Recurrent EIARF and PRES With Severe Renal
Hypouricemia by Compound Heterozygous
SLC2A9
Mutation
abstract
Renal hypouricemia (RHU) is a hereditary disease that predisposes affected people to exercise-induced acute renal failure (EIARF). In most patients with RHU, the disorder is caused by loss-of-function mutations in SLC22A12 (solute carrier family 22, member 12), which encodes urate transporter 1 (URAT1). Patients with RHU without any mutations in the URAT1 gene were recently found to have a mutation in the glu-cose transporter 9 (GLUT9) gene (SLC2A9[solute carrier family 2, mem-ber 9]). Central nervous system complications seem to be rare in pa-tients with RHU withSLC22A12mutations. Here, we report the case of a girl with severe RHU (serum urate: 5.9mol/L [0.1 mg/dL]) associated with recurrent EIARF in whom the disease was caused by a compound heterozygous mutation inSLC2A9, a nonsense mutation in the pater-nal allele (p.G207X in exon 7), and a large duplication (c.1– 2981_1204⫹16502) in the maternal allele detected by reverse-transcription polymerase chain reaction (PCR), semiquantitative PCR, long PCR, and direct sequencing. The episodes of EIARF were compli-cated by posterior reversible encephalopathy syndrome (PRES), which suggested a relationship between PRES and GLUT9 or severe hypouri-cemia. This is the second report of mutations of both alleles ofSLC2A9
that resulted in severe hypouricemia. Our findings indicate that even a nonsense mutation responsible for the heterozygous status ofSLC2A9
did not cause severe hypouricemia, and they lend support to previous speculation that mutations of both SLC2A9alleles cause severe hy-pouricemia. Our case shows that GLUT9, unlike URAT1, may play a spe-cific role in exercise-induced PRES.Pediatrics2011;127:e1621–e1625 AUTHORS:Yuko Shima, MD, PhD,aKandai Nozu, MD, PhD,b
Yoshimi Nozu, BS,bHiroko Togawa, MD, PhD,aHiroshi
Kaito, MD, PhD,bMasafumi Matsuo, MD, PhD,bKazumoto
Iijima, MD, PhD,bKoichi Nakanishi, MD, PhD,aand
Norishige Yoshikawa MD, PhDa
aDepartment of Pediatrics, Wakayama Medical University, Wakayama City, Wakayama, Japan; andbDepartment of Pediatrics, Kobe University Graduate School of Medicine, Kobe, Hyogo, Japan
KEY WORDS
acute renal failure, GLUT9, posterior reversible encephalopathy syndrome, proximal tubule, urate
ABBREVIATIONS
RHU—renal hypouricemia
EIARF—exercise-induced acute renal failure SLC22A12—solute carrier family 22, member 12 URAT1—urate transporter 1
GLUT9—glucose transporter 9
GLUT9S—short-form glucose transporter 9 GLUT9L—long-form glucose transporter 9 SLC2A9—solute carrier family 2, member 9
PRES—posterior reversible encephalopathy syndrome RT-PCR—reverse-transcription polymerase chain reaction
Drs Shima and Nakanishi provided patient management and drafted the manuscript; Dr K. Nozu, Ms Y. Nozu, and Drs Kaito, Matsuo, and Iijima performed gene analysis; and Drs Togawa and Yoshikawa provided patient management.
www.pediatrics.org/cgi/doi/10.1542/peds.2010-2592
doi:10.1542/peds.2010-2592
Accepted for publication Jan 26, 2011
Address correspondence to Koichi Nakanishi, MD, PhD, Department of Pediatrics, Wakayama Medical University, 811-1 Kimiidera, Wakayama City, Wakayama, 641-8509, Japan. E-mail: [email protected]
PEDIATRICS (ISSN Numbers: Print, 0031-4005; Online, 1098-4275).
Copyright © 2011 by the American Academy of Pediatrics
FINANCIAL DISCLOSURE:The authors have indicated they have no financial relationships relevant to this article to disclose.
220150) is an inherited disorder characterized by impaired renal urate reabsorption and subsequent low serum urate levels. It is
associ-ated with severe complications such as exercise-induced acute renal fail-ure (EIARF).1–6 The clinical presenta-tion includes loin pain with nausea and
vomiting several hours later after ex-ercise,4–9and laboratory investigation reveals abnormally increased clear-ance of renal urate. The estimated in-cidence of RHU is 0.12% to 0.72%.10,11
The pathogenesis of EIARF associated with RHU remains unknown. One the-ory suggests that strenuous exercise causes a sequential increase in
free-radical levels. Oxygen free free-radicals seem to play a crucial role in the patho-genesis of ischemic renal failure.12 Therefore, patients with RHU may be at risk of free-radical damage to
the kidney during exercise.1 In most patients with RHU, the disorder is caused by loss-of-function mutations in SLC22A12 (solute carrier family 22, member 12 [GenBank accession
No. AB071863]), which encodes the urate transporter 1 (URAT1) responsi-ble for control of urate reabsorption at the apical side of proximal
tubules.13–15
Patients with RHU without any muta-tions in the URAT1 gene were
re-cently found to have a mutation in the glucose transporter 9 (GLUT9) gene, a transporter of urate from tubule cells, encoded bySLC2A9(solute car-rier family 2, member 9 [GenBank ac-cession No. AF210317]).16–18 SLC2A9 encodes 2 variants of GLUT9: the short form (GLUT9S) on the apical side and the long form (GLUT9L) on the
basolat-eral side.16Moreover, it was reported recently that homozygous SLC2A9
mutations cause severe RHU and EIARF.19
First Episode
An 11-year-old previously healthy girl was admitted to a local hospital with a 2-day history of continuous nausea and abdominal pain starting a few hours after she had participated in a sprint during school sports (admis-sion on day 3 after onset). Her vital signs and results of a physical exami-nation were normal. Laboratory stud-ies revealed elevated concentrations of urea (14.5 mmol/L [40.6 mg/dL]) (normal range: 2.5– 6.8 mmol/L) and creatinine (177 mol/L [2.0 mg/dL]) (normal range: 30 – 61mol/L), which were the maximum values at the time of this first episode. Two days later (day 5), she developed headache and vomiting, and hypertension was evi-dent (142/92 mm Hg [zscore: 3.4/2.6]). Nifedipine was administered once at 10 mg, and her blood pressure was well controlled. On day 7, she com-plained of severe headache, visual dis-turbance, and hallucinations. Hyper-tension (141/93 mm Hg [z score: 3.3/ 2.7]) again became evident, and nifedipine was administered. One hour later, she suffered a generalized con-vulsion that lasted for⬃2 minutes; her eyes deviated to the left side. She was then transferred to our hospital. On admission, her consciousness was slightly clouded (Glasgow Coma Scale: 14). Her body height, weight, and BMI were 155.0 cm (zscore: 1.4), 50.5 kg (z
score: 0.5), and 21.0 (zscore: 1.0), re-spectively. Her blood pressure was 128/75 mm Hg (z score: 2.0/1.1) and pulse rate was 103 beats per minute. The results of other physical examina-tions and her vital signs were normal. Laboratory studies revealed elevated concentrations of urea (11.4 mmol/L [32.0 mg/dL]) and creatinine (106
mol/L [1.2 mg/dL]) and hypouricemia (54 mol/L [0.9 mg/dL]) (normal
range: 5.5%– 8.5%). We knew that the patient had severe hypouricemia (5.9
mol/L [0.1 mg/dL]), because it had been recognized during an examina-tion for enterocolitis that was con-ducted when she was 7 years old. MRI showed an area of high signal intensity in the occipital lobe on T2-weighted and fluid-attenuated inversion recov-ery images and an area of low signal intensity area on T1-weighted images (Fig 1). EIARF with RHU and posterior reversible encephalopathy syndrome (PRES) were diagnosed. Her urine vol-ume was well preserved, and her renal function recovered completely on day 13. Her consciousness gradually be-came clear on day 11 without specific treatments except for maintenance of adequate blood pressure. Follow-up MRI 2 weeks after the first studies (day 21) revealed complete resolution of the abnormal lesions in the occipital lobe, which was compatible with the diagnosis of PRES (Fig 1). She was dis-charged on day 22. Follow-up labora-tory studies revealed normal concen-trations of urea and creatinine (4.6 mmol/L [13 mg/dL] and 35mol/L [0.4 mg/dL], respectively) but severe hy-pouricemia (5.9mol/L [0.1 mg/dL]). Her blood pressure was 112/72 mm Hg (zscore: 0.5/0.8) at nonsick times.
Second Episode
epi-sode, MRI was performed immediately before fluid infusion. The MRI revealed an area of high signal intensity in the same region on T2-weighted and fluid-attenuated inversion recovery images and an area of low signal intensity on T1-weighted images (Fig 1). The maxi-mum concentrations of urea and cre-atinine were 19.3 mmol/L (54.0 mg/dL) and 265 mol/L (3.0 mg/dL), respec-tively, on day 4, but they had recovered completely by day 19. This time, the pa-tient showed no disturbance of con-sciousness or convulsion during the disease course, and her headache im-proved on day 3. She was discharged on day 14. Follow-up MRI on day 21 again showed complete resolution. Follow-up laboratory studies revealed normal concentrations of urea and creatinine (5.0 mmol/L [14 mg/dL] and 35 mol/L [0.4 mg/dL], respectively)
and hypouricemia (5.9 mol/L [0.1 mg/dL]). Her parents’ serum urate lev-els were 202 mol/L (3.4 mg/dL) (mother) and 286mol/L (4.8 mg/dL) (father). We strictly forbade the pa-tient from participating in anaerobic activity.
Mutational Analysis
Mutational analysis revealed that the patient had a compound heterozy-gous mutation inSLC2A9, a nonsense mutation in the paternal allele (p.G207X in exon 7), and a large du-plication (c.1–2981_1204⫹16502) in the maternal allele, detected by reverse-transcription polymerase chain reaction (RT-PCR) and direct sequencing. For detail of the muta-tional analysis, seeSupplemental In-formation, Supplemental Table 2, andSupplemental Figs 2 through 7.
DISCUSSION
This is the second report of mutations of both alleles of SLC2A9with severe RHU. To our knowledge, it is also the first report of a compound heterozy-gous mutation in the GLUT9 gene.
Our findings have several important implications. First, they support the speculation proposed previously that urate efflux may be mediated solely by basolateral GLUT9L.19Although we did not perform a functional analysis of the mutation, it is highly likely that our patient showed loss of function of GLUT9. Considering the lack of impor-tant functional portions and the large duplication of GLUT9, the mutations were thought to have rendered the transporter almost nonfunctional,17–19 which would have explained the pa-tient’s low serum urate level of 5.9
mol/L (0.1 mg/dL). GLUT9S cooper-ates with URAT1 on the apical side of the tubule. Therefore, mutations of the URAT1 gene, even in cases of nonsense homozygous status, would not cause such a low urate level.2In contrast, be-cause GLUT9L is thought to be the only major urate efflux transporter on the basolateral side, loss of function of GLUT9L would cause severe RHU. Therefore, when encountering pa-tients with severe hypouricemia on the order ofⱕ12mol/L (0.2 mg/dL), gene analysis ofSLC2A9seems warranted.
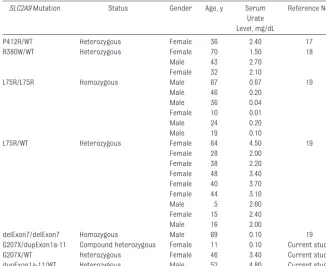
Second, our study revealed that even a nonsense mutation causing a heterozygous status ofSLC2A9did not cause severe RHU. The serum urate level in the patient’s father, who had a nonsense heterozygous mutation (p.G207X), was normal. To date, there has been no report of any such non-sense mutation. Previous reports have documented a total of 4 missense mu-tations (P412R, R380W, R198C, and L75R) and a deletion of exon 7 of
SLC2A9(Table 1). Mutations of only 1 allele cause slight (89 –268 mol/L [1.5– 4.5 mg/dL]), but not severe, hy-First
Onset
Follow-up
Second
Onset
Follow-up
T1WI T2WI FLAIR
FIGURE 1
Brain MRI. MRIs reveal a high signal intensity area in the occipital lobe on T2-weighted (T2WI) and fluid-attenuated inversion recovery (FLAIR) images and a low signal intensity area on T1-weighted images (T1WI) at onset of the first and second episodes (arrows). Follow-up MRIs reveal complete resolution of the abnormal lesions.
pouricemia. These hypouricemia lev-els are similar to the values found in patients with RHU with a URAT1 heterozygous nonsense mutation such as W258X.2Accordingly, when a muta-tion is found in only 1 allele ofSLC2A9
in patients with severe hypouricemia, intensive analysis including transcrip-tion should be performed to reveal whether any other mutation is present in the other allele.
Third, in our patient, the recurrence of EIARF concurrent with PRES was of in-terest. Although we did not obtain di-rect proof that PRES was caused by the severe hypouricemia or a compound
heterozygousSLC2A9mutation, the re-current episodes in our patient at least suggested a relationship between hy-pouricemia itself or anSLC2A9 muta-tion and PRES. PRES was initially de-scribed by Hinchey et al20 as a condition characterized by headache, altered mental function, seizures, and visual disturbance and associated with reversible white matter lesions, predominantly in the posterior region of the cerebral hemispheres on brain computed-tomography scan or MRI. This syndrome is often seen in patients who have acute kidney injury, hyper-tension, or immunosuppression,
espe-believed to be associated with vaso-genic edema caused by destruction of the blood-brain barrier.20It is surpris-ing that no report to date has docu-mented the concurrent manifestation of hypouricemia and PRES, although the URAT1 gene mutation causing hy-pouricemia is not so rare.10,11 This means that the extent of hypouricemia caused by the URAT1 gene mutation is not a risk factor for PRES. On the basis of the findings in our case, 2 interpre-tations can be suggested: (1) severe hypouricemia caused by the GLUT9 gene mutation is a risk factor for exer-tional PRES; and (2) GLUT9 dysfunction is a specific risk factor for exertional PRES. Although there is a possibility that EIARF itself was a major risk factor for PRES in our patient, it seems un-likely, because in the second episode PRES occurred without apparent hy-pertension (fluid excess) or severe el-evation of urea and creatinine levels. It has been reported that GLUT9 is also expressed in the brain.23 GLUT9 may play a specific role in exercise-induced PRES. Accumulation of more cases and investigations will be necessary to confirm this theory.
ACKNOWLEDGMENT
This study was supported by a Grant in Aid for Young Scientists (B-19790720) (to Dr K. Nozu) from the Japan Society for the Promotion of Science.
REFERENCES
1. Ames BN, Cathcart R, Schwiers E, Hochstein P. Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: a hypothesis.Proc Natl Acad Sci USA. 1981;78(11):6858 – 6862
2. Komoda F, Sekine T, Inatomi J, et al. The W258X mutation inSLC22A12is the predom-inant cause of Japanese renal hypourice-mia.Pediatr Nephrol. 2004;19(7):728 –733 3. Anzai N, Kanai Y, Endou H. New insights into
renal transport of urate.Curr Opin Rheu-matol. 2007;19(2):151–157
4. Kikuchi Y, Koga H, Yasutomo Y, et al. Pa-tients with renal hypouricemia with exercise-induced acute renal failure and chronic renal dysfunction.Clin Nephrol. 2000;53(6):467– 472
5. Nakamura A, Niimi R, Yanagawa Y. Renal hy-pouricemia in school-aged children: screening of serum uric acid level before
physical training.Pediatr Nephrol. 2006; 21(12):1898 –1900
6. Takahashi T, Tsuchida S, Oyamada T, et al. Recurrent URAT1 gene mutations and prev-alence of renal hypouricemia in Japanese. Pediatr Nephrol. 2005;20(5):576 –578
7. Tanaka M, Itoh K, Matsushita K, et al. Two male siblings with hereditary renal hy-pouricemia and exercise-induced ARF.Am J Kidney Dis. 2003;42(6):1287–1292 Level, mg/dL
P412R/WT Heterozygous Female 36 2.40 17
R380W/WT Heterozygous Female 70 1.50 18
Male 43 2.70
Female 32 2.10
L75R/L75R Homozygous Male 67 0.67 19
Male 46 0.20
Male 36 0.04
Female 10 0.01
Male 24 0.20
Male 19 0.10
L75R/WT Heterozygous Female 64 4.50 19
Female 28 2.00 Female 38 2.20 Female 48 3.40 Female 40 3.70 Female 44 3.10
Male 5 2.60
Female 15 2.40
Male 16 2.00
delExon7/delExon7 Homozygous Male 69 0.10 19 G207X/dupExon1a-11 Compound heterozygous Female 11 0.10 Current study G207X/WT Heterozygous Female 46 3.40 Current study dupExon1a-11/WT Heterozygous Male 52 4.80 Current study
8. Ohta T, Sakano T, Ogawa T, et al. Exercise-induced acute renal failure with renal hypouricemia: a case report and a review of the literature.Clin Nephrol. 2002;58(4): 313–316
9. Ishikawa I. Acute renal failure with severe loin pain and patchy renal ischemia after anaerobic exercise in patients with or with-out renal hypouricemia.Nephron. 2002; 91(4):559 –570
10. Cheong HI, Kang JH, Lee JH, et al. Mutational analysis of idiopathic renal hypouricemia in Korea. Pediatr Nephrol. 2005;20(7): 886 – 890
11. Wakida N, Tuyen DG, Adachi M, et al. Muta-tions in human urate transporter 1 gene in presecretory reabsorption defect type of fa-milial renal hypouricemia.J Clin Endocrinol Metab. 2005;90(4):2169 –2174
12. Ohta T, Sakano T, Igarashi T, Itami N, Ogawa T. Exercise-induced acute renal failure associ-ated with renal hypouricaemia: results of a questionnaire-based survey in Japan.Nephrol Dial Transplant. 2004;19(6):1447–1453
13. Enomoto A, Kimura H, Chairoungdua A, et al. Molecular identification of a renal urate an-ion exchanger that regulates blood urate levels.Nature. 2002;417(6887):447– 452 14. Ichida K, Hosoyamada M, Hisatome I, et al.
Clinical and molecular analysis of patients with renal hypouricemia in Japan: influence of URAT1 gene on urinary urate excretion.J Am Soc Nephrol. 2004;15(1):164 –173
15. Sperling O. Hereditary renal hypouricemia. Mol Genet Metab. 2006;89(1–2):14 –18
16. Vitart V, Rudan I, Hayward C, et al.SLC2A9is a newly identified urate transporter influ-encing serum urate concentration, urate excretion and gout.Nat Genet. 2008;40(4): 437– 442
17. Anzai N, Ichida K, Jutabha P, et al. Plasma urate level is directly regulated by a voltage-driven urate efflux transporter URATv1 (SLC2A9) in humans.J Biol Chem. 2008; 283(40):26834 –26838
18. Matsuo H, Chiba T, Nagamori S, et al. Muta-tions in glucose transporter 9 geneSLC2A9
cause renal hypouricemia.Am J Hum Genet. 2008;83(6):744 –751
19. Dinour D, Gray NK, Campbell S, et al. Ho-mozygousSLC2A9mutations cause severe renal hypouricemia. J Am Soc Nephrol. 2010;21(1):64 –72
20. Hinchey J, Chaves C, Appignani B, et al. A reversible posterior leukoencephalopathy syndrome. N Engl J Med. 1996;334(8): 494 –500
21. Berden JH, Hoitsma AJ, Merx JL, Keyser A. Severe central-nervous-system toxicity as-sociated with cyclosporin. Lancet. 1985; 1(8422):219 –220
22. Rubin AM, Kang H. Cerebral blindness and encephalopathy with cyclosporin A toxicity. Neurology. 1987;37(6):1072–1076
23. Augustin R, Carayannopoulos MO, Dowd LO, Phay JE, Moley JF, Moley KH. Identification and characterization of human glucose transporter-like protein-9 (GLUT9): alterna-tive splicing alters trafficking.J Biol Chem. 2004;279(16):16229 –16236
DOI: 10.1542/peds.2010-2592 originally published online May 2, 2011;
2011;127;e1621
Pediatrics
Matsuo, Kazumoto Iijima, Koichi Nakanishi and Norishige Yoshikawa
Services
Updated Information &
http://pediatrics.aappublications.org/content/127/6/e1621
including high resolution figures, can be found at:
References
http://pediatrics.aappublications.org/content/127/6/e1621#BIBL
This article cites 23 articles, 6 of which you can access for free at:
Subspecialty Collections
http://www.aappublications.org/cgi/collection/dysmorphology_sub
Dysmorphology
http://www.aappublications.org/cgi/collection/genetics_sub
Genetics
following collection(s):
This article, along with others on similar topics, appears in the
Permissions & Licensing
http://www.aappublications.org/site/misc/Permissions.xhtml
in its entirety can be found online at:
Information about reproducing this article in parts (figures, tables) or
Reprints
http://www.aappublications.org/site/misc/reprints.xhtml
DOI: 10.1542/peds.2010-2592 originally published online May 2, 2011;
2011;127;e1621
Pediatrics
Matsuo, Kazumoto Iijima, Koichi Nakanishi and Norishige Yoshikawa
Yuko Shima, Kandai Nozu, Yoshimi Nozu, Hiroko Togawa, Hiroshi Kaito, Masafumi
Mutation
SLC2A9
Heterozygous
Recurrent EIARF and PRES With Severe Renal Hypouricemia by Compound
http://pediatrics.aappublications.org/content/127/6/e1621
located on the World Wide Web at:
The online version of this article, along with updated information and services, is
http://pediatrics.aappublications.org/content/suppl/2011/04/21/peds.2010-2592.DC1
Data Supplement at:
by the American Academy of Pediatrics. All rights reserved. Print ISSN: 1073-0397.