Catheptic Antigen Processing
Louise Caroline Sealy
A thesis submitted for the degree of Doctor of Philosophy
1996
ProQuest Number: 10106626
All rights reserved
INFORMATION TO ALL USERS
The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed,
a note will indicate the deletion.
uest.
ProQuest 10106626
Published by ProQuest LLC(2016). Copyright of the Dissertation is held by the Author. All rights reserved.
This work is protected against unauthorized copying under Title 17, United States Code. Microform Edition © ProQuest LLC.
ProQuest LLC
789 East Eisenhower Parkway P.O. Box 1346
SUMMARY
For an antigen to be recognized by T cells, it must first be processed into short peptide
fragments and presented by an MHC molecule at the cell surface. In this thesis, the
roles o f cathepsins B, D and E in MHC class II antigen processing are investigated in
human B cells.
A vector-based antisense approach is adopted using EBV-transformed B cell
presentation o f tetanus toxoid to human T cell clones as a model system. A series of
vectors are made containing a portion of each cathepsin gene in a sense or antisense
orientation under the control o f a metallothionein promoter. Stable transfection o f
these constructs produces a panel of EBV-cell lines which can be induced to express
anti-cathepsin or control RNA. Specific reductions in cathepsin protein levels and
activity is monitored in these lines using a variety of biochemical techniques. The
effect o f such reductions on antigen processing capacity is investigated via tetanus
toxoid presentation to T cell clones.
The antigen processing roles o f cathepsins B, D and E are also investigated by treating
B cells with short antisense oligonucleotides. Human B cells expressing the murine
MHC II molecule I-A'" are pre-treated with phosphorothioate oligonucleotides and then
challenged to present hen egg lysosyme (HEL) to murine T cell hybridomas.
The effect of soluble cathepsin inhibitors on antigen processing is also investigated for
both described antigen presentation systems, and a comparison o f the three
investigative approaches is made.
Finally, it is shown that transcription of the cathepsin E gene is upregulated upon B cell
activation (polyclonal activation o f ex vivo tonsillar B cells and PMA-activation o f an
EBV-transformed B cell line). It is also shown that cells that do not usually present
antigen (chondrosarcoma and HeLa cells) can be induced to express cathepsin E when
ACKNOWLEDGEMENTS
Firstly I would like to acknowledge the Arthritis and Rheumatism Council for financial support.
I would like to thank all the members o f the Department o f Immunology for their help. In particular I must mention Joao Kanan, Lee Faulkner, Elaine Hughson, Sunethra Wimalasundera, Mike Binks, Anna Vyakamam, Linda Ho and David Katz.
CONTENTS
Page
Title page 1
Summary 2
Dedication 3
Acknowledgements 4
List of contents 5
List of figures 11
List of tables 14
Chapter 1: Introduction 15
1.1 Antigen processing 16
1.1.1 The MHC II processing pathway 17
l .l .I .l Uptake of exogenous antigen 18 1.1.1.2 Degradation within an acidic compartment 20
1.1.1.3 The role o f invariant chain 21
1.1.1.3 .1 MHC class II assembly 21 1.1.1.3 .2 Occlusion of the peptide binding site 22 1.1.1.3.3 Targeting to endocytic pathway 24 1.1.1.3 .4 Invariant chain-independent antigens 25 1.1.1.4 Peptide binding by MHC class II 25 1.1.1 .5 Export o f peptide;MHC II to the cell surface 26
1.1.2 Epitope Hierarchy 27
1.1.2.1 Selective binding by MHC class II 27 1.1.2.2 Modulation by antigen-binding proteins 28 1.1.2.3 A possible role for cathepsins in the establishment 29
of epitope hierarchy
1.2 Cathepsins 30
1.2.1 Cathepsin B 30
1.2.1.1 Structure 31
1.2.1.2 Synthesis, targeting and activation 33
1.2.1.3 Extracellular cathepsin B 35
1.2.1.4 Multiple cathepsin B transcripts and isoforms 3 5 1.2.1.5 Rational inhibitor design for cathepsin B 36
1.2.2 Cathepsin D 39
1.2.2.1 Structure 41
1.2.2.2 Synthesis and lysosomal targeting 43 1.2.2.3 Transcriptional regulation o f the cathepsin D gene 44 1.2.2.4 Aspartyl proteinase inhibitors 44 1.2.2.5 The cathepsin D knockout mouse 47
1.2.3 Cathepsin E 48
1.2.3.1 Structure 49
1.2.3.2 Tissue distribution and intracellular location 53
1.2.3.3 Activity 54
1.2.3.4 Inhibitors 55
1.2.4 The role o f cathepsins in antigen processing 56
1.2.4.2 Antigen processing 57 1.2.4.2.1 Circumstantial evidence 58 1.2.4.2.2 Evidence from inhibitor studies 59 1.2.4.2.S Evidence from in vitro processing studies 62 1.2.4.2.4 Significance of catheptic cleavage motifs 63
1.3 The Antisense Approach 66
1.3.1 Introduction of an antisense sequence 66 1.3.1.1 The oligonucleotide-based approach 66
1.3.1.2 The vector-based approach 70
1.3.2 Possible target sites 71
1.3.2.1 Transcription 71
1.3.2.2 Splicing 73
1.3.2.3 Translation 73
1.3.3 Attachment of functional groups to oligonucleotides 75
1.3.4 Potential therapeutic use 75
1.3.5 Why choose an antisense approach to study the roles of 76 cathepsins B, D, and E in antigen processing?
Chapter 2: Materials and methods 80
2.1 Cell Biology 80
2.1.1 Cell culture 80
2.1.1.1 Maintenance of human T cell clones 80
2.1.2 Isolation of primary cells 80
2.1.2.1 Isolation of B cells from human tonsils 80 2.1.2.2 Isolation of PBMCs from human blood 82
2.1.3 Activation of cells 82
2.1.3.1 Polyclonal activation of tonsillar B cells 82 2.1.3.2 PMA-treatment of EBV B cell lines 83 2.1.3.3 IFNy-induction of chondrosarcoma cells 83 2.1.3.4 IFNy-induction of HeLa cell mutants 83
2.1.4 Transfection 83
2.1.5 Cloning 83
2.1.6 Treatment of cells with soluble cathepsin inhibitors 84 2.1.7 Treatment of cells with oligonucleotides 84 2.1.7.1 Preparation of oligonucleotides 84 2.1.7.2 Oligonucleotide treatment of cells 85
2.2 Molecular biology 86
2.2.1 Isolation of RNA from cells 86
2.2.2 RT-PCR 86
2.2.2.1 cDNA preparation 86
2.2.2.2 PCR 86
2.2.2.3 “Hot-starf ’ PCR 86
2.2.3 Cloning 88
2.2.3.1 TA cloning 88
2.2.3.2 Ligation into pMEP4 88
2.2.4 Isolation of plasmid DNA 89
2.2.5.1 Production of electrocompetent E.coli 90
2.2.5.2 Electroporation 90
2.2.6 DNA sequencing 90
2.2.7 Northern hybridization 91
2.2.7.1 Northern blotting 91
2.2.7.2 Stripping 92
2.2.7.3 Synthesis o f RNA probes 92
2.3 Protein chemistry 94
2.3.1 The Pierce protein concentration assay 94
2.3.2 Purification of tetanus toxoid 94
2.3.3 Biotinylation 94
2.3.4 '“ l-labeling 94
2.3 . 5 '“‘C-labeling o f haemoglobin 95
2.3.6 Immunoprécipitation 95
2.3.6.1 ^^S-metabolic labeling 95
2.3.6.2 Immunoprécipitation 95
2.3.6 3 SDS-PAGE 96
2.3.6.4 Cathepsin D half life 97
2.3.7 Western Blotting 97
2.3.7.1 Lysis, SDS-PAGE and membrane transfer 97
2.3.7.2 Immunodetection 97
2 3 .1 3 ECL 98
2.3.7.4 Stripping 98
2.4 Histochemical assays 98
2.4.1 Fluorescence methods 98
2.4.1.1 Immunolabeling 98
2.4.1.2 Histochemical fluorescence assays 100
2.4.1.2.1 Cathepsin B 100
2.4.1.2.2 Cathepsin D/E 100
2.4.1.3 FACS analysis 100
2.4.1.4 Confocal microscopy 101
2.4.2 Binding assays 101
2.4.2.1 Biotinylated XT 101
2.4.2 2 ^^^1-labeled XT 101
2 4.2.3 Binding and internalization o f ^^^1-ahulg 101 2.4.3 ^"^C-Hb degradation assay for aspartic proteinase activity 102 2.4.3.1 ^'^C-Hb degradation by cell lysates 102 2.4.3.2 Immunoprécipitation of cathepsin D from cell 102
lysates
2.4.4.X-gal staining for |3-Gal activity 102
2.4.5 MXX assay 102
2.4.6 PCR amplification of plasmid sequences from cell lysates 103
2.5 Antigen presentation assays 103
2.5.1 Presentation of XX by FC7 cells 103
2.5.2 Presentation o f HEL or HEL-derived peptides by 103 WMPX3.3 cells
2.5.2.1 CXLL assays 103.1
2.6 Affiliations 103.1
Chapter 3: Cathepsin regulation in professional and non-professional 105 antigen presenting cells
3.1 Polyclonal activation of tonsillar B cells 106
3.1.1 Activation and characterization of cells 106 3.1.2 Analysis of mRNA levels in resting and activated cells 106 3.1.3 Cathepsin E protein levels in resting and activated cells 109 3.1.4 Cathepsin E activity levels in resting and activated cells 109 3.1.5 Antigen uptake by resting and activated cells 109 3.2 Phorbol ester-activation of EBV-transformed B cells 113 3.2.1 Analysis of cathepsin mRNA levels in PMA-treated 113
and untreated cells
3.2.2 Cathepsin E activity levels in PMA-treated 113 and untreated cells
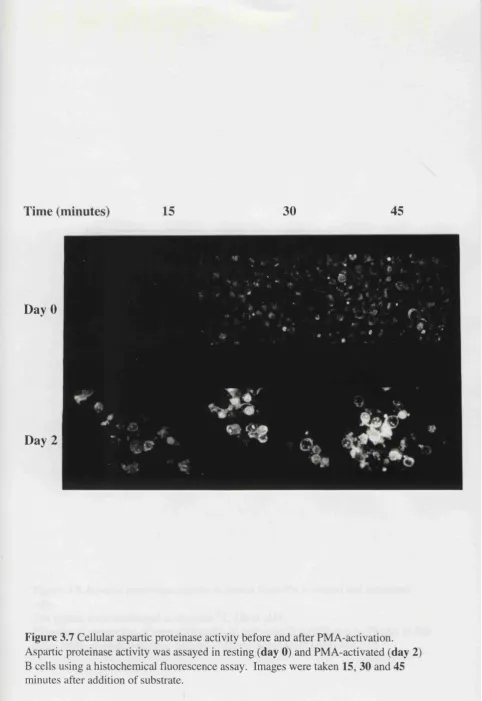
3.2.2.1 Fluorescent substrate assays 113
3.2.2.2 Degradation of ‘"^C-haemoglobin 113 3.3 IFNy-treatment of cells which do not usually present antigen 117
3.3.1 Chondrosarcoma cells 117
3.3.1.1 MHC class II production by IFNy-induced 117 chondrosarcoma cells
3.3.1.2 Cathepsin mRNA levels in IFNy-induced 117 chondrosarcoma cells
3.3.2 HeLa cells 120
3.3.2.1 Cathepsin mRNA levels in mutant HeLa cells 120 upon IFNy-induction
3.4 Discussion 123
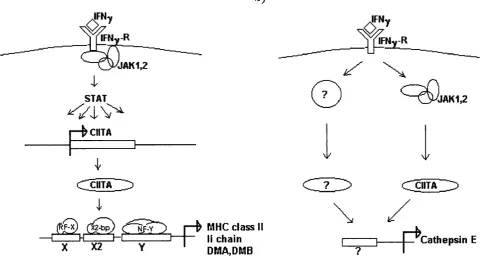
3.4.1 Cathepsin E is upregulated during B cell activation 123 3.4.2 The rate of antigen uptake decreases during B cell activation 126 3.4.3 Cathepsin E expression is induced by IFNy in cells which 126
do not usually present antigen
3.4.4 Constitutive expression of CUT A causes cathepsin E 127 expression in the absence of IFNy
3.4.5 IFNy-induction of cathepsin E does not require JAKl 128
Chapter 4: Characterization of antigen processing by B cells 131
4.1 FC7 is an EBV-transformed B cell line which binds tetanus toxoid 131
4.1.1 Presentation of TT by FC7 cells 134
4.2 WMPT3.3 is a human B cell line which expresses I-A”^ 134 4.2.1 Presentation of HEL and HEL-derived peptides by 134
WMPT3.3 cells
4.3 The characteristics of cathepsins B, D, and E in FC7 cells 138
4.3.1 Localization 138
4.3.2 Comparison of cathepsin D species in FC7 and 138 WMPT3.3 cells
4.3.3 The relative contributions of cathepsin D and cathepsin E to 138 overall aspartic proteinase activity in FC7 cells
4.5 Discussion 147 4.5 .1 The characteristics of cathepsins B, D and E in FC7 cells 147 4.5.2 Cathepsin inhibition using soluble inhibitors 150
Chapter 5: Cathepsin inhibition using a vector-based antisense approach 153 5 .1 Generation of antisense and sense cathepsin clones 153
5.1.1 Construction o f inducible antisense and sense plasmids 153 5.1.2 Production o f transfectant EBV B cells that contain antisense 155 and sense plasmids and can be induced to express the appropriate RNA 5.1.3 Cloning of parental antisense and sense cell lines 160
5.2 Analysis of CBAS and CBS clones 163
5.2.1 Cathepsin B activity in CBAS and CBS clones 163 5.2.2 Cathepsin B protein levels in CBAS and CBS clones 164
5.3 Analysis of CD AS and CDS clones 164
5.3.1 Cathepsin D protein levels in CD AS and CDS clones 164
5.3.1.1 Immunoprécipitation 164
5.3.1.2 FACS analysis and confocal microscopy 168 5.3.2 Cathepsin D activity in CD AS and CDS clones 168
5.4 Analysis o f CEAS and CES clones 170
5.4.1 Cathepsin E protein levels in CEAS and CES clones 170
5 .5 Presentation of tetanus toxoid to T cells 173
5.5.1 Parental antisense and sense lines 173
5.5.2 Antisense and sense clones 173
5.6 Discussion 178
5.6.1 Expression of antisense 178
5.6.2 Efficacy o f antisense sequence 179
5.6 2.1 Secondary structure considerations 179
5.6 2.2 Partial inhibition 181
5 .6.3 Loss of antisense expression with time 181
5 .6.4 A real antisense result? 182
Chapter 6: Cathepsin inhibition using an oligonucleotide response 183
6.1 Ohgonucleotide design 183
6.1.1 Length 183
6.1.2 Target site 184
6.1.3 Backbone modification 184
6.2 The effect of oligonucleotides on antigen processing and presentation 185 6.2.1 HEL-presentation by WMPT3.3 cells following 185
oligonucleotide treatment
6.2.2 Peptide presentation by WMPT3.3 cells following 185 ohgonucleotide treatment
6.2 .3 Encapsulation o f the oligonucleotides in cationic lipids 188 6.2.4 Time course of ohgonucleotide addition 188
6.3 Toxicity of oligonucleotides 188
6.3 .1 MTT assay 188
6.3.2 FACS analysis of ohgonucleotide treated cells 190
6 .4 Specificity of oligonucleotides 190
6.4.1 Control oligonucleotides 190
6.5 Discussion 194
Chapter 7: Conclusions 195
7.1 Alternative strategies 195
7.1.1 Tetracycline-control systems 195
7.1.2 Knockout 196
7.1.3 Protein engineering 198
7.1.4 Overexpression 198
7.2 The role of cathepsins B, D and E in antigen processing 199
References 201
Appendices: 238
Appendix 1: Abbreviations 239
Appendix 2: Vectors 241
2.1 CMVpCal 242
2.2 AVI 243
2.3 pMEP4 244
2.4 pC R II 245
List of Figures
Page
Figure . 1 The MHC class II antigen processing pathway 19
Figure .2 Crystal structure of cathepsin B 32
Figure .3 Procathepsin B primary structure 34
Figure .4 Topological schemes of E-64, CA-030 and CA-074 40
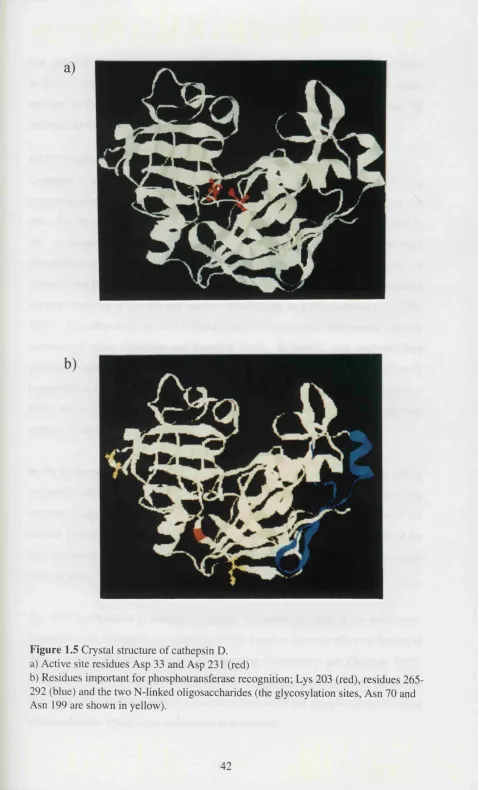
Figure .5 Crystal structure of cathepsin D 42
Figure .6 Side chain interactions between cathepsin D and pepstatin A 46
Figure .7 Procathepsin E primary structure 51
Figure .8 The cathepsin E model 52
Figure .9 Structural relationships between cathepsins and MHC II 65
Figure
Figure
molecules
.10 Oligonucleotide backbone modifications 69
.11 The possible sites of sequence-specific action for antisense 72
nucleic acids
Figure 3.1 FACS analysis of MHC class II expression during 107
SAC-activation of tonsillar B cells
Figure 3.2 Analysis of cathepsin mRNA levels during SAC-activation 108
of tonsillar B cells
Figure 3.3 Cathepsin E protein expression during B cell activation 110
Figure 3.4 Cellular aspartic proteinase activity during B cell differentiation 111
Figure 3.5 Uptake and degradation of '^^l-ahulg by resting and activated 112
cells
Figure 3.6 Analysis of cathepsin mRNA levels during PMA-activation 114
of EBV B cells
Figure 3.7 Cellular aspartic proteinase activity before and after PMA 115
activation
Figure 3.8 Aspartic proteinase activity in lysates from PMA-treated 116
and untreated cells
Figure 3.9 Immunoprécipitation of MHC class II and cathepsin D from 118
resting and IFNy-treated chondrosarcoma cells
Figure 3.10 Analysis of cathepsin mRNA levels during IFNy-induction 119
of chondrosarcoma cells
Figure 3.11 Expression of cathepsin E during IFNy-induction of mutant 121
HeLa cells
Figure 3.12 The role of CUT A in the induction of genes involved in 122
antigen processing
Figure 3.13 Stages of B cell development 124
Figure 4.1 FACS analysis of IgG/IgM expression by FC7 and 132
WMPT3.3 cells
Figure 4.2 TT binding by FC7 cells 133
Figure 4.3 FC7-mediated antigen presentation 135
Figure 4.4 Expression of I-A^ by WMPT3.3 cells 136
Figure 4.5 WMPT3.3-mediated antigen presentation 137
Figure 4.6 Localization of cathepsins B, D and E in FC7 cells 139
Figure 4.7 Immunoprécipitation of cathepsin D from FC7 and 140
WMPT3.3 cells
Figure 4.8 Aspartic proteinase activity in FC7 cell lysates after removal 142
of cathepsin D
Figure 4.9 Estimation of the half life of cathepsin D in FC7 cells 143
Figure 4.10 Effect of soluble cathepsin inhibitors on antigen presentation 145
by FC7 cells
Figure 4.11 Effect of soluble cathepsin inhibitors on antigen presentation 146
by WMPT3.3 cells
Figure 4.12 The proteolytic split sites of rat and human cathepsin D 149
Figure 5.1 Cloning strategy for the construction of inducible sense 154
and antisense plasmids
Figure 5.2 Sequence analysis of the cathepsin fragments used for 156
antisense plasmids
Figure 5.3 Confirmatory restriction digests of sense and antisense 157
plasmids
Figure 5.4 PCR analysis of parental sense and antisense cell lysates 158
without cadmium
Figure 5.7 PCR analysis of cell lysates from sense and antisense clones 162
Figure 5.8 Analysis o f cathepsin B activity in CBAS clones by 165
histochemical fluorescence
Figure 5.9 Analysis of CBAS clones by western blot 166
Figure 5.10 Immunoprécipitation o f cathepsin D from CD AS clones 167
Figure 5.11 Immunofluorescent analysis of cathepsin D inCDAS clones 169
Figure 5.12 Degradation of ^'^C-haemoglobin by CD AS clones 170
Figure 5.13 Flow cytometric analysis o f cathepsin E in CEAS clones 172
Figure 5.14 Presentation of TT by parental sense and antisense lines 174
Figure 5.15 Presentation o f TT by CBAS clones 175
Figure 5.16 Presentation of TT by CD AS clones 176
Figure 5.17 Presentation o f TT by CEAS clones 177
Figure 6.1 The effect o f antisense oligonucleotides on 186
WMPT3.3-mediated presentation o f HEL
Figure 6.2 The effect o f antisense oligonucleotides on 187
WMPT3.3-mediated presentation o f HEL-derived peptides
Figure 6.3 The time course o f oligonucleotide treatment 189
Figure 6.4 The effect of sense oligonucleotides on WMPT3.3-mediated 191
presentation of HEL
Figure 6.5 The effect o f sense oligonucleotides on WMPT3 3-mediated 192
presentation of HEL-derived peptides
Figure 6.6 Cathepsin protein levels in oligonucleotide-treated cells 193
Figure 6.7 The effect o f oligonucleotides on cell viability 193 .1
Figure 6.8 FACS analysis o f ohgonucleotide-treated cells 193.2
Figure 7.1 The reverse Tet system 197
List of Tables
Page
Table 1.1 Inhibitors of cathepsin B 37
Table 1.2 Inhibitors of cathepsin D and cathepsin E 45
Table 1.3 Inhibitor studies 60
Table 1.4 In vitro processing studies 61
Table 2.1 Cell lines: characteristics and culture conditions 81
Table 2.2 Oligonucleotides and cycling conditions used for PCR 87
Table 2.3 Synthesis of RNA probes 93
Table 2.4 Antibodies used to preload protein A sepharose for 96
immunoprécipitation
Table 2.5 Antibodies and conditions used for FACS analysis 99
CHAPTER 1
INTRODUCTION
For an antigen to be recognized by T cells, it must first be processed into small
fragments and presented in conjunction with an MHC molecule on the surface of an
antigen presenting cell. The enzymes involved in antigen processing for presentation
by MHC class II molecules are unknown, but several members of the cathepsin family
of proteinases have been implicated. The aim of this thesis was to analyze the
properties of cathepsins B, D and E in B cells, and to examine their role in antigen
processing by blocking the production of each enzyme individually using an antisense
approach. The introduction therefore covers three topics: the MHC class II antigen
processing pathway, the characteristics of cathepsins B, D and E, and the antisense
approach.
Chapter 1.1 Antigen processing
Unlike B cells, which can only recognize epitopes that are accessible on the antigen in
its native form, T cells have the capacity to recognize internally-derived epitopes that
are usually concealed by protein folding. T cells are therefore very powerful in
immune defence as they prevent foreign entities from escaping detection by
modification of their external epitopes to be more like those o f the host or by hiding
inside host cells.
For a T cell to recognize such an internal epitope, the antigen must first be internalized
and degraded into fragments by an antigen presenting cell. The derived peptides are
bound by MHC-encoded class I and class II molecules which transport and display
them on the cell surface. Endogenously-derived antigens, such as viral proteins, are
predominantly presented by MHC class I molecules, which can be expressed on nearly
all cell types. Presentation of exogenously-derived antigens usually occurs in
conjunction with MHC class II molecules, whose expression is limited to so-called
professional antigen presenting cells but can be induced on other cell types by the
cytokine y-interferon. MHC class I- and MHC class Il-associated antigen presentation
have different pathways o f antigen processing. A multisubunit catalytic assembly
known as the proteasome is thought to be responsible for antigen degradation in the
class I pathway, although some associated trimming activity may also be involved
(Elliott et al 1995). No equivalent specialized degradative machinery has been found for the class II pathway. The most widely accepted view is that it is mediated by the
concerted action o f various members of the cathepsin family o f proteinases, which also
fijlfill other intracellular functions.
In general, MHC class II molecules present antigens to helper (CD4+) T cells. The
MHC II:peptide complex interacts with the TCR CD4 on the T cell surface and causes
the T cell to secrete various cytokines. IL-2 is produced which induces proliferation
activate B cells. Hence antigen processing for presentation to helper T cells is central
to triggering of a coordinated immune response.
MHC Il-restricted antigen presentation is also important in T cell development.
During maturation in the thymus, T cells undergo two selective processes; positive
selection, in which they are screened for self MHC restriction, and negative selection
which eliminates those cells that are specific for self peptides bound to self MHC.
Antigen processing and presentation by MHC class II is important for both positive
(von Boehmer 1994) and negative (Nossal 1994) selection, and disruption of antigen
processing can seriously alter the T cell repertoire.
Finally, some autoimmune diseases result from recognition of self molecules by T cells
which then mount an anti-self immune response (Lanzavecchia 1995). For example,
type II collagen is the arthritogenic antigen for collagen-induced arthritis, an
experimental autoimmune disease which mimics the inflammatory articular
manifestations of rheumatoid arthritis (RA) (Wooley et al 1985). Hence in humans, RA may be caused by aberrant processing of type II collagen (Londei et at 1989). It is therefore of interest to study MHC II antigen processing to know more about normal
immune responses, T cell selection and certain autoimmune diseases.
1.1.1 The MHC II processing pathway
Three cell types are principally responsible for antigen presentation via the MHC class
II pathway: dendritic cells, B cells and macrophages. Dendritic cells have abundant
MHC class II surface expression (Crowley et al 1989) and express certain costimulatory molecules constitutively (Larsen et al 1992) making them potent APC. It has been proposed that dendritic cells are the only antigen presenting cell type that
can prime naive T cells (Levin et al 1993), so that B cells and macrophages can only act as APC for memory or activated T cells. This is still a matter for debate (Fuchs
and Matzinger 1992, Lin et al 1991), particularly with respect to the capacity of dendritic cells to prime responses against intact protein antigens (Constant et al 1995a, 1995b). B cells and macrophages only express certain costimulatory molecules upon
activation. B cells are particularly efficient at presentation of soluble antigens such as
bacterial toxins, whereas macrophages can actively ingest microbes and particulate
antigens.
Although it has been known that antigens can be presented in conjunction with MHC
class II for many years, the antigen processing pathway is still not completely
characterized. Much research effort is currently being devoted to identification of
proteins and compartments that may be involved in the pathway and elucidation of
their functional roles. A schematic representation of the currently accepted view is
shown in Figure 1.1.
l . l . I . l Uptake of exogenous antigen
All cell types have the capacity to take up antigen constitutively from the fluid phase by
micropinocytosis. Invagination of the plasma membrane occurs forming a clathrin-
coated pit and enclosing the antigen in a small intracellular vesicle (-0.1 pm).
However, micropinocytosis is slow and inefficient and professional APCs have other
strategies for antigen uptake which reflect their specialized roles in vivo.
Dendritic cells (Sallusto et al 1995) and macrophages (Racoosin and Swanson 1993) can capture large amounts of antigen by macropinocytosis, where large vesicles (0.5-
3pm) are taken up in a process mediated by membrane ruffling which is driven by the
cytoskeleton. Large amounts of fluid are taken up by this process and macrosolutes
can be concentrated in a MHC-II and proteinase-rich compartment. Macrophages are
also particularly adept at non-specifically internalizing antigen by phagocytosis where
the foreign particle is surrounded by pseudopods which fuse to form a phagocytic
vacuole (Unanue 1984).
Dendritic cells can also capture antigen via the mannose receptor (MR), which is
expressed at high levels on the cell surface (Sallusto et al 1995). A wide variety of antigens can be internalized by binding the MR, and it offers a degree of selectivity to
7
^,
\o
ER
EE LE?
1. Receptor-m ediated endocytosis 2. Synthesis o f M H C class II 3. Peptide loading
MHC
TGN
2
KEY
Antigen
RIHC class n Peptide
nilgG
Igof Ig^ CLIP
B cells can internalize antigen specifically via membrane-bound Ig (mig) (Lanzavecchia
1985, Abbas et al 1985). B cells express clonally distributed mIg molecules that are non-covalently associated with two heterodimeric chains Ig a and Igp, forming the B
cell receptor (BCR). Receptor-mediated endocytosis via the BCR is highly efficient
because mig acts as a high-affinity receptor, but also because mig specifically targets
bound antigen to the intracellular compartment where peptide-MHC II association
occurs (West et al 1994). The cytoplasmic tails of both Ig a and Igp have been shown to mediate efficient mig-antigen internalization, but Ig a seems to be responsible for
targeting the antigen to the peptide-loading compartment (Bonnerot et al 1995). The transmembrane region of mig itself may also be important in the targeting process
(Mitchell et al 1995). After internalization of mIg-antigen complexes, mig is either degraded along with antigen in the endosomal/lysosomal pathway (Lanzavecchia 1999)
or it recycles back to the cell surface where it can bind additional antigen (Watts and
Davidson 1988, Davidson et al 1990).
Antigen acquisition via surface immunoglobulin is 10^ to lO'^-fold more efficient than
for B cells with an irrelevant surface antibody (Lanzavecchia 1990). Therefore for a
CD44- T-cell to provide useful help for antibody production, it must focus on a B cell
with the proper specificity. The fact that the antigen is attached to Ig influences its
degradation as well as its uptake mechanism (section 1.1.2).
Cells with Fc receptors, such as macrophages, can use \cytophilic antibody to capture
antigen and internalize the Ig:antigen complex in an analogous manner to B cells. The
increased efficiency of presentation of antigens internalized by receptor-mediated
endocytosis may potentially be exploited (Scardino et al 1994) and could be useful for immunotherapeutic approaches.
1.1.1.2 Degradation within an acidic compartment
The antigen-containing vesicle passes along the endocytic pathway and becomes
progressively more acidic, taking on the characteristics of an early and then a late
processing (section 1.2.4) which are present in endosomal and lysosomal
compartments. Cathepsins B and D and MHC class II molecules have been co
localised in an early endocytic compartment in human B lymphoblastoid cells
(Guagliardi et al 1990) suggesting that this is the site for antigen degradation. Although proteinases have been identified in low density endosomes and lysosomes of
B cells, no processed peptide or peptide-class II complexes were found in the
lysosomes when pulsed with antigen (Barnes and Mitchell 1995). Moreover, blocking
intracellular transport of internalized antigen from low density endosomes to lysosomes
does not inhibit the generation of processed antigen in this system (Barnes and Mitchell
1995). Antigen associated with mig in B cells therefore seems to be efficiently
processed in an endosomal compartment without the contribution of proteinases
present within denser organelles. MHC:peptide association occurs optimally at acidic
pH (4.5) (Mouritsen et al 1992a), which is also consistent with this event occurring in an endosomal or lysosomal compartment. However, there is now evidence that
peptide binding occurs within a distinct compartment within B cells which is distinct
from endosomes and lysosomes (section 1.1.1.4).
1.1.1.3 The role of invariant chain
MHC class II proteins assemble in the ER as a stoichiometric complex with a non-
polymorphic type II integral membrane protein known as the invariant chain (li)
(Cresswell et al 1990). The nine-chain complex, formed by the association of three class II aP dimers with a preformed li trimer, is transported through the Golgi. Within
the trans-Golgi network it appears to segregate from the default transport pathway to
the plasma membrane (Peters et al 1991) and is targeted to the endocytic compartment. Invariant chain plays a critical role in MHC class II assembly, shielding
of the peptide-binding site (Romagnoli and Germain 1994) and targeting of MHC class
II to endocytic compartments (Lotteau et al 1990, Simonsen et al 1993).
1.1.1.3.1 MHC class II assembly
Class Il-expressing li-negative fibroblasts (Layet and Germain 1991, Anderson and
Miller 1992) and B lymphocytes from li-deficient mice (Viville et al 1993, Bikoff et al 1993) show decreased assembly of class II a and p chains. The class II dimers that do
form are inefficiently transported to the cell surface and appear to be empty (unstable).
Invariant chain seems to act as a framework for assembly of class II a p dimers (Lamb
and Cresswell 1992, Roche et al 1991), and promotes correct post-translational processing (Schaiff et at 1991). Calnexin may also be important for class II assembly (Schreiber et al 1994). In kinetic studies it is found in association with free p chain within three minutes of class II synthesis. Five minutes later it is associated with the
assembling a p li complex (Anderson and Cresswell 1994). Calnexin appears to
dissociate when the third a p dimer is added to the li trimer and coincides with egress
of the complex from the ER (Anderson and Cresswell 1994). Therefore calnexin may
function to retain a and p chains and assembling a p ii in the ER and also stabilize the
assembly intermediates. A recent study involving unglycosylated E (which is
unrecognizable by calnexin) has shown that inhibition of the E-calnexin interaction
enhances E breakdown but does not prevent the assembly of apE complexes
(Romagnoli and Germain 1995). This data would suggest that calnexin is not an
absolute requirement for assembly of class II:E nonamers but acts primarily to retain E
in the ER and inhibit its degradation.
1.1.1.3.2 Occlusion of the peptide binding site
The peptide binding site of MHC class II has a very open design (section 1.1.2.1)
which permits it to interact with segments of intact proteins. Invariant chain blocks the
binding site to avoid persistent, unproductive interaction with intact proteins in the ER
(Roche and Cresswell 1990, Teyton et al 1990). When the MHC II-E complex reaches the endosomal compartment, a combination of acidic pH and specific
proteinases (section 1.2.4.1) progressively cleaves the COOH-terminal portions of E
(Blum and Cresswell 1988) so only a portion is left attached to the class II molecule.
Invariant chain possesses a short internal segment, the CLIP (class Il-associated
invariant chain peptide (Riberdy et al 1992)) which has been shown to associate with murine and human class II molecules (Hunt et al 1992, Chiez et al 1993) and compete with antigenic peptide for class II binding. CLIP consists of a nested set of peptides
derived from amino acids 80-104 of E. It is now widely accepted that CLIP occludes
biosynthesis, promoting the formation of stable dimers. Alignment studies suggest that
CLIPs are designed for promiscuous binding in the groove o f many MHC class II
molecules by taking advantage o f one or more supermotifs (Malcherek et al 1995). Binding of CLIPs to MHC class II is reversible and fairly weak, in an analogous way to
anchor-amputated ligands, allowing them to bind many different MHC alleles but none
with high affinity.
Removal of CLIP from class II a p is catalyzed by DM (Sloan et al 1995, Denzin and Cresswell 1995, Sherman et al 1995) a heterodimeric transmembrane glycoprotein encoded by a gene that maps to the class II region of the MHC (Morris et al 1994, Fling et al 1994). A number of mutant cell lines have been characterized containing lesions in DM a or DMp genes which are defective in presentation of intact protein
antigens but can present exogenously supplied peptides (Mellins et al 1990). Essentially all o f the class II molecules at the cell surface of mutant cells are associated
with CLIPs (Riberdy et al 1992, Sette et al 1992). Transfection of DM genes into such mutants restores the wild-type phenotype (Morris et al 1994). Mice lacking H2- M (the murine equivalent of DM) have recently been generated (Fund-Leung et al
1996, Martin et al 1996, Miyazaki et al 1996). Cells fi'om these animals express normal amounts of class II at the cell surface, but it is predominantly bound by CLIP.
Such cells are unable to present proteins to class Il-restricted T cells and show reduced
capacity to present exogenous peptides. The observed phenotype o f DM-deficient
human and murine cells is consistent with DM having an essential role in removing
CLIP from class II molecules.
There is also some evidence that DM can enhance the release o f non-CLIP peptides
from MHC class II molecules and facilitate the binding of other peptides (Sloan et al 1995). DM may act as a peptide "editor", catalyzing the release o f pre-bound peptides
and ensuring that only long-lived class Il-peptide complexes escape to the cell surface
(Sloan et al 1995). DM is thought to function catalytically, as its concentration is relatively low but its half life is long (Denzin et al 1994). DM-associated CLIP removal has an acidic pH optimum consistent with its occurrence in a lysosome- or
endosome-like compartment in vivo (section 1.1.1.4) (Denzin and Cresswell 1995).
Antibody blocking experiments suggest that CLIP-removal requires a transient
interaction between HLA-DM and the MHC II-CLIP complex.
1.1.1.3.3 Targeting to endocytic pathway
Invariant chain can localize to the endocytic compartment independently of class II
molecules, due to a signal sequence in the amino-terminal chain of the molecule
(Bakke et al 1990, Lotteau et al 1990, Romagnoli et al 1993). Also co-expression of li has been shown to enhance localization of class II molecules to endocytic
compartments (Lamb et al 1991) suggesting that li possesses an endosomal sorting signal which can direct the lixlass II complex. Deletion of the N-terminus results in
the expression of li alone (Bakke et al 1990, Lotteau et al 1990) or of class ILIi complexes (Roche et al 1992, Anderson et al 1993) at the cell surface. There are two signals in the amino-terminus, one at residues 7 and 8 and the other involving residues
15-17 which may belong to a family of dileucine-based intracellular sorting motifs
(Pieters et al 1993, Odorizzi et al 1994). In addition to the two signals in the cytosolic chain that promote endosomal retention (Loss and Sant 1993, NeeQes and Ploegh
1992) there may be another signal in the transmembrane region of li (Odorizzi et al 1994). It is possible that these multiple signals found at different positions on li are
recognized at different points along the biosynthetic pathway. Some signals will be
removed early during proteolysis, but those in the N-terminus (the last portion to
dissociate from the class 11 molecule) may function after initial proteolysis.
There are two alternatively spliced forms of the li gene product; p31 and p41. The
p41 isoform is mainly responsible for enhancing antigen presentation (Peterson and
Miller 1992). To date, the only detectable biochemical difference between p31 and
p41 is that amino-terminal fragments of li (plO and p i 2) remain associated with class
11 molecules for extended times in cells expressing p41, compared with those
expressing p31 (Fineschi et al 1995). It has recently been suggested that p41 acts as a proteinase inhibitor allowing the generation of p i 2, and also controls the processing of
1.1.1.3 .4 Invariant chain-independent antigens
In studies of both li-negative mice and transfected cell lines, li has been shown to
enhance the presentation of some, but not all, antigens. Some antigens appear to be
presented equally well by class II whether li is present or not (Viville et al 1993, Nadimi et al 1991, Peterson et al 1992). In these cases the class II molecule must be able to access the endocytic pathway without the assistance o f invariant chain. The
mechanism by which this occurs is controversial. One possibility is that the MHC II
molecule is newly synthesized and reaches the endosome by an as yet unknown
mechanism (Swier and Miller 1995). The other possibility is that class II can recycle
from the cell surface. Ultrastructural studies have provided evidence that MHC II can
be internalized from the cell surface into endocytic compartments (Harding et al 1990). MHC class II appears to be exported as a class Il-Ii complexes and then rapidly
internalized into early endosomes (Roche et al 1993, Romagnoli et al 1993). Truncation of the cytosolic tail of class II inhibits internalization from the cell surface
and inhibits presentation of the li-independent antigen influenza haemagglutinin (Pinet
et al 1995). Presentation o f the li-dependent matrix protein from the same virus particle is unaffected, providing strong evidence for li-independent antigen
presentation by recycled class H molecules.
1.1.1.4 Peptide binding by MHC class H
A novel compartment, distinct from endosomes and lysosomes, has recently been
identified and is thought to be the site of MHC class II: peptide binding (Amigorena et al 1995). The MHC (or CIIV) is a class Il-rich compartment with an unusual multimembraneous structure. It was first described by immunoelectron microscopy in
human B cells (Peters et al 1991) and subsequently in mouse macrophages (Harding and Geuze 1993) and B cells (Qiu et al 1994, Amigorena et al 1994) using subcellular fractionation. Recently, immunogold labeling of ultrathin cryosections has revealed the
presence of MllC-like compartments in dendritic cells (Kleijmeer et al 1995). MIIC is a site for both the proteolytic cleavage o f invariant chain from class II molecules and
the binding of peptide (Amigorena et al 1995, Xu et al 1995). DM resides exclusively in the MIIC (Sanderson et al 1994, Denzin et al 1994) due to a MllC-targeting motif in the DMp chain (Marks et al 1995). In DM negative cells, class Il-Ii complexes
accumulate in large acidic vesicles which contain MllC-like markers but have a
different morphology (Riberdy et al 1994).
Interestingly, the potential to develop MIIC does not seem to be restricted to
professional antigen presenting cells. Transfection with genes encoding class II a and
p chains causes the generation of MllC-like compartments (Calafat et al 1994) and MIIC have also been identified in melanoma cells (Tulp et al 1994).
MIIC may not be the only site for MHC-peptide binding. Recent evidence from high-
resolution density gradient protocols indicates that class II molecules traffic through
many different compartments in the endocytic route and that peptide can be bound to
class II in multiple locations (Casatellino and Germain 1995).
Peptide binding is enhanced at low pH (Mouritsen et al 1992) and the optimal pH is similar to that found in MIIC compartments (about pH 4.5). It has recently been
shown that some MHC II molecules undergo a conformational change in the pH range
where antigen binding is enhanced (Boniface et al 1996, Runnels et al 1996) suggesting that they may transiently exist in a partially unfolded state which facilitates
peptide binding. The intermediate unfolded state may also be important for CLIP
release.
1.1.1.5 Export of peptideiMHC II to the cell surface
In macrophages, vesicles from class II enriched phagolysosomes are thought to
provide transport for MHC IPpeptide complexes to the plasma membrane (Harding
and Geuze 1992). MIIC compartments in B cells have a limiting outer membrane
enclosing characteristic membrane vesicles, both of which express class II. It has
recently been shown by immunoelectron microscopy that the hmiting membrane can
fuse with the plasma membrane and release the internal MHC-containing vesicles
1.1.2 Epitope Hierarchy
Cell surface presentation of peptides derived from a single antigen is not random and
tends to be dominated by one or two frequently occurring epitopes. The dominance of
a particular peptide will depend on many factors, such as relative affinity for binding
the MHC molecule, spatio-temporal point of generation and resistance to degradation.
The epitope hierarchy for an antigen can change with progression of an immune
response which has important implications for self-tolerance and autoimmunity.
Dominant determinant regions that are processed and presented efficiently will act as
good inducers of tolerance, while determinant regions that are inefficiently processed
and presented will be poor tolerogens. After a primary immune response to the
immunodominant peptide, epitope "spreading" may occur to cryptic self-determinants
(Lehmann et al 1992). Presentation of sub-dominant epitopes of self proteins may trigger autoimmunity as T cells will not have been tolerized to this epitope.
1.1.2.1 Selective binding by MHC class II
MHC class II is a heterodimer of two transmembrane subunits, a (33kDa) and (3
(29kDa). X-ray crystallography has revealed that each subunit has two domains
(Brown et al 1993). The two membrane distal domains of the a and p subunits combine to form a peptide binding region, each subunit contributing one helix and four
P strands. In the centre of this region lies a peptide-binding groove. The ends of the
binding site are open allowing class Il-bound peptides to extend beyond both termini.
Consequently class Il-associated peptides have a broad range of lengths, typically 12-
24 residues (Rudensky et al 1991, Chicz et al 1992, Hunt et al 1992). The extensive sequence polymorphism characteristic of MHC class II maps largely to the peptide
binding site. The binding groove is surrounded by pockets which have different size
and chemical characteristics which exert strong preferences for interacting with certain
amino acid side chains. The specific distribution of pockets for each MHC allele
influence the range of peptides that optimally bind. Within each range there are usually
one to three anchor residues which occur with high frequency in specific peptide
positions (Falk et al 1991, Hammer et al 1993). At other positions in the peptide certain residues will be favoured or disfavoured but are not compulsory for MHC
binding. Side chain contacts differ between different alleles, and determine the peptide
sequence specificity or “m otif’ for each MHC class II. The motifs characteristic for
several class II alleles have been characterized by analysis of eluted, naturally-bound
peptides (Newcomb and Cresswell 1993a, Chicz et al 1993, Rudensky et al 1991) and by selection of large class Il-bound peptide repertoires using M13 peptide display
libraries (Hammer gf a/ 1992, 1993, 1994)
1.1.2.2 Modulation by antigen-binding proteins
Epitope hierarchy can be influenced by antigen-binding proteins such as IgG,
complement fragments (C3,C4) or az-macroglobulin. A single bound antibody has
been shown to enhance presentation of one determinant and suppress presentation of
another (Simitsek et al 1995). Biochemical analysis revealed that both the boosted and the suppressed epitopes he within the antibody's "footprint", but the mechanism of
action is unknown. The same effect is observed if the antigen is bound and internalized
via mig, or if it binds soluble antibody and is taken up "piggy-back" fashion by FcR (or
another mIg) into B cells or macrophages. The antigen determinants can therefore be
enhanced or suppressed as a direct consequence of antibody modulation. Hence the
prevailing antibody specificity can skew the T cell response towards or away from
particular determinants, and may even promote recognition of cryptic epitopes.
Covalent binding of an antigen to the complement protein C3b has been shown to
delay antigen processing leading to more gradual generation of immunogenic peptides
and increased and prolonged stimulation of specific T cell proliferation (Jaquier-Sarlin
et al 1995). Selectively delaying production of a particular epitope may bias the MHC Il-peptide selection towards its competing epitopes as their relative concentration in
the peptide-binding compartment is increased. Interestingly inhibitor and in vitro proteolysis studies indicate that free and C3b-bound antigen are degraded differently
by cathepsins B and D, suggesting that these enzymes may be responsible for the
1.1.2.3 A possible role for cathepsins in the establishment of epitope hierarchy If cathepsin enzymes are involved in antigen processing, they are likely to play a
pivotal role in establishing the hierarchy of epitopes. An individual cathepsin will
degrade an antigen into a characteristic set of fragments. Peptides that span the
fragment junctions will be cleaved and therefore disfavored, those lying within the
fragments will survive. The concerted action of two or more cathepsin enzymes will
destroy all epitopes except those that he between the sites of both enzymes. Also if a
cathepsin preferentially cleaves some sites in the antigen, then peptides released by
such cleavages may be produced first and have an advantage for MHC E-binding.
During an immune response the balance of cathepsin enzymes in the processing
compartment may change, causing quantitative and qualitative changes in the antigen
cleavage pattern. Hence, cathepsins may be involved in alteration of the peptide
repertoire available for MHC II binding and contribute to epitope spreading.
In support of this hypothesis, it has recently been shown that the epitope hierarchy of
HEL can be altered by introduction of a dibasic sequence, which is a favoured
processing site for endopeptidases such as cathepsin B (section 1.2.1.1, Takahashi et al 1989). Introduction of a dibasic sequence to the flanking region of a cryptic HEL
epitope can dramatically enhance its expression, whereas introduction of a dibasic
sequence at one end of the immunodominant epitope results in loss of expression
(Deng et al, unpublished data). Hence, introduction of a putative catheptic cleavage motif can boost or suppress expression of antigenic determinants and therefore alter
the immunodominance of an epitope.
1.2 Cathepsins
Proteinases are classified on the basis of their catalytic mechanism as belonging to one
of four classes: aspartic proteinases, cysteine proteinases, serine proteinases and
metalloproteinases (Kay 1982).
Serine proteinases have a uniquely active serine side chain at their active centre and
their catalytic mechanism involves covalent binding of substrates to this residue (Kraut
1977). Aspartic and cysteine proteinases have crucial aspartate and cysteine residues
in their active sites and will be discussed more fully in the next sections.
Metalloproteinases have metal ions (usually zinc) at the active centre. The metal ions
are integral to the structure of these proteinases and are thought to enhance the
nucleophilicity of H2O and polarize the peptide bond to be cleaved prior to
nucleophilic attack (Holmes and Matthews 1982, Bond and Benyon 1985).
Generally serine proteinases are found in secretory granules or specialized granules
(such as azurophil granules) (Bond and Butler 1987). Metalloproteinases are
associated with the ER, the plasma membrane, mitochondria or the cytosol. Only
some members of the aspartic and cysteine proteinase families are located in the
endosomal/lysosomal compartment thought to be the site of antigen processing
(section 1.1.1.2). Of the aspartic proteinases, cathepsins D and E are located in this
compartment and both of these enzymes were chosen for study in this project. Of the
cysteine proteinases, cathepsins B, L, H, N, S, M and T are found in the
endosomal/lysosomal compartment, and cathepsin B was chosen for further study as it
was deemed to have the most evidence supporting its role in antigen processing and
presentation (section 1.2.4)
1.2.1 Cathepsin B
Cathepsin B is a lysosomal cysteine proteinase belonging to the papain superfamily
Cathepsin B has been implicated in a variety of physiological processes such as general
protein turnover (Shaw and Dean 1980), bone resorption (Délaissé et al 1991), cartilage proteoglycan breakdown (Buttle and Saklatava 1992), generation of renin
from its proenzyme (Wang et al 1991), processing of thyroxin (Dunn et al 1991) and antigen processing (Guagliardi et al 1990). It has also been implicated in several pathological conditions, in particular arthritis (Esser et al 1994, Buttle et al 1993, Trabant et al 1991), muscular dystrophy (Katunuma and Kominami 1987) and tumour invasion and metastasis (Sloane 1990).
1.2.1.1 Structure
The three dimensional structure o f cathepsin B has been solved using enzyme from
human liver (Musil et al 1991) and a recombinant rat form of the enzyme (Jia et al 1995). It is roughly disc-shaped with a diameter of 50Â, a thickness of 30Â and a
marked incision representing the active site cleft (Figure 1.2). The polypeptide chain is
folded into two distinct domains which interact with one another through an extended
polar interface. The active site has a cysteine (Cys 29) and a histidine (His 199)
residue which are absolutely required for catalysis (Figure 1.2a, Hasnain et al 1992). The crystal structure of rat cathepsin B complexed with the reversible blocking reagent
pyridyl disulphide showed two large conformational changes for the residues Cys 29
and His 199 indicating potential flexibility of these side chains (Jia et al 1995). Cathepsin B exhibits optimal activity towards most substrates in slightly acidic media
(pH6); at pH values above 7.0 its activity falls sharply, irreversible inactivation occurs
due to deprotonation o f the active site His 199 (Turk et al 1994).
Overall the 3D structure o f cathepsin B is largely superimposable on that of papain, the
stereotypical cysteine proteinase. However, it has some unique features which reflect
its capacity to act as a dipeptidylpeptidase and its substrate specificity. In particular,
cathepsin B has a large insertion consisting o f an 18 residue surface loop which blocks
the primed site o f the active site cleft and is probably responsible for exopeptidase
activity. Peptide substrates are favoured which have two residues C-terminal to the
scissile bond. It is thought that two residues His 110 and His 111 in the "occluding
loop" provide positively charged anchors for the C-terminal carboxylate group of such
polypeptide substrates (Figure 1.2b). Cathepsin B has also
Figure 1.2 Crystal structure of cathepsin B.
a) Active site residues; Cys 29 (red) and His 199 (green)
been crystallized in complex with the specific inhibitors CA-074 (Yamamoto et al 1992a) and CA-030 (Turk et al 1995). CA-074 and CA-030 are E-64 derivatives which are designed to be selective for cathepsin B (section 1.2.1.5). They both have
an -Ile-Pro-OH chain replacing the terminal guanidinobutylamine of E-64, which
mimics a carboxy-terminal dipeptide. By mimicking the substrate PI' and P2' residues
CA-030 revealed the ST and S2' sites of cathepsin B in the crystal structure and
confirmed the role of His 110 and His 111 as receptors for the C-terminal carboxylic
group.
Cathepsin B is also unique in its capacity to accept basic residues in the PI and P2
positions. The region expected to define the S2 pocket contains a negatively charged
residue Glu 245 which is thought to interact with positive (particularly arginine) or
large hydrophobic (phenylalanine) P2 residues. The importance of Glu 245 in
substrate interaction is clearly illustrated by site-directed mutagenesis (Hasnain et al 1993).
1.2.1.2 Synthesis, targeting and activation
Cathepsin B is synthesized as a 45kDa inactive preproenzyme which is substituted with
two N-linked oligosaccharide units, one in the pro-region and one at Asn 113 (Figure
1.3). In the cis-Golgi the mannose residues on the carbohydrate groups are
phosphorylated by N-acetylglucosamine phosphotransferase. The marked proenzyme
is recognized by the mannose-6-phosphate receptor which diverts lysosomal enzymes
from the secretory pathway to the endocytic pathway. The importance of mannose-
6-phosphate in cathepsin B targeting is reinforced by the observation that I-cell
fibroblasts secrete 95% of cathepsin B precursor whereas normal fibroblasts secrete
less than 5% (Hanewinkel et al 1987). In I-cell disease, synthesis of mannose-6- phosphate is hindered due to a phosphotransferase deficiency resulting in secretion of
biosynthetic precursors of most lysosomal enzymes (Creek and Sly 1984).
-62 -42 _1 1 Cys29 47 50 113 254 280 pro region m ature enzym e
two-chain form m ade by cleavage and lo s s of a dipeptide
th re e dipeptides rem oved se q u en tial^
Procathepsin B is converted into the active single chain form of the mature enzyme by
limited proteolysis in an endosomal or prelysosomal compartment. Investigation of the
mechanism o f proenzyme processing has been greatly facilitated by the expression of
recombinant proenzyme in the yeast S.cerevisiae (Rowan et al 1992). In this system procathepsin B is activated by autocatalytic unimolecular processing (Mach et al 1993). Finally three dipeptides are sequentially removed by active (single chain)
cathepsin B to yield the mature protein (Rowan et al 1993). Mature cathepsin B can exist in a single chain (30kDa, 254 amino acids) or a double chain form (heavy chain
25kDa residues 50-254, hght chain 5kDa residues 1-47) made by enzymatic cleavages
between residues 47 and 50 with the loss of a dipeptide (Figure 1.3). Processing into
the two-chain form occurs in the lysosomes in a cell type specific manner (Mach et al 1992).
1.2.1.3 Extracellular cathepsin B
Only small amounts o f the latent proenzyme are secreted by default into the
extracellular fluid, although secretion of newly synthesized procathepsin B can be
elevated in response to viral transformation (Achkar et al 1990), inflammation (Mort et al 1984), or malignant dedifferentiation (Qian et al 1989). Mature cathepsin B would be expected to be inactivated extracellularly due to its instability at neutral pH and the
presence o f secreted cysteine proteinase inhibitors, such as cystatins. However,
cathepsin B can form a non-covalent complex with its pro-region (Mach et al 1994) which enables the enzyme to lie dormant in a stable form until activated by
acidification. There is thought to be an acidic microenvironment around living cells,
for example in the extracellular matrix o f cartilage. Procathepsin B has been shown to
be activated in vitro by cathepsin D or pepsin at pH3 (Dalet-Fumeron et al 1993) and by cathepsin G or elastase at neutral pH (Burnett et al 1995).
1.2.1.4 Multiple cathepsin B transcripts and isoforms
Multiple cathepsin B RNA transcripts, ranging between 1.5 to 5.0 kb in size, have been
reported in murine and human tissues. The major 2.2 kb transcript is present in normal
and cancerous tissues, and has been fully sequenced by two groups (Fong et al 1986, Chan et al 1986). Larger cathepsin B transcripts (for example, 4 and 5 kb) have been
identified which have extended 3' and/or 5' UTRs. In one report such transcripts are
reported as tumour-specific, arising from the use of alternative splice sites unique to
malignant tissues (Qian et al 1991); other reports argue that cancer cells have increased but not exclusive expression of longer mRNAs (Moin et al 1989, Sloane 1990, Mumane et al 1991). The variant messages differ in their rates of translation (Gong et al 1993) suggesting that cathepsin B expression may be regulated at the level of mRNA processing. In support of this, variant cDNAs from both normal (Tam et al
1994) and cancerous (Cao et al 1994) cells have been found to contain a lObp insertion in the 3 -UTR which permits the formation of a highly stable stem-loop
structure. Stem-loop structures can provide binding sites for regulatory proteins which
alter mRNA stability (Klausner et al 1993).
Alternative mRNA transcripts can also affect the mature protein and produce an
isoform. In adenocarcinoma cells cDNA has been isolated which encodes cathepsin B
with an extra N-glycosylation site (Cao et al 1994). In addition to altering the structure and function of cathepsin B, such a modification could affect mannose-6-
phosphate mediated trafficking of the enzyme.
Cathepsin B also has multiple forms generated by differential post-translational
processing and glycosylation of precursor protein (Buttle et al 1988, Keppler et al 1988).
1.2.1.5 Rational inhibitor design for cathepsin B
The properties of some commonly used cathepsin B inhibitors are summarized in Table
1.1.
Cystatins are naturally occurring polypeptide inhibitors of cysteine proteinases.
Cystatin C is ubiquitous in human tissues and body fluids and has a general protective
function preventing connective tissue destruction by intracellular enzymes escaping
Inhibitor
Characteristics
Method of
action
Specificity
Reference
E-64 Epoxysuccinyl peptide Irreversible active site blocking All cysteine proteinasesHanada et al 1978
CA-074, CA- 030
E-64 derivatives Specific for
CB
Murata et al 1991
Leupeptin Peptide aldehyde Reversible active site blocking All cysteine and serine proteinases Umezawa and Aoyagi 1983
Peptidyl diazo- and fluoro- methyl ketones and methanes, acyloxymethyl- ketones, nitriles and a|3- unsaturated esters Chemically modified peptides Active site blocking/ inactivation All cysteine proteinases
Watanabe et al 1979, Buus and Werdelin 1986, Smith et al 1988a,b, Krantz 1994, Liang and Abeles 1987, Govardhan and Abeles 1996 P-hydroxymercuri benzoate (pHMB) Small non-peptide based molecule Inactivation of active site cysteine Bond and Butler 1987
Alkylating reagents e.g. iodoacetate, iodo acetamide, N-ethylmaleimide ot2-macroglobulin Plasma protein Mw 725,000
Entrapment All 4 classes of proteinase
Buttle et al 1991 Cystatins Naturally occurring polypeptide inhibitors Specific
blocking of the active site
All cysteine proteinases
Musil et al 1991
Table 1.1 Inhibitors of cathepsin B
A summary of some commonly used cysteine proteinase inhibitors and their properties.
are physiological and stable in body fluids. They effectively inhibit most endogenous
cysteine proteinases (cathepsins B, H, L, and S) but it may be possible to increase their
specificity by introducing rational modifications now that the structure basis for
specificity is understood (Hall et al 1995). Such an approach may be technically difficult as cystatins are relatively large complex molecules (cystatin C is 120 residues).
Also, cystatins are predisposed to have a reduced affinity for cathepsin B compared to
other cysteine proteinases as the occluding loop interferes with cystatin binding (Musil
et al 1991).
A number of peptide-based inhibitors have been developed which inactivate the active
site cysteine, such as peptidyl diazomethyl ketones (Watanabe et al 1979, Buus and Werderlin 1986), acyloxymethyl ketones (Smith et al 1988b, Krantz et al 1988), nitriles (Liang et al 1987) and ap-unsaturated esters (Govardhan and Abeles 1996). Such reagents can be used to inhibit cathepsin B but they will also inhibit other
members of the cysteine proteinase family. The specificity of peptidyl diazo- and
fiuoro-methyl ketones can be increased by using amino acids that are favoured as
substrates for each enzyme. For example Z-Phe-Ala-CHzF is selective for cathepsin B
and Z-Phe-Phe-CHN2 is selective for cathepsin L (Green et al 1981, Kirschke and
Shaw 1981); however the degree of specificity is still fairly low and such inhibitors
would probably cross-react significantly. Also there are potential problems with using
diazomethanes intracellularly as they can be mutagenic and have toxic affects on
protein synthesis.
The most commonly used inhibitors for cathepsin B are leupeptin and E-64. Leupeptin
is a peptide aldehyde which inhibits cysteine proteinases by forming a hemithioacetal
with the active site cysteine residue (Umezawa and Aoyagi 1983). It has very broad
specificity, inhibiting both cysteine and serine proteinases (Dean 1979). E-64 (Hanada
et al 1978) is an irreversible inhibitor of plant and mammalian cysteine proteinases (Barrett 1986). It reacts exclusively with active site cysteine residues to form