CELLS EXHIBITING STRONG P16INK4A PROMOTER ACTIVATION IN VIVO DISPLAY FEATURES OF SENESCENCE
Jie-Yu Liu
A dissertation submitted to the faculty at University of North Carolina at Chapel Hill in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the
Curriculum in Genetics and Molecular Biology in the UNC Graduate School.
Chapel Hill 2019
©2019 Jie-Yu Liu
ABSTRACT
Jie-Yu Liu: Cells exhibiting strong p16INK4a promoter activation in vivo display features of senescence
(Under the direction of Norman E. Sharpless)
The activation of cellular senescence throughout the lifespan promotes tumor suppression, whereas the persistence of senescent cells contributes to aspects of aging. This theory has been limited, however, by an inability to identify and isolate individual senescent cells within an intact organism. Toward that end, we generated a murine reporter strain by ‘knocking-in’ a fluorochrome, tandem-dimer Tomato (tdTom), into exon 1α of the endogenous p16INK4a locus. We used this allele (p16tdTom) for the enumeration, isolation and characterization of individual p16INK4a-expressing cells (tdTom+). The half-life of the knocked-in transcript was shorter than that of the
endogenous p16INK4a mRNA, and therefore reporter expression better correlated with p16INK4a promoter activation than p16INK4a transcript abundance. The frequency of tdTom+ cells increased with serial passage in cultured murine embryonic fibroblasts from p16tdTom/+mice. In adult mice, tdTom+ cells could be readily detected at low
transcripts encoding SA-secretory phenotype (SASP) phenotype. These results indicate that cells harboring activation of the p16INK4a promoter accumulate with aging and
ACKNOWLEDGEMENTS
The journey through my PhD has been challenging. I did not expect it would be that tough and take me this long. Despite being bumpy, it was fulfilling toward the end. I am grateful to have many wonderful people support me through my PhD life.
Firstly, I would like to express my gratitude to my advisor, Dr. Norman Sharpless for his patience, encouragement and immense knowledge. He trained me to be an independent researcher and allowed me to conduct research freely and regardless of expense. Ned was extremely supportive while I was struggling with my research
projects. I cannot remember how many hours we spent coming up with new plans in his office. He believed in me and made me believe in myself. His guidance helped me in all the time of research and preparing of the manuscript.
Besides my advisor, I would like to thank the rest of my dissertation committee for their insightful and constructive comments. They are always supportive and
encouraging, and never give me hard time in the committee meetings. Their feedback incented me to think about my research from different perspectives. Special thanks to Dr. Vicki Bautch and Dr. Bob Duronio, they provided additional support and guidance after Ned left for National Cancer Institute.
I thank all my fellow labmates for the stimulating discussion and technical
make to PNAS. George is actually the one who initiated my project by making the reporter construct. He was my mentor when I first rotated in this lab. It turned out that he continued to mentor me throughout my PhD trainingeven though we worked on different projects. George is not only a great mentor, but also a very good friend. Our coffee hour was the only time I felt relaxed in the lab. Brian is also a terrific mentor. He’s warm, gentle and patient. He always provides helpful feedbacks on my project. He is the one who found this allele can be valuable for cartilage research. Besides
research, I am grateful to Dr. Chenlin Zhou for the mental support. She is a good listener, and she is there for me whenever I am frustrated. I always feel more positive after talking to her.
Last but not the least, I would like to thank my family in Taiwan and my husband
for supporting me spiritually throughout my life in general. They always encourage me
to dream and support any decision I make. This accomplishment would not have been
TABLE OF CONTENTS
LIST OF TABLES ... xi
LIST OF FIGURES ... xii
LIST OF ABBREVIATIONS ... xiii
CHAPTER 1: General Introduction ... 1
1.1 Senescence in Cancer and Aging ... 1
1.2 Markers of Senescence ... 4
1.3 The roles of p16INK4a in Cancer and Aging ... 10
1.4 The roles of p16INK4a in macrophage biology ... 12
1.5 Summary ... 14
REFERENCES ... 16
CHAPTER 2: Cells exhibiting strong p16INK4a promoter activation in vivo display features of senescence ... 26
2.1 Overview ... 26
2.2 Introduction ... 27
2.3 Results ... 29
2.3.1 Generation and Characterization of the p16tdTom Allele ... 29
2.3.2 The p16tdTom Allele Reports Promoter Activation ... 31
2.3.3 Characterization of p16INK4a Transcriptionally Active Cells In Vitro ... 33
2.3.4 Enumeration of p16INK4a-Activated Cells In Vivo ... 35
2.3.5 Characterization of p16INK4a-Activated Macrophages ... 36
2.5 Materials and Methods ... 60
2.5.1 Animals ... 60
2.5.2 Generation of the p16tdTomAllele ... 60
2.5.3 Isolation and culture of MEFs ... 60
2.5.4 Quantitative real-time PCR ... 60
2.5.5 Single-cell quantitative real-time PCR ... 61
2.5.6 Tissue dissociation and Flow cytometry analysis ... 61
2.5.7 Alginate bead experiment ... 62
2.5.8 In vivo bioluminescent imaging ... 62
2.5.9 In vitro and In vivo cell sorting ... 63
2.5.10 In vitro cell growth assay ... 63
2.5.11 In vitro and In vivo EdU incorporation ... 63
2.5.12 In vitro phagocytosis assay ... 64
2.5.13 In vitro treatment ... 64
2.5.14 SA-β-gal staining ... 64
2.5.15 mRNA stability assay ... 64
2.5.16 RNA sequencing and analysis ... 64
2.5.17 Gene Set Enrichment Analysis (GSEA) ... 65
2.5.18 Statistical Analysis ... 65
REFERENCES ... 66
CHAPTER 3: GENERAL DISCUSSION ... 71
3.1 Summary ... 71
3.2 Overview of p16INK4a reporter systems ... 71
3.3 p16INK4a promoter activity vs. p16INK4 transcript abundance ... 73
LIST OF TABLES
Table S2.1. Expression of SASPs in tdTom+ peritoneal macrophages
LIST OF FIGURES
Figure 2.1. Design and validation of the p16tdTom allele ... 45
Figure 2.2. Promoter activity of the p16tdTom allele ... 46
Figure 2.3. p16INK4a-activated cultured MEFs exhibit senescence phenotypes. ... 47
Figure 2.4. Age-dependent increase in the frequency of p16INK4a-activated cells in different tissues. ... 48
Figure 2.5. Reduced proliferation and high SA-β-gal activity of p16INK4a -activated peritoneal macrophages ... 49
Figure 2.6. Gene expression profile of p16INK4a-activated peritoneal macrophages ... 50
Figure S2.1. Validation of the p16tdTom allele design ... 51
Figure S2.2. Influence of a retained Neomycin selection cassette on p16tdTom expression ... 52
Figure S2.3. Transcript stability of tdTom and p16INK4a ... 53
Figure S3.4. Gene expression profile of p16INK4a-activated MEFs ... 54
Figure S3.5. Transient p16INK4a activation in peripheral blood cells (PBCs) ... 55
Figure S3.6. Expression of tdTom in peritoneal lavage cells ... 56
LIST OF ABBREVIATIONS ActD: actinomycin D
BMDM: bone-marrow derived macrophage DAPI: 4′,6-diamidino-2-phenylindole DDR: DNA damage response ECM: extracellular matrix EdU: 5-ethynyl-2′-deoxyuridine
FACS: Fluorescence-activated cell sorting FLP: flippase
FRT: flippase recognition target GSEA: gene set enrichment analysis GWAS: genome-wide association studies IAT: inguinal adipose tissue
IFN: interferon IL: Interleukin
LPS: lipopolysaccharide LUC: luciferase
MEF: murine embryonic fibroblasts NDF: neonatal dermal fibroblast ORF: open reading frame
PBTL: peripheral blood T lymphocytes Rb: retinoblastoma
SASP: senescence-associated secretory phenotype SNP: single nucleotide polymorphisms
CHAPTER 1: General Introduction 1.1 Senescence in Cancer and Aging
Cellular senescence was first discovered in 1961 by Hayflick and Moorhead who noticed that normal human fibroblasts had finite replicative lifespan in culture (1). This limited proliferative capacity was later on shown to arise from the shortened telomeres, a consequence of multiple rounds of cell division and was subsequently referred to as replicative senescence. Senescence represents a state of durable cell-cycle arrest that is resistant to oncogenic or mitogenic challenge and distinct from other forms of growth arrest such as quiescence and terminal differentiation. When replication-competent cells receive cellular stresses such as telomeres dysfunction (2, 3), persistent DNA damage (4, 5), oncogene activation (6, 7) and oxidative damage (8, 9),the senescence program is activated and maintained via two critical tumor suppressor pathways, p53/p21 and/ or p16INK4a/Rb (retinoblastoma) (10-12). Both p21 and p16INK4a are cyclin-dependent
kinase (CDK) inhibitors (13, 14). They negatively regulate cell-cycle progression through Rb hypophosphorylation to block the proliferation of stressed and potentially oncogenic cells. Although the relative contributions of these two effector pathways to senescence depend on the cell type and the type of senescence-inducing stimuli, both may
initial growth arrest, however, it is the p16INK4a/Rb pathway that is required to maintain the growth-arrest state. While senescence is primarily characterized in proliferative cells, recent studies have suggested that various terminally differentiated cell types can also initiate a senescence program (17-19). For example, chondrocytes show extremely low proliferative rates in vivo but still express high levels of p16INK4a and display other features of senescence (17). However, the role of postmitotic cellular senescence on tissue function are not well understood.
Senescence has both beneficial and detrimental effects on health depending on the contexts. For example, senescent cells can promote beneficial tissue remodeling during embryonic development (20, 21) and wound healing (22, 23). As a stress-response program, senescence undoubtedly provides a barrier to tumorigenesis in mammals. It’s well established that normal cells undergo senescence in response to oncogene activation (6); likewise, loss of tumor suppressor genes can trigger
senescence (24-26). Furthermore, mice lacking 16INK4a or p53 are prone to develop cancer (27, 28). Mice deficient for both 16INK4a and p53 have exhibited an even higher incidence of cancer and worse tumor-free survival (29). Conversely, mice carrying an extra copy of 16INK4a or p53 are more cancer-resistant (30, 31). Consistent with these observations in animal models, p53/p21 or/ and p16INK4a/Rb pathways are very
frequently disrupted in a wide spectrum of human cancers (32). These observations underscore the importance of cellular senescence to tumor suppression. While
Aging is defined as a decline of tissue function and regenerative capacity in response to physiologic demands. Senescence contributes to mammalian aging through at least two mechanisms: in a cell-autonomous fashion, senescent cells lose their replicative capacity, thereby depleting the pool of cycling cells especially stem and progenitor cells to impair tissue homeostasis and regeneration (33). On the other hand, senescent cells produce a variety of cytokines and chemokines to disrupt tissue
structure, promote chronic inflammation and even induce paracrine senescence in healthy neighboring cells (34, 35). As earlier noted, senescent cells accumulate in tissues with aging and at the sites of age-associated pathology such as osteoarthritis, pulmonary fibrosis and atherosclerosis (36-38). Yet, it remains unclear whether this age-dependent accumulation of senescent cells is due to increased production or decreased clearance of senescent cells, or both. The causal role of senescent cells in aging has been suggested by modulating the abundance of senescent cells in vivo. For example, tobacco use, which is strongly linked to aging, augments the number of senescent cells in both human and mice (39, 40). Perhaps most strikingly, targeted clearance of senescent cells, using either genetic or pharmacological approach,
increases at old ages. It could be argued, therefore, that a senescence program is activated with age, resulting in not only aging but also tumor prevention. In the meantime, oncogenic mutations also accumulate progressively throughout life, and aged stroma becomes more favorable for cancer growth. This may explain the paradox of why aged organisms are more susceptible to cancer despite the protective
mechanisms of senescence.
1.2 Markers of Senescence
Given the prominence of senescence in cancer and aging, there has been long-standing interest in the identification and characterization of senescent cells in an intact adult organism. While senescent cells enter a state of durable growth arrest, they are metabolically active and resistant to apoptosis (44). In addition, senescent cells
become flat and enlarged in vitro (45), whereas their morphological changes in vivo are poorly determined. Moreover, senescence is variably associated with expression of senescence-associated β-galactosidase (SA-β-gal), and CDK inhibitors (especially
p16INK4a), senescence-associated heterochromatin foci (SAHF), persistently activated DNA damage response (DDR), shortened telomeres, elaboration of cytokines that comprise the senescence-associated secretory phenotype (SASP). A couple more senescence features have recently been noted but they are still under investigation. These include accumulation of lipofusin (46, 47) and loss of nuclear high-mobility group box 1 (HMGB1) or lamin B1 (48-50). Despite the fact that senescent cells are
identify senescent cells in aged tissues prevents us to further study their precise role in age-related pathologies and hinders the clinical development of “senolytics” that
selectively ablate senescent cells. Below, the caveats of several commonly used senescence markers are discussed.
While senescent cells are characterized by durable cell cycle arrest, quiescence and terminal differentiation are two other forms of growth arrest that is distinct from senescence. Quiescence is a reversible non-dividing cellular state in which quiescent cells can re-enter the cell division cycle when released from contact inhibition or mitogen deprivation. Although both senescence and terminal differentiation are
characterized by stable cell cycle exit, they are different in several ways. Senescence is a cellular stress response, whereas terminal differentiation is a programmed
developmental process. Unlike senescent cells, terminally differentiated cells often exhibit distinct function from their progenitor cells and have different but well-defined morphological features. Despite that durable cell cycle arrest is a hallmark of
senescence, it’s difficult to distinguish one from the others in vivo by the proliferative status.
SA-β-gal is the most commonly used marker of senescence due to its ease of detection. Endogenous β-galactosidase is encoded by the GLB1 gene and is an essential lysosomal enzyme with optimal activity at pH 4.0-4.5. Senescent cells often exhibit increased levels of β-galactosidase; therefore, SA-β-gal activity can be detected at suboptimal pH 6.0 with the artificial substrates (52-54). Although this marker is
physiological conditions with exuberant lysosomal synthesis. For examples, active phagocytes and osteoblasts appear to have higher activity of β-galactosidase (55, 56). Expression of β-galactosidase is also upregulated during autophagy (57-59).
Additionally, β-galactosidase activity is induced in contact-inhibited quiescent cells (60) and tissues from mice receiving ionizing radiation (61). Lastly, expression of SA-β-gal is dispensable for senescence, and any biological process that enhances lysosomal biogenesis will result in the positive blue stains of SA-β-gal. Interpreting the expression of SA-β-gal in vivo, therefore, requires extra caution, especially when looking at tumors and inflammatory conditions in which phagocytes may accumulate.
Senescence is associated with the expression of CDK inhibitors including
p16INK4a and p21. While p21 induces cell cycle withdrawal by inhibiting G1 CDK activity (13, 62), its role in senescence remains controversial. It’s clear that p21 expression is not specific to senescent cells as p21 is upregulated to produce a reversible cell-cycle arrest for DNA repair in response to transient DNA damage. Further, inactivation of p21 does not abrogate senescence in vitro (63). Although p21 plays a role for initiation of senescence in some settings, its expression is not persistent in senescent cells (16, 64, 65). Therefore, p21 cannot serve as a specific marker of senescence. p16INK4a
not characterized by p16INK4a expression (3, 15, 68, 69) and at least in some conditions, absence of p16INK4a expression can be compensated by upregulation of other INK4 family proteins (e.g. p15INK4B) (70-73). By contrast, high level of p16INK4a expression can be detected in non-senescence settings such as Rb-inactivated tumors (74-77), a result of negative feedback loop. In turn, growth arrest can be abolished by inactivating
downstream effectors of senescence despite the initiation of senescence program. Some senescent cells are characterized by chromatin changes including DNA damage foci and SAHFs. DNA damage is closely associated with many forms of senescence. For example, shortened/uncapped telomeres are recognized as DNA damage which induces replicative senescence. Oncogene activation causes DNA
damage due to altered DNA replication, leading to oncogene-induced senescence. DNA damage can be triggered by either intrinsic (e.g., oxidative stress, telomere attrition) or environmental genotoxic insults including ultraviolet (UV), ionizing radiation and
chemotherapies. Once DNA damage is generated in proliferating cells,
ataxia-telangiectasia mutated (ATM)-p53-p21 signaling is activated to induce growth arrest. If DNA damage is promptly fixed, cells resume normal proliferation; however, if DNA damage is severe and can’t be fixed, persistent DDR activation eventually triggers cellular senescence. DNA-damage foci are frequently found in senescent cells through
staining of γH2AX or p53-binding protein 1 (p53BP1), which is the sensor and mediator
breaks express these markers as well so most cells expressing the markers of DNA damage may not be senescent. Therefore, identifying senescent cells by DNA damage foci has little specificity. Cells undergo extensive chromatin remodeling in the
senescence state, which appears as punctate nuclear 4′,6- diamidino-2-phenylindole (DAPI) staining, termed SAHFs. SAHFs are dense chromatin structure resulting from repression of E2F-targeted genes in human senescent cells and associated with
enrichment of heterochromatin protein 1 (HP1) and trimethylation of Lys9 on histone H3 (H3K9me3) (85). Using SAHFs as a senescence marker may only applicable in human as mouse cells preferentially form pericentromeric foci that are not resulting from senescence (86, 87). However, formation of SAHFs is cell-type dependent and
relatively specific to oncogene-induced senescence (88, 89). Besides, it’s dispensable for senescence. SAHFs thereby have limited usage as a senescence marker.
Senescent cells exhibit a distinct secretome termed SASP, consisting of growth factors, enzymes that degrade extracellular matrix (e.g., Matrix metalloproteinases), immune modulators (e.g., chemokines) and pro-inflammatory cytokines. As the most intriguing senescence phenotype, SASP has been shown to spread senescence to neighboring cells, promote tissue repair (23, 90) and alter tissue architecture (91, 92) in a paracrine manner. This senescence-associated inflammatory network is regulated by
several signaling pathways including NOTCH1 (93), NF-κB (94), p38MAPK (95) and
to measure, expression of SASP is not a reliable senescence marker as the SASP components vary depending on the cell types and the forms of senescence. In addition, evidence for the causal role of any specific SASP factor in aging is not strong. More importantly, most if not all of the SASP factors are often produced in non-senescence occasions such as infections, inflammation and ongoing malignancies.
Telomere attrition is highly linked to senescence. Telomeres, the tandem repeats protecting chromosome ends, shorten with each cell division because DNA
polymerases are unidirectional; thereby the ends of linear DNA cannot be completely replicated. In mice, many cells express telomerase that can add telomeric DNA repeats de novo to prevent telomere deprotection (102, 103). However, normal human somatic cells lack telomerase activity (104). Consequently, telomere attrition triggers DDR, leading to cellular senescence (105-107). The contribution of telomere shortening to senescence and aging has been demonstrated by human GWAS and murine studies. GWAS have linked telomerase to various age-related diseases including cancers and idiopathic pulmonary fibrosis (108). Cultured human cells can bypass replicative
independent of telomere attrition, and telomere shortening and dysfunction can occur in non-senescence settings.
1.3 The roles of p16INK4a in Cancer and Aging
The gene encoding p16INK4a is located within the CDKN2A locus (also known as the INK4A–ARF locus) on human chromosome 9p21.3. This locus also specifies another critical tumor suppressor protein, p14ARF (or p19ARF in mouse) by alternative
transcriptional start sites (111). p16INK4a and ARF have unique exon 1 but common exon 2 and 3. Because of the alternative reading frames, p16INK4a and ARF do not share any amino acid homology and have distinct molecular functions. p16INK4a is a selective CDK4/6 inhibitor. The binding of p16INK4a to CDK4/6 induces an allosteric change that disrupts the interaction of CDK4/6 with Cyclin D (14). Consequently, CDK4/6-mediated phosphorylation of Rb-family proteins is abrogated, maintaining Rb proteins in an active, hypophosphorylated state to bind E2Fs leading to a G1 cell-cycle arrest. ARF instead regulates p53 through inhibition of the MDM2 E3 ligase activity, which in turn prevents p53 proteasomal degradation (112-114). Significantly, two key tumor suppressor
Expression of p16INK4a is highly linked to cancer and aging in mammals. p16INK4a is frequently inactivated in a wide spectrum of human cancers by point mutation, small deletion or promoter methylation (119). While p16INK4a knockout mice are predisposed to cancers, overexpression of the Ink4-Arf locus in mice greatly reduced the incidence of spontaneous tumorigenesis (30). These findings suggest that 16INK4a is a critical tumor suppressor, and loss of p16INK4a allows would-be cancer cells to bypass
senescence. In addition to its prominent function in cancer, p16INK4a is one of the most robust aging biomarkers. Expression of p16INK4a markedly increases with aging in both rodent and human tissues, and it’s highly dynamic; being low or undetectable in healthy young tissues, but raising sharply in many aged or injured tissues such as wounding (39, 120, 121). Supporting these observations, the level of p16INK4a activation positively
correlates with age in a knock-in p16LUCreporter mouse model (67). Several studies have suggested that p16INK4a is not merely a biomarker but also contributes to age-associated pathologies. Murine models with p16INK4a deficiency or overexpression demonstrated that age-induced accumulation of p16INK4a compromises the replicative potential and function in certain cell types, especially self-renewing compartments
including Hematopoietic stem cells (HSCs), neural stem cells (NSCs) and pancreatic β
-cells (122-127). Old p16INK4a-deficient HSCs exhibited an enhanced repopulating ability to serially transplant mice (122). Similarly, p16INK4a deficiency partially abrogated age-associated decline in neural progenitor function and neurogenesis (125). Moreover, old p16INK4a-deficient mice had enhanced islet regeneration and improved diabetes-specific
survival after chemical ablation of β-cells (123). A caveat to these studies is the use of
from cell-non-autonomous mechanisms. Conditional inactivation and overexpression of p16INK4a, however, confirmed the cell-autonomous role of p16INK4a in aging of T
lymphocytes and pancreatic β-cells (123, 124). Further evidence for a crucial role of
p16INK4a in aging comes from selective elimination of p16INK4a–positive cells using the INK-ATCC mouse model, where, caspase-8 is activated upon exposure to the synthetic drug and induces apoptosis in p16INK4a–expressing cells. Systemic clearance of
p16INK4a–expressing cells mitigated some age-associated phenotypes and improved the healthy lifespan of progeroid and physiologically aged mice (41, 42), suggesting that age-related accumulation of p16INK4a contributes to some aspect of aging. Furthermore, these murine findings are underscored by unbiased genome-wide association studies (GWAS). GWAS has linked susceptibility single nucleotide polymorphisms (SNPs) on chromosome 9p21 near the CDKN2a/b locus with multiple age-related diseases including cancers, atherosclerosis, type 2 diabetes, and Alzheimer’s disease (108). Expression of p16INK4a correlates with some of the disease-associated SNP genotypes, suggesting that p16INK4a is involved in the development of certain degenerative diseases associated with aging in humans. Despite that, it’s not well understood whether or how these SNP variants may regulate p16INK4a transcription. One proposed mechanism is that these SNPs are located in the regulatory regions of p16INK4a such as enhancer elements, thereby altering the expression of p16INK4a transcript (128).
1.4 The roles of p16INK4a in macrophage biology
homeostasis and tissue repair (129). Macrophages display great plasticity in terms of phenotypes and functions to adapt to different tissue environments. Despite being overly simplistic, macrophages have been conventionally classified into two different subsets, termed M1 and M2, depending on the local cytokine milieu. The activated (M1)
macrophages are induced by interferon-gamma (IFN-γ) or lipopolysaccharide (LPS) to
promote T helper type 1 (Th1) responses by producing pro-inflammatory cytokines (e.g., interleukin-1β (IL-1β), IL-6, IL-12, IL-23, and TNF-α), reactive oxygen and nitrogen intermediates to eliminate pathogens and virus-infected cells as well as cancer cells. By contrast, in response to Th2 cytokines such as IL-4 and IL-13,
alternatively activated (M2) macrophages participate in Th2 responses and have anti-inflammatory functions by secreting anti-anti-inflammatory cytokines (e.g., IL-10 and
TGF-β), involved in parasite clearance, allergy, tissue repair and tumor progression. A link between 16INK4a and macrophage biology has been described where expression of p16INK4a was shown to cause potent growth arrest in macrophages (130). Moreover, GWAS demonstrated an association of the CDKN2A locus with increased risk for cardiovascular diseases and type 2 diabetes in which macrophages have
expression in macrophage function may be related to its role independent of cell cycle control such as M1/M2 polarization. Interestingly, bone-marrow derived macrophages (BMDM) had increased p16INK4a expression during in vitro differentiation, and loss of p16INK4a did not affect the cell-cycle status (70). Further, p16INK4a-deficient BMDMs resemble M2 subtype by producing significantly less inflammatory cytokines and upregulation of M2-associated genes, a model supported by a study using human monocyte-derived macrophages (134). Notably, recent studies have shown that macrophages were attracted in response to SASP components and exhibited some
senescence features including p16INK4a and SA-β-gal expression (135); yet, these
senescence markers were modulated by M1- and M2-inducing agents (136),
supporting the role of p16INK4 expression in macrophage polarization. Together, these data further highlight the complex role of p16INK4a in macrophage biology.
1.5 Summary
Inexorable accumulation of senescent cells throughout the lifespan not only drives aging but promotes age-related diseases. It’s believed that enumeration of senescent cells in vivo will allow us to predict physiological age and the susceptibility of age-associated diseases. Generation of mouse alleles that selectively eliminate
the most urgent, the development of “senolytics” requires the in vivo enumeration and characterization of senescent cells. To address this gap in our understanding, I have developed an in vivo reporter allele that allows the identification and isolation of single cells featuring high-level activation of the p16INK4a promoter, the most widely used in
REFERENCES
1. Hayflick L (1965) The Limited in Vitro Lifetime of Human Diploid Cell Strains. Exp Cell Res 37(3):614-636.
2. Allsopp RC, et al. (1992) Telomere Length Predicts Replicative Capacity of Human Fibroblasts. P Natl Acad Sci USA 89(21):10114-10118.
3. Herbig U, Jobling WA, Chen BPC, Chen DJ, & Sedivy JM (2004) Telomere
shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol Cell 14(4):501-513.
4. Meng AM, Wang Y, Van Zant G, & Zhou DH (2003) Ionizing radiation and busulfan induce premature senescence in murine bone marrow hematopoietic cells. Cancer Res 63(17):5414-5419.
5. Bartkova J, et al. (2006) Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 444(7119):633-637.
6. Serrano M, Lin AW, McCurrach ME, Beach D, & Lowe SW (1997) Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16(INK4a). Cell 88(5):593-602.
7. Zhu JY, Woods D, McMahon M, & Bishop JM (1998) Senescence of human fibroblasts induced by oncogenic Raf. Gene Dev 12(19):2997-3007.
8. Parrinello S, et al. (2003) Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol 5(8):741-747.
9. Yaswen P & Stampfer MR (2002) Molecular changes accompanying senescence and immortalization of cultured human mammary epithelial cells. Int J Biochem Cell B 34(11):1382-1394.
10. Campisi J (2013) Aging, Cellular Senescence, and Cancer. Annu Rev Physiol 75:685-705.
11. Campisi J & di Fagagna FD (2007) Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Bio 8(9):729-740.
12. Rodier F & Campisi J (2011) Four faces of cellular senescence. J Cell Biol 192(4):547-556.
14. Serrano M, Hannon GJ, & Beach D (1993) A New Regulatory Motif in Cell-Cycle Control Causing Specific-Inhibition of Cyclin-D/Cdk4. Nature 366:704-707.
15. Beausejour CM, et al. (2003) Reversal of human cellular senescence: roles of the p53 and p16 pathways. Embo J 22(16):4212-4222.
16. Stein GH, Drullinger LF, Soulard A, & Dulic V (1999) Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol Cell Biol 19(3):2109-2117.
17. Diekman BO, et al. (2018) Expression of p16INK4a is a biomarker of chondrocyte agingbut does not cause osteoarthritis. Aging Cell 17(4).
18. Farr JN, et al. (2016) Identification of Senescent Cells in the Bone Microenvironment. J Bone Miner Res 31(11):1920-1929.
19. Jurk D, et al. (2012) Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 11(6):996-1004. 20. Munoz-Espin D, et al. (2013) Programmed Cell Senescence during Mammalian
Embryonic Development. Cell 155(5):1104-1118.
21. Storer M, et al. (2013) Senescence Is a Developmental Mechanism that Contributes to Embryonic Growth and Patterning. Cell 155(5):1119-1130.
22. Demaria M, et al. (2014) An Essential Role for Senescent Cells in Optimal Wound Healing through Secretion of PDGF-AA. Dev Cell 31(6):722-733.
23. Jun JI & Lau LF (2010) The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat Cell Biol 12(7):676-685.
24. Chen ZB, et al. (2005) Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 436(7051):725-730. 25. Courtois-Cox S, et al. (2006) A negative feedback signaling network underlies
oncogene-induced senescence. Cancer Cell 10(6):459-472.
26. Young AP, et al. (2008) VHL loss actuates a HIF-independent senescence programme mediated by Rb and p400. Nat Cell Biol 10(3):361-369.
27. Sharpless NE, et al. (2001) Loss of p16(Ink4a) with retention of p19(Arf) predisposes mice to tumorigenesis. Nature 413(6851):86-91.
29. Sharpless NE, et al. (2002) p16(INK4a) and p53 deficiency cooperate in tumorigenesis. Cancer Res 62(10):2761-2765.
30. Matheu A, et al. (2004) Increased gene dosage of Ink4a/Arf results in cancer resistance and normal aging. Gene Dev 18(22):2736-2746.
31. Garcia-Cao I, et al. (2002) 'Super p53' mice exhibit enhanced DNA damage response, are tumor resistant and age normally. Embo J 21(22):6225-6235. 32. Hahn WC & Weinberg RA (2002) Mechanisms of disease: Rules for making
human tumor cells. New Engl J Med 347(20):1593-1603.
33. Sharpless NE & Depinho RA (2007) How stem cells age and why this makes us grow old. Nat Rev Mol Cell Bio 8(9):703-713.
34. Acosta JC, et al. (2013) A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol 15(8):978-990. 35. Nelson G, et al. (2012) A senescent cell bystander effect: senescence-induced
senescence. Aging Cell 11(2):345-349.
36. Chang E & Harley CB (1995) Telomere Length and Replicative Aging in Human Vascular Tissues. P Natl Acad Sci USA 92(24):11190-11194.
37. Price JS, et al. (2002) The role of chondrocyte senescence in osteoarthritis. Aging Cell 1(1):57-65.
38. Vasile E, Tomita Y, Brown LF, Kocher O, & Dvorak HF (2001) Differential expression of thymosin beta-10 by early passage and senescent vascular
endothelium is modulated by VPF/VEGF: evidence for senescent endothelial cells in vivo at sites of atherosclerosis. Faseb J 15(2):458-466.
39. Liu Y, et al. (2009) Expression of p16(INK4a) in peripheral blood T-cells is a biomarker of human aging. Aging Cell 8(4):439-448.
40. Sorrentino JA, et al. (2014) p16(INK4a) reporter mice reveal age-promoting effects of environmental toxicants. J Clin Invest 124(1):169-173.
41. Baker DJ, et al. (2016) Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 530(7589):184-189.
42. Baker DJ, et al. (2011) Clearance of p16(Ink4a)-positive senescent cells delays ageing-associated disorders. Nature 479(7372):232-236.
44. Childs BG, Baker DJ, Kirkland JL, Campisi J, & van Deursen JM (2014) Senescence and apoptosis: dueling or complementary cell fates? Embo Rep 15(11):1139-1153.
45. Yang HS & Hinds PW (2003) Increased ezrin expression and activation by CDK5 coincident with acquisition of the senescent phenotype. Mol Cell 11(5):1163-1176. 46. Evangelou K, et al. (2017) Robust, universal biomarker assay to detect senescent
cells in biological specimens. Aging Cell 16(1):192-197.
47. Salmonowicz H & Passos JF (2017) Detecting senescence: a new method for an old pigment. Aging Cell 16(3):432-434.
48. Davalos AR, et al. (2012) p53-dependent release of Alarmin HMGB1 is a central mediator of senescent phenotypes. Cancer Res 72.
49. Freund A, Laberge RM, Demaria M, & Campisi J (2012) Lamin B1 loss is a senescence-associated biomarker. Mol Biol Cell 23(11):2066-2075.
50. Shimi T, et al. (2011) The role of nuclear lamin B1 in cell proliferation and senescence. Gene Dev 25(24):2579-2593.
51. Sharpless NE & Sherr CJ (2015) Forging a signature of in vivo senescence. Nat Rev Cancer 15(7):397-408.
52. Dimri GP, et al. (1995) A Biomarker That Identifies Senescent Human-Cells in Culture and in Aging Skin in-Vivo. P Natl Acad Sci USA 92(20):9363-9367. 53. Kurz DJ, Decary S, Hong Y, & Erusalimsky JD (2000) Senescence-associated
beta-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J Cell Sci 113(20):3613-3622.
54. Lee BY, et al. (2006) Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell 5(2):187-195.
55. Bursuker I, Rhodes JM, & Goldman R (1982) Beta-Galactosidase - an Indicator of the Maturational Stage of Mouse and Human Mononuclear Phagocytes. J Cell Physiol 112(3):385-390.
56. Kopp HG, Hooper AT, Shmelkov SV, & Rafii S (2007) beta-galactosidase staining on bone marrow. The osteoclast pitfall. Histol Histopathol 22(9):971-976.
57. Dorr JR, et al. (2013) Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature 501(7467):421-425.
59. Young ARJ & Narita M (2010) Connecting autophagy to senescence in pathophysiology. Curr Opin Cell Biol 22(2):234-240.
60. Itahana KC, Judith & P Dimri, Goberdhan (2007) Methods to detect biomarkers of cellular senescence: The senescence-associated beta-galactosidase assay. Methods in molecular biology 371(1):21-23.
61. Coates PJ, Lorimore SA, Rigat BA, Lane DP, & Wright EG (2001) Induction of endogenous beta-galactosidase by ionizing radiation complicates the analysis of p53-LacZ transgenic mice. Oncogene 20(48):7096-7097.
62. Xiong Y, et al. (1993) P21 Is a Universal Inhibitor of Cyclin Kinases. Nature 366(6456):701-704.
63. Pantoja C & Serrano M (1999) Murine fibroblasts lacking p21 undergo
senescence and are resistant to transformation by oncogenic Ras. Oncogene 18(35):4974-4982.
64. Gire V & Dulic V (2015) Senescence from G2 arrest, revisited. Cell Cycle 14(3):297-304.
65. Johmura Y, et al. (2014) Necessary and Sufficient Role for a Mitosis Skip in Senescence Induction. Mol Cell 55(1):73-84.
66. Baker DJ, Weaver RL, & van Deursen JM (2013) p21 Both Attenuates and Drives Senescence and Aging in BubR1 Progeroid Mice. Cell Rep 3(4):1164-1174. 67. Burd CE, et al. (2013) Monitoring Tumorigenesis and Senescence In Vivo with a
p16(INK4a)-Luciferase Model. Cell 152(1-2):340-351.
68. Rheinwald JG, et al. (2002) A two-stage, p16(INK4A)- and p53-dependent keratinocyte senescence mechanism that limits replicative potential independent of telomere status (vol 22, pg 5157, 2002). Mol Cell Biol 22(19):6930-6930. 69. Yamakoshi K, et al. (2009) Real-time in vivo imaging of p16(Ink4a) reveals cross
talk with p53. J Cell Biol 186(3):393-407.
70. Cudejko C, et al. (2011) p16(INK4a) deficiency promotes IL-4-induced polarization and inhibits proinflammatory signaling in macrophages. Blood 118(9):2556-2566. 71. Krimpenfort P, et al. (2007) p15(Ink4b) is a critical tumour suppressor in the
absence of p16(Ink4a). Nature 448(7156):943-U946.
72. Kuilman T, et al. (2008) Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 133(6):1019-1031.
74. Khleif SN, et al. (1996) Inhibition of cyclin D-CDK4/CDK6 activity is associated with an E2F-mediated induction of cyclin kinase inhibitor activity. P Natl Acad Sci USA 93(9):4350-4354.
75. Nakao Y, et al. (1997) Induction of p16 during immortalization by HPV 16 and 18 and not during malignant transformation. Brit J Cancer 75(10):1410-1416.
76. Shapiro GI, et al. (1995) Reciprocal Rb Inactivation and P16(Ink4) Expression in Primary Lung Cancers and Cell-Lines. Cancer Res 55(3):505-509.
77. Witkiewicz AK, Knudsen KE, Dicker AP, & Knudsen ES (2011) The meaning of p16(ink4a) expression in tumors Functional significance, clinical associations and future developments. Cell Cycle 10(15):2497-2503.
78. Di Micco R, et al. (2006) Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 444(7119):638-642. 79. Fumagalli M, Rossiello F, Mondello C, & di Fagagna FD (2014) Stable Cellular
Senescence Is Associated with Persistent DDR Activation. Plos One 9(10).
80. Alimonti A, et al. (2010) A novel type of cellular senescence that can be enhanced in mouse models and human tumor xenografts to suppress prostate
tumorigenesis. J Clin Invest 120(3):681-693.
81. McConnell BB, Starborg M, Brookes S, & Peters G (1998) Inhibitors of cyclin-dependent kinases induce features of replicative senescence in early passage human diploid fibroblasts. Curr Biol 8(6):351-354.
82. Rodier F, et al. (2009) Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol 11(8):973-979. 83. Bakkenist CJ & Kastan MB (2003) DNA damage activates ATM through
intermolecular autophosphorylation and dimer dissociation. Nature 421(6922):499-506.
84. Soutoglou E & Misteli T (2008) Activation of the cellular DNA damage response in the absence of DNA lesions. Science 320(5882):1507-1510.
85. Narita M, et al. (2003) Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 113(6):703-716.
86. Bartholdi MF (1991) Nuclear-Distribution of Centromeres during the Cell-Cycle of Human-Diploid Fibroblasts. J Cell Sci 99:255-263.
88. Di Micco R, et al. (2011) Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat Cell Biol 13(3):292-302.
89. Kosar M, et al. (2011) Senescence-associated heterochromatin foci are
dispensable for cellular senescence, occur in a cell type- and insult-dependent manner, and follow expression of p16(ink4a). Cell Cycle 10(3):457-468.
90. Krizhanovsky V, et al. (2008) Senescence of activated stellate cells limits liver fibrosis. Cell 134(4):657-667.
91. Parrinello S, Coppe JP, Krtolica A, & Campisi J (2005) Stromal-epithelial interactions in aging and cancer: senescent fibroblasts alter epithelial cell differentiation. J Cell Sci 118(3):485-496.
92. Tsai KKC, Chuang EYY, Little JB, & Yuan ZM (2005) Cellular mechanisms for low-dose ionizing radiation-induced perturbation of the breast tissue
microenvironment. Cancer Res 65(15):6734-6744.
93. Hoare M, et al. (2016) NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat Cell Biol 18(9):979-992.
94. Chien YC, et al. (2011) Control of the senescence-associated secretory
phenotype by NF-kappa B promotes senescence and enhances chemosensitivity. Gene Dev 25(20):2125-2136.
95. Freund A, Patil CK, & Campisi J (2011) p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. Embo J 30(8):1536-1548.
96. Herranz N, et al. (2015) mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat Cell Biol 17(9):1205-1217. 97. Laberge RM, et al. (2015) MTOR regulates the pro-tumorigenic
senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol 17(8):1049-1061.
98. Narita M, et al. (2011) Spatial Coupling of mTOR and Autophagy Augments Secretory Phenotypes. Science 332(6032):966-970.
99. Coppe JP, Kauser K, Campisi J, & Beausejour CM (2006) Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J Biol Chem 281(40):29568-29574.
101. Liu D & Hornsby PJ (2007) Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res 67(7):3117-3126.
102. Bodnar AG, et al. (1998) Extension of life-span by introduction of telomerase into normal human cells. Science 279(5349):349-352.
103. Collins K & Mitchell JR (2002) Telomerase in the human organism. Oncogene 21(4):564-579.
104. Kim NW, et al. (1994) Specific Association of Human Telomerase Activity with Immortal Cells and Cancer. Science 266(5193):2011-2015.
105. di Fagagna FD, et al. (2003) A DNA damage checkpoint response in telomere-initiated senescence. Nature 426(6963):194-198.
106. Takai H, Smogorzewska A, & de Lange T (2003) DNA damage foci at dysfunctional telomeres. Curr Biol 13(17):1549-1556.
107. Fumagalli M, et al. (2012) Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat Cell Biol 14(4):355-365.
108. Jeck WR, Siebold AP, & Sharpless NE (2012) Review: a meta-analysis of GWAS and age-associated diseases. Aging Cell 11(5):727-731.
109. Jaskelioff M, et al. (2011) Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature 469(7328):102-106.
110. Aubert G, Hills M, & Lansdorp PM (2012) Telomere length measurement-Caveats and a critical assessment of the available technologies and tools. Mutat Res-Fund Mol M 730(1-2):59-67.
111. Quelle DE, Zindy F, Ashmun RA, & Sherr CJ (1995) Alternative Reading Frames of the Ink4a Tumor-Suppressor Gene Encode 2 Unrelated Proteins Capable of Inducing Cell-Cycle Arrest. Cell 83(6):993-1000.
112. Kamijo T, et al. (1998) Functional and physical interactions of the ARF tumor suppressor with p53 and Mdm2. P Natl Acad Sci USA 95(14):8292-8297. 113. Pomerantz J, et al. (1998) The Ink4a tumor suppressor gene product, p19(Arf),
interacts with MDM2 and neutralizes MDM2's inhibition of p53. Cell 92(6):713-723.
115. Beroukhim R, et al. (2010) The landscape of somatic copy-number alteration across human cancers. Nature 463(7283):899-905.
116. Bignell GR, et al. (2010) Signatures of mutation and selection in the cancer genome. Nature 463(7283):893-898.
117. Kamijo T, et al. (1997) Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19(ARF). Cell 91(5):649-659.
118. Chandler H & Peters G (2013) Stressing the cell cycle in senescence and aging. Curr Opin Cell Biol 25(6):765-771.
119. LaPak KM & Burd CE (2014) The Molecular Balancing Act of p16(INK4a) in Cancer and Aging. Mol Cancer Res 12(2):167-183.
120. Krishnamurthty J, et al. (2004) Ink4a/Arf expression is a biomarker of aging. J Clin Invest 114(9):1299-1307.
121. Zindy F, Quelle DE, Roussel MF, & Sherr CJ (1997) Expression of the p16(INK4a) tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene 15(2):203-211.
122. Janzen V, et al. (2006) Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16(INK4a). Nature 443(7110):421-426.
123. Krishnamurthy J, et al. (2006) p16(INK4a) induces an age-dependent decline in islet regenerative potential. Nature 443(7110):453-457.
124. Liu Y, et al. (2011) Expression of p16(INK4a) prevents cancer and promotes aging in lymphocytes. Blood 117(12):3257-3267.
125. Molofsky AV, et al. (2006) Increasing p16(INK4a) expression decreases forebrain progenitors and neurogenesis during ageing. Nature 443(7110):448-452.
126. Signer RAJ, Montecino-Rodriguez E, Witte ON, & Dorshkind K (2008) Aging and cancer resistance in lymphoid progenitors are linked processes conferred by p16(Ink4a) and Arf. Gene Dev 22(22):3115-3120.
127. Sousa-Victor P, et al. (2014) Geriatric muscle stem cells switch reversible quiescence into senescence. Nature 506(7488):316-321.
128. Jarinova O, et al. (2009) Functional Analysis of the Chromosome 9p21.3 Coronary Artery Disease Risk Locus. Arterioscl Throm Vas 29(10):1671-1677. 129. Wynn TA, Chawla A, & Pollard JW (2013) Macrophage biology in development,
130. Randle DH, Zindy F, Sherr CJ, & Roussel MF (2001) Differential effects of
p19(Arf) and p16(Ink4a) loss on senescence of murine bone marrow-derived preB cells and macrophages. P Natl Acad Sci USA 98(17):9654-9659.
131. Liu Y, et al. (2009) INK4/ARF Transcript Expression Is Associated with Chromosome 9p21 Variants Linked to Atherosclerosis. Plos One 4(4).
132. Kuo CL, et al. (2011) Cdkn2a Is an Atherosclerosis Modifier Locus That Regulates Monocyte/Macrophage Proliferation. Arterioscl Throm Vas 31(11):2483-2492. 133. Childs BG, et al. (2016) Senescent intimal foam cells are deleterious at all stages
of atherosclerosis. Science 354(6311):472-477.
134. Fuentes L, et al. (2011) Downregulation of the tumour suppressor p16(INK4A) contributes to the polarisation of human macrophages toward an adipose tissue macrophage (ATM)-like phenotype. Diabetologia 54(12):3150-3156.
135. Hall BM, et al. (2016) Aging of mice is associated with p16(Ink4a)- and beta-galactosidase-positive macrophage accumulation that can be induced in young mice by senescent cells. Aging-Us 8(7):1294-1315.
CHAPTER 2: Cells exhibiting strong p16INK4a promoter activation in vivo display features of senescence1
2.1 Overview
The accumulation of senescent cells over a lifetime causes age-related
pathologies; however, the inability to reliably identify senescent cells in vivo has
hindered clinical efforts to employ this knowledge as a means to ameliorate or reverse
aging. Here, we describe a novel reporter allele, p16tdTom, enabling the in vivo
identification and isolation of cells featuring high-level activation of the p16INK4a promoter.
Our findings provide an insight into the functional and molecular characteristics of
p16INK4a-activated cells in vitro and in vivo. We show that such cells accumulate with
aging or other models of injury, and that they exhibit clinically targetable features of
cellular senescence.
1This chapter has been published at “PNAS” in 2019: Liu J.-Y., Souroullas G.P., Diekman B.O.,
Krishnamurthy J., Hall B.M., Sorrentino J.A., Parker J.S., Sessions G.A., Gudkov A.V., Sharpless N.E.
2.2 Introduction
Cellular senescence refers to a specific form of highly durable cell cycle arrest of previously proliferation-competent cells that is resistant to mitogenic stimulation and accompanied by persistent DNA damage response. Senescence is an important tumor suppressor mechanism, and is believed to contribute to organismal aging (1, 2). A senescence response is triggered by a variety of genotoxic stresses including shortened telomeres, exposure to DNA damaging agents and oncogenic insult (1, 3). While
senescence is primarily characterized in replication-competent cells, recent studies have suggested that largely post-mitotic cell types can also initiate a senescence program (4, 5). In addition to growth arrest, senescence is variably associated with the expression of cyclin-dependent kinase (CDK) inhibitors (especially p16INK4a),
senescence-associated beta-galactosidase (SA-β-gal) activity, and the elaboration of
cytokines that comprise the SA-secretory phenotype (SASP) (3, 6). Given the
prominence of senescence in cancer and aging, there has been great interest in the identification and characterization of senescent cells in an intact adult organism.
aging (8, 9) or after certain sorts of tissue injury (10-12). Murine studies suggest that accumulation of p16INK4a leads to an age-related loss of replicative capacity in select tissues, thereby causing some phenotypic aspects of aging (13-16). The clearance of p16INK4a-expressing cells attenuates age-associated phenotypes and improves the healthy lifespan of progeroid and physiologically aged mice (17, 18). These murine results are underscored by a remarkable string of associations of the CDKN2a/b locus (encoding the p16INK4a, ARF, and p15INK4b transcripts) with human age-related
phenotypes by genome-wide association studies (19, 20).
In prior work, activation of the p16INK4a promoter has been used to suggest
senescence in vivo. Our lab and others have placed reporter genes (e.g. luciferase (LUC)) under the control of the p16INK4a promoter by either transgenic (10, 17, 21, 22) or
knock-in approaches (23). These reporter alleles have been employed to demonstrate that the p16INK4a promoter activity increases during wounding, inflammation,
tumorigenesis or aging in vivo in tissues. While valuable for studies at the tissue or organ level, these alleles have been limited in their ability to detect and isolate individual cells with strong activation of the p16INK4a promoter in vivo. To study individual p16INK4a
-activated cells, we have generated a fluorescence-based reporter allele with tandem-dimer Tomato (tdTom) knocked into the endogenous p16INK4a locus. This allele enables the identification and isolation of p16INK4a-activated cells (tdTom+) at the single-cell level
from cultured cells and in vivo. Using this allele, we quantified tdTom+ cells in several tissues with aging or in the setting of inflammation, and isolated these cells for
2.3 Results
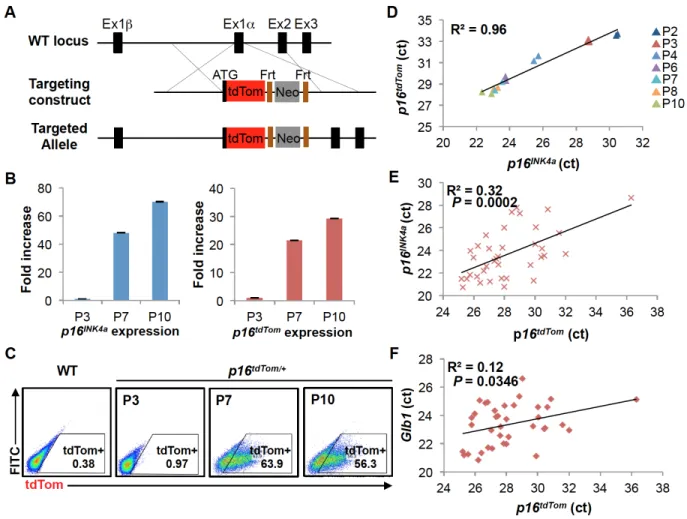
2.3.1 Generation and Characterization of the p16tdTom Allele
To study individual p16INK4a-expressing cells from in vivo sources, we knocked an open reading frame (ORF) encoding a fluorescent reporter protein (tandem dimer
Tomato or tdTom) into the endogenous first exon (exon 1α) of p16INK4a through
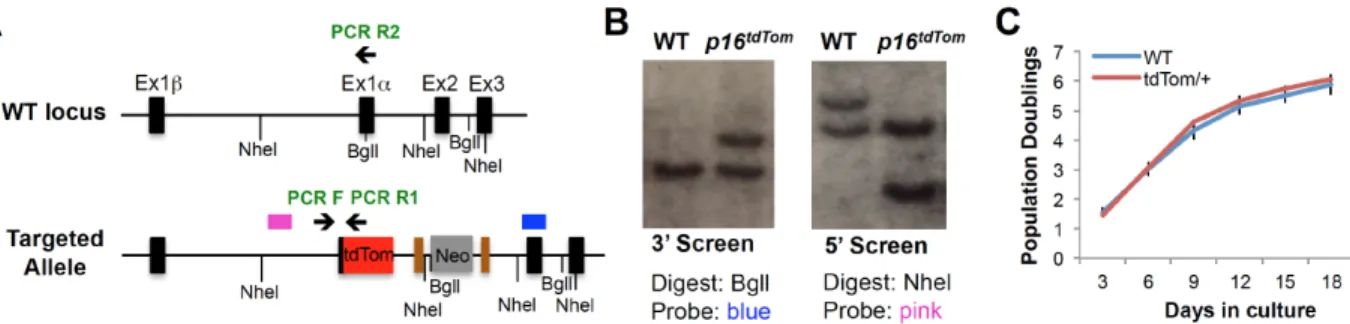
homologous recombination (Fig. 2.1A). The targeted allele (p16tdTom) was designed to be null for p16INK4a expression, yet with unperturbed expression of the Arf transcript, as well as retention of cis-regulatory elements around the Cdkn2a (or Ink4a/Arf) locus. A stop codon and poly-A signal were included at the end of the knocked-in tdTom ORF, and therefore the targeted mRNA would not be expected to produce a message that splices to exon 2. Importantly, an FRT-flanked neomycin selection cassette under the regulation of a strong PGK promoter was knocked into the first intron to allow for ES cell selection (Fig. 2.1A). Correct homologous targeting was verified by PCR, sequencing and Southern blot (Fig. S2.1A-B).
Prior efforts in our lab failed to produce a usable single-cell reporter allele through similar approaches. Despite correct knock-in targeting, alleles featuring
compromising tissue reporter fidelity (25, 26), we elected to characterize the p16tdTom
allele in cells with both a retained neomycin selection cassette and after flipase (FLP)-mediated excision.
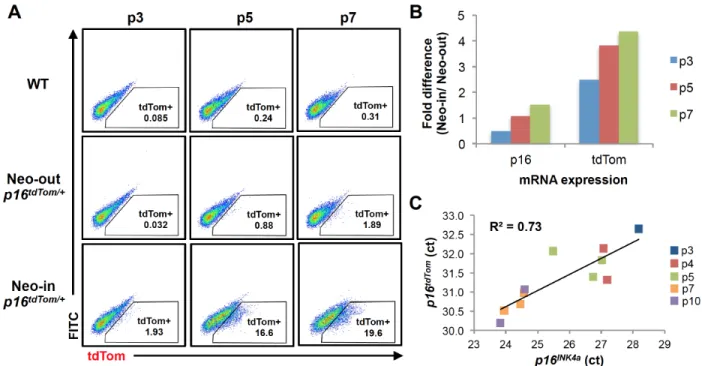
We assessed the allele function in cultured murine embryonic fibroblasts (MEFs) heterozygous for the tdTom knock-in (p16tdTom/+). In line with prior results (23, 27, 28), p16tdTom/+ and wild-type (WT) MEFs have comparable growth characteristics at early
passage (Fig. S2.1C). As expected (29, 30), serial passage induced increasing expression of endogenous p16INK4a mRNA produced from the WT locus and tdTom transcript from the p16tdTom allele (Fig. 2.1B). The frequency of tdTom+ cells identified
by flow cytometry also increased with passage (Fig. 2.1C). While the frequency of tdTom+ MEFs increased with passage in cells derived from mice retaining (“Neo-in”) or without (“Neo-out”) the neomycin cassette, the frequency of tdTom+ cells in Neo-in cultures was an order of magnitude higher. For example, we show a comparison of MEFs at passage 7 (~25 days in culture) where 1.9% of Neo-out cells and 19.6% of Neo-in cells were tdTom+ (Fig. S2.2A). To determine which allele was more faithful to endogenous p16INK4a expression, we measured tdTom and p16INK4a transcript levels in
heterozygous cells (Neo-in/WT or Neo-out/WT) by quantitative RT-PCR (qRT-PCR). As was the case for tdTom protein, the tdTom mRNA increased with passage in cells derived from both Neo-in or Neo-out mice, but mRNA expression was more dynamic with passage in Neo-in cells (Fig. S2.2B). Correspondingly, tdTom mRNA expression in Neo-in/WT cells strongly correlated with p16INK4a expression when measured in multiple
detection, and therefore correlated less well with endogenous p16INK4a mRNA (Fig.
S2.2C, R2 = 0.73). Given the higher fidelity and stronger signal using the Neo-in allele, we elected to pursue all subsequent experiments using this version, and henceforth “p16tdTom” will designate the Neo-in allele. To test the functionality of this single-cell
reporter, we assessed the expression of tdTom and p16INK4a on the single cell level. By single-cell qRT-PCR, we noted a significant association between tdTom and p16INK4a
expression (Fig. 2.1E, R2 = 0.32; p-value= 0.0002). Additionally, we found tdTom levels
significantly correlated with β-galactosidase (Glb1) levels as well (Fig. 2.1F, R2= 0.12;
p-value= 0.0346).
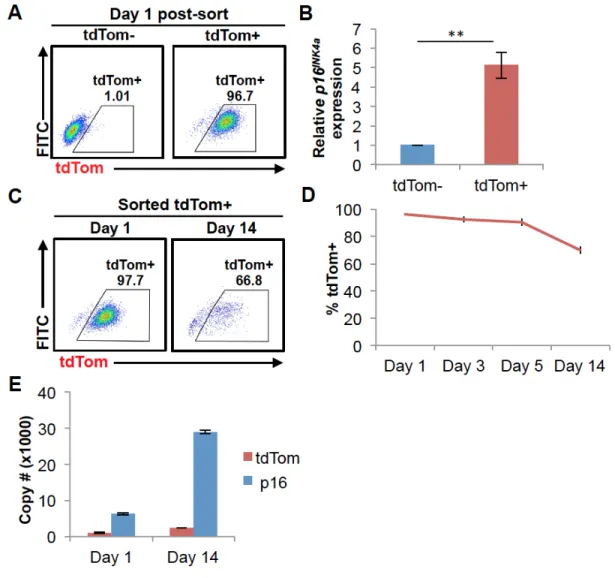
2.3.2 The p16tdTom Allele Reports Promoter Activation
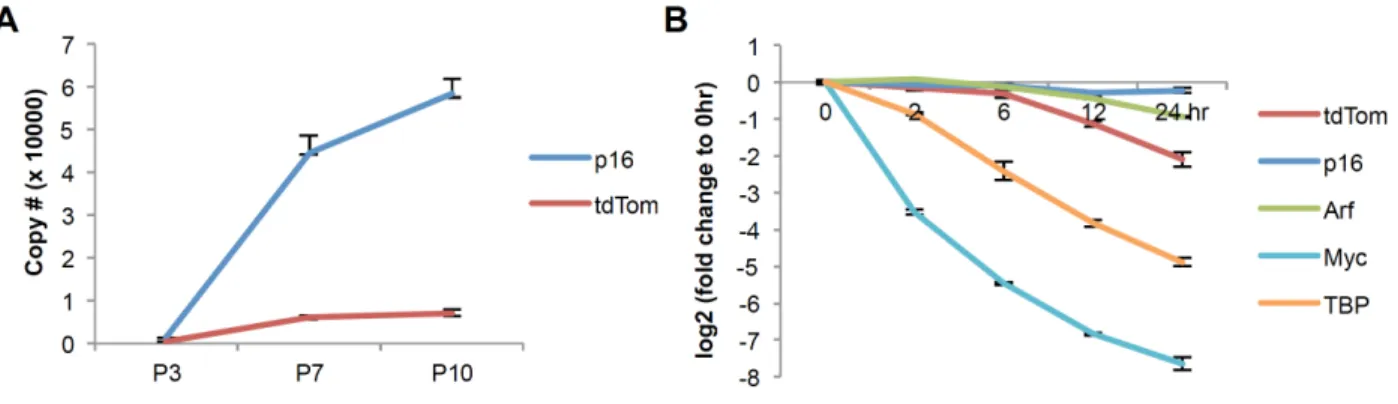
Although expression of the reporter transcript strongly correlated with that of the WT allele (Fig. 2.1D-E), the absolute level of tdTom was lower than that of the native p16INK4a transcript in cultured p16tdTom/+ MEFs (Fig. S2.3A). This pattern was also noted
in the unpublished p16INK4a-GFP and p16INK4a-CRE alleles, as well as our published p16LUC allele (23). To explain this recurrent finding that expression of the endogenous
allele was reproducibly 5- to 10-fold higher than the knocked-in allele in heterozygous cells, we considered the possibility that mRNA stability differs between the various knocked-in ORFs versus the endogenous p16INK4a transcript. Hara and Peters reported
actinomycin D (ActD) treatment followed by qRT-PCR quantification to determine the mRNA half-life of the endogenous p16INK4a and Arf mRNAs as well as the tdTom mRNA in p16tdTom/+ MEFs (Fig. S2.3B). As expected, the p16INK4a and Arf transcripts showed little change for up to 24 hours after ActD treatment, whereas the tdTom transcript exhibited a half-life of ~12 hours (Fig. S2.3B). The decreased stability of these knocked-in transcripts occurs despite the knocked-inclusion of a 3’ poly-A signal derived from SV40 which augments transcript stability in other systems (34). These observations indicate that the
lower expression of transcripts knocked into exon 1α of the Cdkn2a locus, and
commensurately weak expression of reporter proteins, reflects the fact that the
knocked-in transcripts do not recapitulate the extraordinary stability of the endogenous p16INK4a mRNA.
We next examined the levels of p16INK4a mRNA in p16tdTom/+ cells that were either tdTom+ or tdTom-. We sorted MEF cultures using fluorescent activated cell sorting (FACS) based on tdTom expression (Fig. 2.2A) and measured p16INK4a mRNA levels in each fraction (Fig. 2.2B). We noted a moderate enrichment of p16INK4a mRNA in tdTom+
MEFs (~5-fold), but p16INK4a mRNA was readily detectable in tdTom- fractions at all passages tested. Given the observed differences in p16INK4a versus tdTom transcript half-life, we analyzed their relative expressions in MEF cultures for up to 14 days post-sorting. When analyzed in this way, we noted that expression of tdTom protein and p16INK4a mRNA began to diverge with long-term culture. At day 1 post-sort, virtually all
as opposed to overgrowth of the culture by tdTom- cells for two reasons. First, the proliferation rate in the tdTom+ cultures remained low for two weeks after sorting, which would not be consistent with an expansion of proliferating tdTom- cells. Moreover, during the period of culture, tdTom+ MEFs exhibited a significant further accumulation of p16INK4a transcript (Fig. 2.2E), which would not be expected with expansion of tdTom- cells. Of note, levels of tdTom mRNA in cells sorted as tdTom+ on day 1 were little changed over 14 days of culture (Fig. 2.2E). These results suggest that while tdTom protein expression has ‘peaked’ at the time of FACS and then decreases in some tdTom+ cells, levels of p16INK4a continues to sharply rise in the same cells for weeks
after sorting. We believe these results have significant implications as to the
interpretation of what the p16tdTom reporter allele actually ‘reports’. Specifically, these
results suggest that tdTom positivity is a better proxy for high-level activation of the p16INK4a promoter rather than total abundance of the p16INK4a transcript. This in turn indicates that some cells with formerly strong activation of the p16INK4a promoter could
appear tdTom- a few weeks later (as in Fig. 2.2C), despite high-level expression of the p16INK4a transcript (and presumably protein).
2.3.3 Characterization of p16INK4a Transcriptionally Active Cells In Vitro Using the p16tdTom allele, we next turned to the question of the functional properties of cultured cells featuring high-level p16INK4a promoter activation. FACS-isolated tdTom+ MEFs showed lower rates of growth (Fig. 2.3A) and decreased
incorporation of a thymidine analog, 5-ethyl-2'-deoxyuridine (EdU, Fig. 2.3B) compared
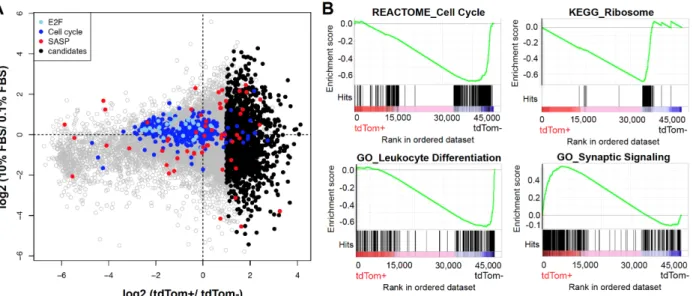
performed an unbiased analysis of RNA expression by next-generation sequencing (RNA-seq) in tdTom+ versus tdTom- cells with or without serum starvation (0.1% FBS for 48 hours). While serum starvation induced a decline in the expression of mRNAs associated with proliferation (cell cycle, E2F) as expected, an even greater reduction in the expression of proliferative mRNAs was noted in tdTom+ compared to tdTom- cells (Fig. S2.4A). Gene Set Enrichment Analysis (GSEA) of tdTom+ versus tdTom- MEFs also demonstrated enrichment of signatures associated with proliferation (e.g. cell cycle and ribosomal transcripts) in tdTom- cells consistent with their increased rates of
proliferation. Additionally, GSEA demonstrated differential expression of many
signatures associated with developing tissue lineages (e.g. neural, cardiac, cutaneous and hematopoietic (e.g. synaptic signaling and leukocyte development shown in Fig. S2.4B). Given that MEF cultures are derived from disaggregated whole murine embryos, we believe this finding reflects an increased propensity to activate p16INK4a expression in certain tissue types (e.g. brain, heart and skin), but not others (e.g. leukocytes). For the RNA-seq analyses, we developed a list of SASP-transcripts compiled from several sources studying senescence in a variety of human or murine cell types (35-38) (Table S2.1). Using this list, there was no association of SASP transcript expression with either serum starvation or tdTom expression in MEFs (Fig. S2.4A). These findings could
indicate that p16INK4a-activated, hyporeplicative MEF cultures expressing SA-β-gal are
2.3.4 Enumeration of p16INK4a-Activated Cells In Vivo
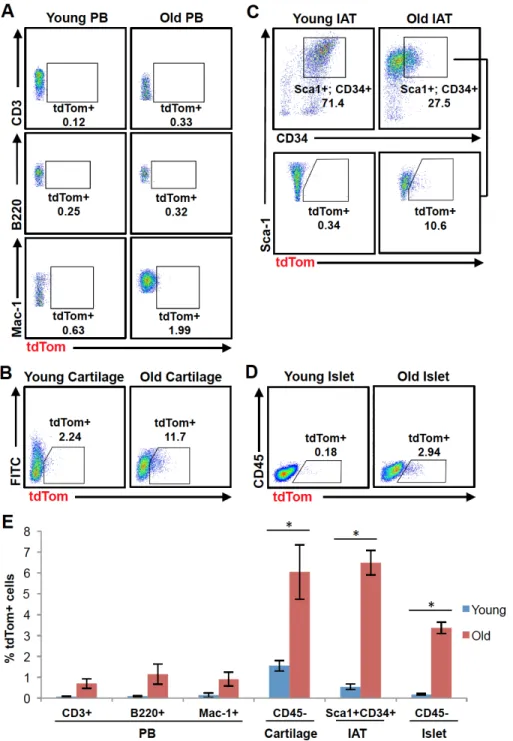
Although evidence suggests that the in vivo accumulation of senescent cells contributes to age-associated tissue dysfunction, the frequency of senescent cells within different aged tissues is unclear. To address this issue, we examined the percentage of tdTom+ cells from tissues harvested from young (8-12 weeks) or old (100-120 weeks) p16tdTom/+ mice. As p16INK4a mRNA is readily detected in murine or human peripheral
blood T cells and increases with aging (9, 16), we examined the frequency of tdTom+ cells in peripheral blood CD3+ (T cells), B220+ or Mac-1+ (myeloid cells) populations every 6 months through phlebotomy. In these compartments, there was only a minimal increase in the frequency of tdTom+ cells with aging (Fig. 2.4A). Of note, a subset of mice (n=6), displayed a transient, high-level increase in the frequency of tdTom+ cells in peripheral blood at the time of routine phlebotomy (Fig. S2.5). These transient ‘flares’ of p16INK4a expression in peripheral blood occurred in otherwise well-appearing mice and generally resolved within 1 month of initial observation. Up to 30-40% of mononuclear blood cells were found to be tdTom+ during these episodes, and all 6 observed cases showed a sharp increase within the Mac-1+ population. We have noted similar flares of luciferase activity in p16LUC/+ mice (23) and reasoned these episodes might represent a transient, sub-clinical inflammatory state (e.g. an occult viral infection). However, we were unable to provoke such responses by administering p16tdTom/+ mice toll-like receptor agonists (e.g. polyinosinic:polycytidylic acid or lipopolysaccharide (LPS)). These data suggest that while expression of p16INK4a mRNA is abundant in T cells from
Next, we examined activation of the p16INK4a promoter in non-hematopoietic
tissues with aging. We focused on tissues where prior work has suggested increased p16INK4a mRNA expression with aging (4, 14, 18, 39-41). We made single-cell
preparations of each tissue from young and old p16tdTom/+ mice and then employed
immunophenotyping and gating schemes where appropriate to focus on specific tissue fractions of interest (e.g. CD45- cells from cartilage or pancreatic islets, and
Sca1+CD34+ progenitors from adipose). We observed significant increases in the frequency of tdTom+ cells with aging in single cells derived from articular cartilage, inguinal adipose tissue (IAT), and pancreatic islets (Fig. 2.4B-E). The percentage of tdTom+ cells increased approximately 4 to 18 fold in these tissue compartments when comparing young and old mice, suggesting that the frequency of individual
chondrocytes, white adipose progenitors and beta cells having high-level activation of the p16INK4a promoter increases with aging.
2.3.5 Characterization of p16INK4a-Activated Macrophages
While we were able to identify significant numbers of p16INK4a-expressing cells
from several tissues with aging, the low frequency of tdTom+ cells (<10%) and difficulty of isolating these fractions prevented us from further functional and molecular
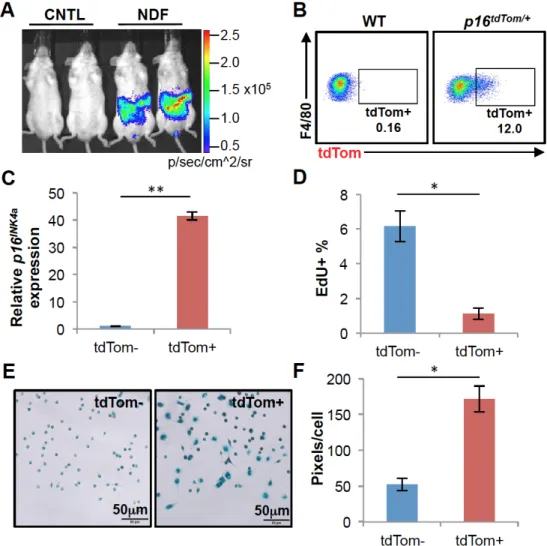
characterization. Therefore, we turned to a recently described inflammatory model to induce high-level p16INK4a expression in activated macrophages in vivo (42). Toward that end, we implanted quiescent neonatal dermal fibroblast (NDF)-containing alginate beads into p16LUC/+ or p16tdTom/+ mice via intraperitoneal injection. Prior work has shown
including IL-6 and IL-8, in turn leading to a large influx of inflammatory cells (42). As reported, NDF beads induced a strong luminescent signal in the abdomen of p16LUC/+ mice by 3 weeks post-injection (Fig. 2.5A). Flow cytometric analysis of cells in the peritoneal lavage of p16tdTom/+ mice 3 weeks after implanting NDF beads showed a
strong induction of tdTom expression in macrophage (Mac-1+F4/80+) populations (Fig. 2.5B), but not other lavage cell types (e.g. T cells, B220+ cells and eosinophils, Fig. S2.6). To characterize peritoneal macrophages with high-level p16INK4a promoter
activation, we isolated Mac-1+F4/80+ cells by FACS based on tdTom expression. Using this approach, we observed a much greater enrichment of p16INK4a mRNA expression in
tdTom+ vs. tdTom- macrophages (40-fold, Fig. 2.5C) compared to that seen in MEFs (5-fold, Fig. 2.2B). This likely reflects much greater homogeneity among the sorted macrophages compared to mixed MEF cultures. As was the case for MEFs, tdTom+ macrophages exhibited a marked reduction in EdU incorporation (Fig. 2.5D) and
increased SA-β-gal activity (Fig. 2.5E-F). It is worth noting that SA-β-gal activity has
been considered an unreliable marker of senescence in vivo, especially in this cell type (43, 44). These results show that a substantial fraction of macrophages induced in response to NDF-loaded beads exhibit features of senescence: activation of the
p16INK4a promoter, reduced proliferation and expression of SA-β-gal activity.
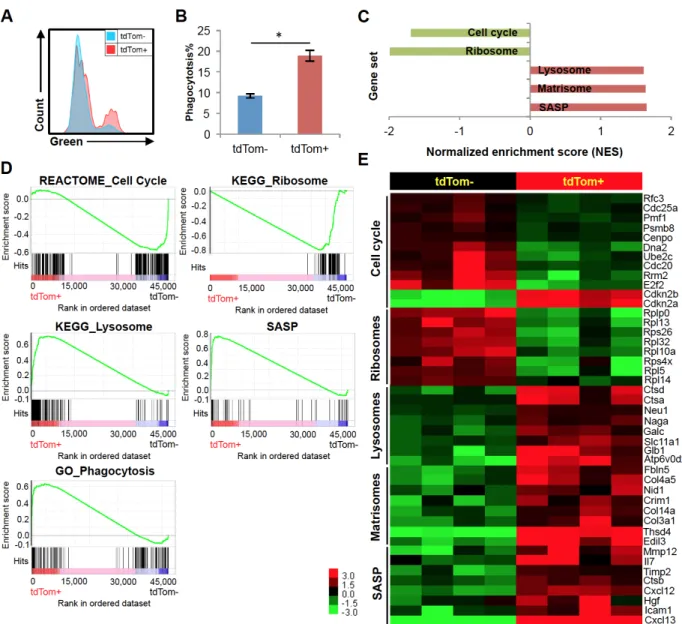
with regard to macrophage polarity (e.g. CD80, CD206 and MHCII). Moreover, we did not find a modulation of p16INK4a promoter activity by M1/M2 polarizing agents including LPS and interleukin 4 (IL-4) in either tdTom+ or tdTom- macrophages (Fig. S2.7). However, in vitro phagocytosis assays showed that tdTom+ macrophages exhibited greater phagocytic activity than tdTom- cells (Fig. 2.6A-B). This demonstration of altered or even increased cellular function is reminiscent of findings in other cell types in the setting of high-level p16INK4a expression (e.g. increased insulin secretion from p16INK4a -expressing pancreatic beta cells (47) and increased cell killing in senescent T cells (46, 48)).
In order to study the underlying mechanisms and genes responsible for the response of p16INK4a-activated macrophages to NDF-beads, we performed RNA-seq of
tdTom+ versus tdTom- peritoneal macrophages. We identified 456 transcripts being upregulated and 118 transcripts downregulated in tdTom+ macrophages (P<0.01). Through gene set enrichment analysis (GSEA), we identified several gene signatures related to the cell cycle, senescence and macrophage functions (Figure 2.6C-E). Specifically, consistent with the decreased proliferation of these cells (Fig. 2.5D), tdTom+ macrophages exhibited a profound decline in the expression of transcripts associated with cell cycle traversal and ribosomal proteins. Even though expression of a few “cell cycle”-classified genes was increased in tdTom+ cells, these were largely inhibitors of the cell cycle such as p16INK4a/Arf (Cdkn2a) and p15INK4b (Cdkn2b, Fig. 2.6E). Macrophages with high-level activation of the p16INK4a promoter also exhibited
increased expression of lysosomal mRNAs, consistent with the observed increase in β