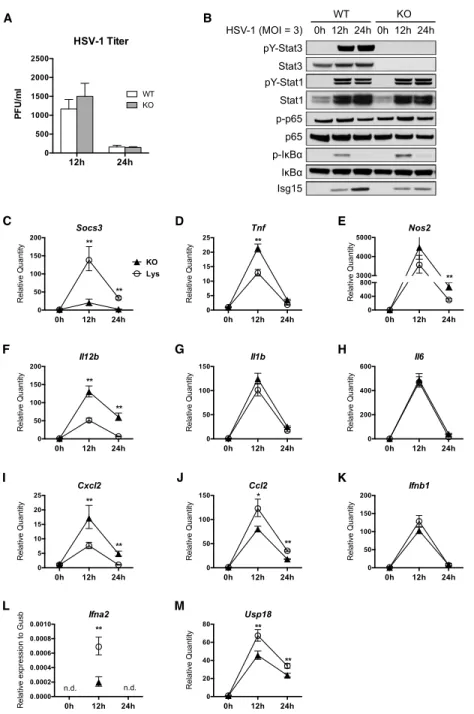
non-permissive to productive HSV-1 infection (4,5). To determine if STAT3 plays a role in the intrinsic resistance of macrophages to HSV-1, BMMs from wild-type or knockout mice were infected with HSV-1 at a multiplicity of infection (MOI) of 3, and infectious viral particles in the supernatant were measured at 12 and 24 hours post infection (hpi) by plaque assay. Similar to wild-type BMMs, STAT3 knockout BMMs produced limited amount of infectious viral
particles produced (Figure 2.1A). Thus, Stat3 deletion in the macrophages does not impair their intrinsic resistance to HSV-1.
To further investigate the response of BMMs to HSV-1, we examined the signaling events and host gene expression following HSV-1 infection. HSV-1 induced robust tyrosine phosphorylation and activation of STAT1 and STAT3 in wild-type BMMs, and STAT1 activation appeared to be normal in the absence of STAT3 (Figure 2.1B). Phosphorylation of IκBα (p-IκBα) was induced, and phospho-p65 (p-p65) was induced to a lesser extent, indicating activation of the NF-κB pathway by HSV-1 infection (Figure 2.1B). Importantly, p-IκBα level was higher in knockout BMMs, indicating enhanced NF-κB activity, while the induction of ISG15, an interferon-stimulated gene (ISG), was reduced in knockout BMMs (Figure 2.1B). HSV-1 also induced the expression of several genes involved in signaling and immune
regulation (Figure 2.1C-M). Of note, induction of SOCS3, a STAT3 inhibitor highly upregulated by active STAT3 (32), was blunted in knockout macrophages (Figure 2.1C), confirming the lack of STAT3 functionality in knockout BMMs.
It has been reported that STAT3-null macrophages express higher levels of
pro-inflammatory genes in response to LPS and bacterial infection (26,30). Similarly, we found that during HSV-1 infection, knockout BMMs expressed more TNFα, inducible nitric oxide synthase (iNOS), and IL-12b than wild-type BMMs (Figure 2.1D-F), but not other inflammatory
cytokines such as IL-1β and IL-6 (Figure 2.1G-H). Knockout BMMs also expressed more
CXCL2 (Figure 2.1I), a neutrophil chemoattractant responsible for neutrophil mobilization to the site of HSV-1 infection (33). On the contrary, induction of CCL2, a monocyte chemoattractant and a STAT3 target gene, was decreased in knockout BMMs (Figure 2.1J). These data show that
viral infection. We also examined the expression of type I interferons and ISGs. While
expression of IFNβ was unaffected (Figure 2.1K), HSV-1-induced IFNα expression was severely diminished in the absence of STAT3 (Figure 2.1L). Moreover, expression of two ISGs, namely ISG15 and USP18, was decreased in the knockout BMMs (Figure 2.1B, 2.1M), indicating that while STAT3 is dispensable for IFNβ expression, it is required for IFNα expression and optimal expression of ISGs in response to HSV-1 infection.
Taken together, our data showed that in vitro HSV-1 infection activates STAT1 and STAT3 in BMMs. Loss of STAT3 did not impair STAT1 activation or the intrinsic ability of BMMs to limit HSV-1 replication. However, Stat3 knockout BMMs had significantly lower expression of IFNα and ISGs, suggesting that effective induction of IFNα and ISGs in BMMs during HSV-1 infection is dependent on STAT3.
2.3.3 Myeloid Stat3 knockout mice are more susceptible to HSV-1 infection
(Supplemental Figure 2.2). These results suggest that STAT3 in myeloid cells have a protective role during lethal systemic HSV-1 infections and prompted further investigation.
Next, we challenged the mice with a lower dose of HSV-1. Mice were infected with 1x107 pfu of HSV-1 by tail vein injection. Both groups of mice displayed symptoms including hunched posture, signs of hind limb weakness, and significant weight loss starting at 2 days post infection (dpi). No statistical difference in survival was observed (Figure 2.2B). However, Stat3
knockout mice experienced significantly worse weight loss over the course of infection (Figure 2.2C). To determine the impact of myeloid-specific Stat3 deletion on the innate immunity against HSV-1 at the early stage of infection, we examined the viral loads in the brain and the spleen at 2 dpi. Stat3 knockout mice had significantly higher viral loads in the brain at 2 dpi (Figure 2.2D). Viral loads were also marginally higher in the spleens of knockout mice at 2 dpi (Figure 2.2E). These data showed that Stat3 knockout mice are more susceptible to HSV-1 infection, and the differences are discernible at the earlier stages of infection.
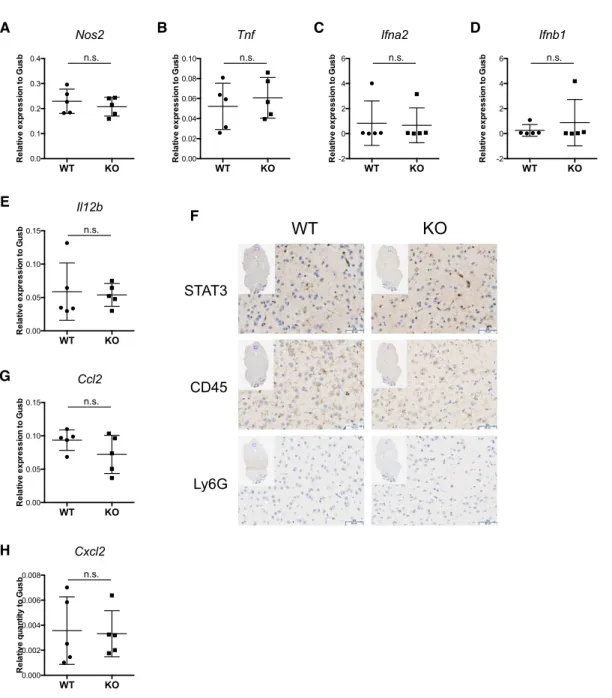
2.3.4 Susceptibility of Stat3 knockout mice is not caused by immunopathology in the brain
To investigate the mechanisms contributing to the higher brain viral loads in knockout mice, we examined the expression of immunoregulatory genes in the brain. Factors including IFNα, IFNβ, IFNγ, IL-12, TNFα, and NO are known to be critical in inhibiting HSV-1
replication in various settings in vitro and in vivo (10-13,35,36). While IFNγ expression in the brain was below the detection threshold (data not shown), iNOS, TNFα, IFNα, IFNβ, and IL-12 were not differentially expressed in the brains of wild-type and knockout mice (Figure 2.3A-E), and thus cannot account for the higher viral loads in the brains of STAT3 knockout mice.