Selective Inhibition of Bacterial Tryptophanyl-tRNA
Synthetases by Indolmycin Is Mechanism-based
*
Received for publication, September 5, 2015, and in revised form, November 3, 2015 Published, JBC Papers in Press, November 9, 2015, DOI 10.1074/jbc.M115.690321
Tishan L. Williams‡1, Yuhui W. Yin§, and Charles W. Carter, Jr.‡
From the‡Department of Biochemistry and Biophysics, University of North Carolina, Chapel Hill, North Carolina 27599-7260 and§Department of Pharmacology and Toxicology, University of Texas Medical Branch, Galveston Texas 77555-0144
Indolmycin is a natural tryptophan analog that competes with tryptophan for binding to tryptophanyl-tRNA synthetase (TrpRS) enzymes. Bacterial and eukaryotic cytosolic TrpRSs have comparable affinities for tryptophan (Km⬃2M), and yet
only bacterial TrpRSs are inhibited by indolmycin. Despite the similarity between these ligands,Bacillus stearothermophilus (Bs)TrpRS preferentially binds indolmycin ⬃1500-fold more tightly than its tryptophan substrate. Kinetic characterization and crystallographic analysis of BsTrpRS allowed us to probe novel aspects of indolmycin inhibitory action. Previous work had revealed that long range coupling to residues within an allosteric region called the D1 switch of BsTrpRS positions the Mg2ⴙion in a manner that allows it to assist in transition state stabilization. The Mg2ⴙion in the inhibited complex forms sig-nificantly closer contacts with non-bridging oxygen atoms from each phosphate group of ATP and three water molecules than occur in the (presumably catalytically competent) pre-transi-tion state (preTS) crystal structures. We propose that this altered coordination stabilizes a ground state Mg2ⴙ䡠ATP config-uration, accounting for the high affinity inhibition of BsTrpRS by indolmycin. Conversely, both the ATP configuration and Mg2ⴙcoordination in the human cytosolic (Hc)TrpRS preTS structure differ greatly from the BsTrpRS preTS structure. The effect of these differences is that catalysis occurs via a different transition state stabilization mechanism in HcTrpRS with a yet-to-be determined role for Mg2ⴙ. Modeling indolmycin into the tryptophan binding site points to steric hindrance and an inabil-ity to retain the interactions used for tryptophan substrate rec-ognition as causes for the 1000-fold weaker indolmycin affinity to HcTrpRS.
The accumulation of resistance in pathogenic organisms over time and with prolonged drug use necessitates the contin-ued development of new anti-infective therapeutics. Such developments can include modifications to current drugs that are active against exploited targets while counteracting current resistance mechanisms or novel compounds targeted against
underexploited targets. One group of enzyme targets that has been validated but remains underexploited is the class of ami-noacyl-tRNA synthetases (aaRSs).2Aminoacyl-tRNA syntheta-ses maintain the fidelity of the genetic code by ensuring the charging of tRNA with its cognate amino acid via the following two-step reaction.
aaRS⫹ATP⫹aa¡aaRS䡠aa-AMP⫹PPi
REACTION 1
aaRS䡠aa-AMP⫹tRNA¡aaRS⫹aa-tRNA⫹AMP
REACTION 2
All aaRS enzymes bind ATP and activate a specific amino acid by catalyzing the formation of an aminoacyl 5⬘-adenylate (aa-AMP) during the first step. This is followed by transfer of the activated amino acid to the 3⬘-end of the correct tRNA. Structural and mechanistic differences among the different aaRS enzymes as well as orthologs of individual synthetases make it possible to selectively modulate the activity of specific synthetases,e.g.prokaryotic over eukaryotic TrpRS (1). This makes the aaRS enzymes attractive targets for novel anti-infec-tive therapeutics.
Any compounds intended for clinical use must be much less inhibitory against the eukaryotic orthologs of its intended tar-get. Naturally occurring aminoacyl-tRNA synthetase inhibitors include indolmycin (TrpRS), granaticin (LeuRS), mupirocin (IleRS), and ochratoxin A (PheRS) (1– 4). Of these, mupirocin displays the required selectivity for prokaryotic over eukaryotic IleRS and has been developed for the treatment of infections in humans (5).
Indolmycin produced byStreptomyces griseusdisplays selec-tive inhibition for prokaryotic TrpRS (9 nM; Escherichia coli)
over eukaryotic TrpRS (4 mM;Bos taurus) (6). Problems with off-target effects on tryptophan metabolism have prevented its clinical use (7). However, if we could understand the molecular basis for the observed inhibition and selectivity we could exploit this information for the rational design of antibiotics targeted against TrpRS on pathogens.
Structurally, tryptophan and indolmycin are quite similar with a heterocyclic indole moiety at the root of each ligand (Fig.
*This work was supported by National Institutes of Health Grant GM90406 through the NIGMS (to C. W. C.) and an NIGMS diversity supplement (to T. W.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (code5DK4) have been deposited in the Protein Data Bank (http://wwpdb.org/).
1To whom correspondence should be addressed. Fax: 919-966-2852; E-mail:
2The abbreviations used are: aaRS, aminoacyl-tRNA synthetase; TrpRS,
tryp-tophanyl-tRNA synthetase; Bs,B. stearothermophilus; Hc, human
cytoplas-mic; LTN, tryptophanamide; IND, indolmycin; PreTS, pre-transition state; Bt,B. taurus; TEV, tobacco etch virus; OXA, methylamino-substituted oxa-zolinone ring.
1). Indolmycin differs from tryptophan in the following ways. (i) The carbon that is functionally equivalent to Cis substituted with a methyl group. (ii) The carbonyl carbon is part of an oxazolinone ring. (iii) The hydroxyl and amine groups of tryp-tophan are replaced by the nitrogen and oxygen atoms of the oxazolinone ring, respectively. (iv) The -NH-CH3 moiety attached to the oxazolinone ring does not have functionally equivalent atoms in tryptophan.
BsTrpRS is one of the most extensively characterized TrpRS enzymes. Mechanistically a Mg2⫹ion is linked to what appears
to be a dissociative transition state for tryptophan activation (8 –10). During catalysis, the Mg2⫹ion helps compensate for the increased negative charge that develops on the PPileaving group, resulting from breaking the␣P–O–P bond. The Mg2⫹ ion must move to be catalytically competent, but no protein-metal interactions have been observed in any of the BsTrpRS crystal structures determined. Instead, a remote allosteric loca-tion, the D1 switch, must undergo significant conformational change to promote the Mg2⫹ion to a catalytically competent position. The metal moves closer to the PPi leaving group, whose charge is further stabilized in the transition state by the KMSKS loop. ATP binding is required for the conformational switching between the open and closed states that allows for catalysis. ATP-dependent induced fit closing of the active site brings ATP ⬃4 Å closer to tryptophan in a predominantly translational movement mediated by relative movement of the catalytic and anticodon-binding domains.
In the absence of ATP, tryptophan binding is promoted by hydrophobic van der Waals interactions,-interactions with Phe5, and a hydrogen bond between the indole nitrogen and Asp132of the specificity helix. When both substrates bind, the tryptophan substrate undergoes a rotational movement that brings the indole ring deeper into the binding pocket and results in more stabilizing interactions between tryptophan and active site residues. This change is facilitated by the inward movement of the specificity helix that is not observed when only tryptophan is bound.
ATP-dependent induced fit rearrangement of the active site facilitates proper ATP positioning in BsTrpRS, and molecular dynamics simulations demonstrate that tryptophan is required to achieve the requisite movement of the␣P in HcTrpRS (11, 12). Even a modest substitution of tryptophanamide in place of trypto-phan prevents the repositioning of ATP. These findings support
the idea that HcTrpRS is intrinsically better at discriminating between tryptophan and its structural analogs than is BsTrpRS.
HcTrpRS uses different structural elements for substrate rec-ognition than its prokaryotic orthologs (13). Such elements include an extended N terminus with a1-2 hairpin structure shown to have a role in ATP binding as well as the amino acid activation reaction in HcTrpRS (14). In contrast to BsTrpRS, it is tryptophan binding that leads to induced fit rearrangement of the active site in HcTrpRS. There are a greater number of bind-ing determinants for tryptophan recognition as eight direct and water-mediated hydrogen bonds with polar side chains stabi-lize tryptophan in the active site. It has been proposed that amino acid activation proceeds via an associative transition state in HcTrpRS with an unclear role of Mg2⫹in the catalytic
transition state (11). However, comparison of the pre-transi-tion (Protein Data Bank code 2QUI) and product states (Pro-tein Data Bank code 2QUJ) shows that, as with BsTrpRS, the␣P of ATP must move 5.3 Å to be in a position for nucleophilic attack by tryptophan.
Despite mechanistic and structural differences, BsTrpRS and HcTrpRS have comparable tryptophan binding affinities. How-ever, these inherent differences between prokaryotic and eukaryotic TrpRS enzymes promote the binding of indolmycin to prokaryotic TrpRSs⬃1500-fold while protecting eukaryotic TrpRSs from such inhibition by a comparable amount. Deter-mining the structure of BsTrpRS bound by Mg2⫹䡠ATP and indolmycin allowed us to probe the structural basis for indol-mycin inhibition and selectivity. Specifically, we examined this structure along with the catalytically relevant structures of BsTrpRS and HcTrpRS deposited in the Protein Data Bank to answer the following questions. 1) What are the structural con-sequences of binding indolmycin? 2) Why is indolmycin a tight inhibitor of prokaryotic TrpRS? 3) Why is indolmycin not an inhibitor of eukaryotic cytosolic TrpRSs?
Experimental Procedures
Construction of pet28-His-BsTrpRS Vector—The full-length BsTrpRS sequence was PCR-amplified from a pet11 struct made previously in the laboratory. PCR primers con-tained restriction sites for BamHI and HindIII. The resultant PCR product was digested with BamHI and HindIII. A three-way ligation among the PCR product (BamHI/HindIII), dou-ble-stranded oligo encoding for the TEV site (NdeI/BamHI), and pet28b (NdeI/HindIII) yielded an expression vector for His-TEV-BsTrpRS.
Expression and Purification of His-BsTrpRS—BsTrpRS was expressed by autoinduction with BL21(DE3)pLysS cells at 37 °C (15). The cells were pelleted at 4500 rpm for 30 min, resus-pended in lysis buffer, and frozen at ⫺20 °C. Upon thawing, cells were sonicated and centrifuged (16,000 rpm, 4 °C, 1 h). His-BsTrpRS was captured from the lysate on nickel-nitrilotri-acetic acid resin and eluted with 0.3Mimidazole. Purified
pro-tein was cleaved overnight with TEV while dialyzing out the imidazole. The cleaved protein mixture was passed back over a nickel-nitrilotriacetic acid column to capture both uncleaved protein and His-TEV protease.
Active Site Titration—Active sites were titrated by following the loss of ATP to determine the fraction of molecules compe-FIGURE 1.Functional equivalences of tryptophan and indolmycin.
tent for catalysis (16, 17). The reaction was performed at 37 °C in a final reaction mixture containing 5000 cpm/l [␥-32P]ATP, 10MATP, 0.5 mMtryptophan, 5 mMMgCl2, and 0.05 unit/l inorganic pyrophosphatase. The reaction was initiated with enzyme at a final concentration of 3M. At various time points
between 10 s and 30 min, 3l of the reaction were added to 6l of quench buffer (sodium acetate, pH 5.3, 1% SDS) and placed on ice. Three microliters of each quenched reaction were spot-ted onto a cellulose-PEI TLC plate and run in 0.75MKH2PO4,
pH 3.5 with 4Murea to separate32Piand [␥
-32P]ATP. Plates
were developed using a Typhoon imager and analyzed with ImageJ (18) and JMP (19).
Michaelis-Menten Kinetics—The incorporation of [32P]PP i into ATP was tracked either by TLC or filter binding after puri-fication on charcoal (17). Reactions contained [32P]PP
i(5000 cpm/l for TLC and 400 cpm/l for the filter assay), 0.1MTris,
pH 8.0, 70 mM-mercaptoethanol, 5 mMMgCl2, 10 mMKF, 2
mMPPi, 2 mMATP, and tryptophan ranging from 0.3 to 100M. Reactions were initiated with enzyme at a final concentration of 30 nM.
Indolmycin Inhibition Assays—Inhibition assays were per-formed as described above; Michaelis-Menten experiments for tryptophan were performed in the presence of stoichiometric amounts of indolmycin (a gift from Pfizer, ca. 1994) to enzyme. Indolmycin to enzyme ratios of 1:5, 1:1, and 5:1 were used, and results were fitted to a competitive inhibition model (1) using JMP (19). Non-linear regression to Equation 1 allowed for determination ofKmfor tryptophan andKifor indolmycin.
Rate⫽ 关Tryptophan兴⫻kcat
冉
1⫹关Indolmycin兴Ki ⫹关
Tryptophan兴
冊
(Eq. 1)
Differential Scanning Fluorimetry (Thermofluor)—The ef-fects of ATP, tryptophanamide (LTN), and indolmycin on the thermal stability of BsTrpRS were assessed by thermofluor. We showed separately3 that differential scanning fluorimetry detects a conversion of TrpRS into a molten globule form that fully denatures only at higher temperature. The following sat-urating ligand concentrations were used to ensure a predomi-nance of conformations corresponding to those observed in crystal structures: 5 mMATP, 5 mMMgCl2, 10 mMLTN, and 600Mindolmycin. All reactions contained 8MBsTrpRS, 50
mMNaCl, 5 mM-mercaptoethanol, 50 mMHepes, pH 7.5, and
0.15% SYPRO Orange in a final volume of 20l. Fluorescence intensities were determined using an Applied Biosystems 7900HTFast Real Time PCR instrument, and data were ana-lyzed with MATLAB (Mathworks) with routines developed by Visinets, Inc. The software was built as a pipeline of several m-files connected to provide full analysis of the data, including thermodynamic characterization and presentation of statistics. Fluorescence at each data point along a melting curve is assumed to be the sum of contributions from two states with probabilitiesp1andp2established by thermodynamic equilib-rium between the two states.
F共t兲⫽共a1⫹b1T兲⫻p1⫹共a2⫹b2T兲⫻p2 (Eq. 2)
wherea1anda2are adjustable parameters representing inter-cepts,b1andb2are the slopes of the linear dependences of the initial and final states, and Tis the Kelvin temperature. The pipeline consists of the following three parts.
Part A is reading the data from high throughput, 384-well, real time PCR files and transforming them into a matrix con-sisting of four columns: (i) number of the well from which temperature-dependent readings were taken, (ii) an index rep-resenting the protein variant, and finally the data, (iii) temper-ature and (iv) fluorescence readings.
Part B is fitting the thermofluor data to a thermodynamic model (Equations 3 and 4).
F⫽F0⫹a1⫹b1T⫹共a2⫹b2T兲e
⫺⌬G共T兲/RT
1⫹e⫺⌬G共T兲/RT
⫽F0⫹共a2⫹b2T兲⫹
共a1⫺a2兲⫹共b1⫺b2兲T 1⫹e⫺⌬G共T兲/RT (Eq. 3)
where⌬Gis the Gibbs energy difference between the two states ande⫺⌬G(T)/RT is the Boltzmann factor that determines the state probabilitiesp1andp2.
⌬G⫽⌬H共Tm兲⫹⌬cp⫻共T⫺Tm兲⫺T
冋
⌬S共Tm兲⫹cpln冉
TTm
冊册
(Eq. 4)
where ⌬H and ⌬S are the enthalpy and entropy changes between the states,cpis the heat capacity at temperatureT, and ⌬cpis the heat capacity change between the two states at the melting temperatureTm.
Part C is independent determination ofTmassuming that the state probabilitiesp1andp2can be estimated from distances between the intersection of the melting curve with vertical lines connecting the extrapolated linear final and initial slopes. Data initially worked up using both methods B and C agreed closely, and the analysis reported here follows C.
Crystallization, Data Collection, and Structure Determina-tion—Crystals of seleno-Met-substituted BsTrpRS in complex with ATP, Mg2⫹, and indolmycin were grown by vapor diffu-sion against a reservoir of 1.4Mpotassium citrate and 0.1M
Hepes, pH 7.4. Crystals were cryoprotected in Fomblin-Y and passed in a nitrogen airstream before plunging into liquid nitro-gen. Data were collected remotely at Southeast Regional Col-laborative Access Team (Beamline ID-22) using inverse beam geometry at 0.979 Å to obtain experimental phases from the Bijvoet differences and processed with XDS (20). PHENIX (21) and Coot (22) were used for phase determination, to interpret the map, and to iteratively refine the final structure (Protein Data Bank code 5DK4).
Results
BsTrpRS Binds Indolmycin ⬃1500⫻ More Tightly than Tryptophan—Indolmycin is a competitive inhibitor ofBacillus stearothermophilus and other bacterial TrpRS enzymes that competes with tryptophan for binding to the active site of the enzyme. By conducting Michaelis-Menten experiments at
increasing tryptophan concentrations in the presence of differ-ent indolmycin concdiffer-entrations and fitting all 64 data points simultaneously to Equation 1, we were able to determine Km,tryptophan(3M) andKi,indolmycin(2 nM) (Table 1). As these experiments were carried out under exchange conditions (23), we determined the standard free energy,⌬G0⫽ ⫺
RTlnK, at 310 K for tryptophan and indolmycin binding to be 7.8 and 12.3 kcal/mol, respectively. This translates to a free energy differ-ence of 4.5 kcal/mol between the affinities of the catalytic and inhibited complexes for the indole-containing ligand. To deter-mine what factors account for the observed difference in binding free energy, we determined the structure of the BsTrpRS䡠ATP䡠indolmycin ternary complex and then con-ducted differential scanning fluorimetric experiments in the presence of various ligands.
Indolmycin and ATP Form a Ternary Complex with BsTrpRS— Extensive crystallization studies conducted on BsTrpRS have revealed three distinct conformational states: an open confor-mation (ligand-free, tryptophan, low ATP (12, 24)), a closed pre-transition state (high ATP, ATP⫹tryptophanamide (9, 12)), and a closed product conformation (Trp-5⬘-AMP (25, 26)).
A previously unpublished structure of BsTrpRS bound to Mg2⫹䡠ATP and indolmycin was never deposited (27). Never-theless, that structure was the first example of a series of sub-sequently solved structures that have been described as “pre-transition state” (PreTS) structures (Protein Data Bank codes 1MAU and 1M83 (12)). In these structures, the initial ATP binding site in the small domain composed of the N-terminal
␣-helix and the anticodon-binding domain closes on the remainder of the Rossmann fold, bringing the nucleotide
␣-phosphate from 6.7 Å away to within van der Waals contact distance of the tryptophan carboxyl oxygen (12).
The new structure presented here is at higher resolution (1.9 versus2.4 Å), and the experimental phases greatly enhanced the quality of electron density maps (Table 2 and Fig. 2). Details of the new structure, such as the orientation of the ribose and the metal position, are quite similar to those observed in deposited PreTS structures 1MAU and 1M83. Detailed differences that appear functionally relevant are discussed below.
Indolmycin Induces New Contacts with Active Site Side Chains—Indolmycin makes contacts with the side chains of His43, Asp132, and Gln147as well as two water molecules (Fig. 3A). The interaction between O␦2 of Asp132in the specificity helix and the nitrogen atom of the indole ring is observed when tryptophan (3.1 Å), tryptophanamide (3.0 Å), or indolmycin (2.9 Å) is bound. The addition of the oxazolinone group to the ligand allows for stabilizing interactions with His43and Gln147 with functionalities on either side of the ring, which have the effect of fixing the rotation of the ring (Fig. 3B). N␦1 of His43can
donate and/or accept a hydrogen bond from N2 (methylamino group) of indolmycin. In addition to these hydrogen bonds, His43can form a salt bridge with O␦2 of Asp132(2.8 Å). The amide group of Gln147forms two hydrogen bonds, one with the carbonyl oxygen of the oxazolinone ring (3.0 Å) and another with O␦2 of Asp146(3.0 Å). O␦1 of Asp146makes a highly con-served hydrogen bond with the 2⬘-OH group of ATP (2.7 Å). These side chain interactions in the conserved GEDQ motif link the indolmycin and ATP binding sites while reinforcing the linkage between opposite sides of the indole-binding pocket.
Structurally, indolmycin binding also prevents the Tyr125 rotamer switch that occurs upon the enzyme going from its open to closed conformation (Fig. 4,AandB). During the cat-alytic cycle, Tyr125changes hydrogen-bonding partners from His150(2.7 Å) in the open conformation to the␣-amino group (2.4 Å) of the tryptophan substrate in the closed PreTS. The tryptophanyl-adenylate intermediate is stabilized by two polar contacts with the hydroxyl group of Tyr125(Protein Data Bank code1I6K). The inhibited state maintains the side chain inter-action between Tyr125and His150(2.8 Å) observed in the open state.
Structural Modifications Induced by Indolmycin to the Mg2⫹䡠ATP Configuration—Superposition of the inhibited
structure onto the closed PreTS structure (Protein Data Bank code 1MAU) gives a root mean square deviation of 0.28 Å for 323 C␣pairs. For comparison, the two closed PreTS structures,
TABLE 1
Steady-state kinetic analysis of indolmycin inhibition
Non-linear regression analysis of 64 data points from steady-state kinetics experiments confirms indolmycin as a tight binding competitive inhibitor of BsTrpRS. PPi exchange assays performed in the presence of saturating Mg2⫹䡠ATP with varying concentrations of tryptophan and indolmycin show that BsTrpRS binds indolmycin⬃1450 times more tightly than tryptophan. The difference in free energy between the catalytic and inhibited complexes is⬃4.5 kcal/mol.
kcat ⌬Gkcat Km,tryptophan ⌬GKm Ki,indolmycin ⌬GKi Km,tryptophan/Ki,indolmycin s⫺1 kcal/mol
M kcal/mol M kcal/mol
31.6⫾0.8 ⫺2.1⫾0.01 3.0E⫺06⫾6.4E⫺07 7.8⫾0.1 2.0E⫺09⫾5.2E⫺10 12.3⫾0.1 1.5E⫹03
TABLE 2
Data collection and refinement statistics for crystals of the BsTrpRS䡠indolmycin䡠Mg2ⴙ䡠ATP complex
Crystallographic data, including experimental phases, of selenomethionylated BsTrpRS crystals were collected on Beamline ID-22. The final structure was deter-mined to 1.9-Å resolution and refined to anRwork/Rfreeof 16.9/18.9%. r.m.s., root mean square.
Data collection
Space group P43212
Cell constants
a, b, c (Å) 62.04, 62.04, 219.06
␣,,␥(°) 90.00, 90.00, 90.00 Resolution (Å) 43.02–1.9 Completeness (%)a
99.7 (97.9) CC1/2 (%)a,b
99.6 (93.2)
Rmeas(%)
a
15.3 (61.9) MeanI/Ia
12.6 (3.3) Number of reflections 464,942
Multiplicity 15.5
Refinement
Rwork/Rfree(%) 16.6/18.7 Fo, Fccorrelation 0.96
r.m.s. bonds 0.005
r.m.s. angles 0.994
Ramachandran favored (%) 97.0 Ramachandran outliers (%) 0.0 Average B, all atoms (Å2) 32.0
Clash score 2.2
Protein Data Bank code 5DK4 aHighest resolution shell is shown in parentheses.
bCC1/2 is the percentage of correlation between intensities from random
1MAU and 1M83, have a root mean square deviation 0.17 Å for 328 C␣pairs. The greatest structural difference between the PreTS and inhibited states occurs around Glu103–Ala120with a root mean square deviation of 0.63 Å for these 18 residues. This mobile loop, which contains Gln107and Lys111, is more open by ⬃0.5 Å in the inhibited structure as measured from the␥P of ATP to the␣-carbons of residues Gln107, Lys111, and Lys115. The carbonyl oxygen of the Gln107side chain accepts a hydro-gen bond from the water molecule coordinated to the Mg2⫹ion (2.8 Å) and another from N⑀2 of Gln147 (3.0 Å) in the pre-transition state. Steric clashing with the constrained Tyr125 rotamer prevents Gln107from switching rotamers in the inhib-ited state. As such, the interaction with Gln147is not observed. Instead, the side chain of Gln107accepts a hydrogen bond at O⑀1 from a water molecule (3.0 Å) and donates a hydrogen bond at N⑀2 to another water molecule (3.3 Å).
In the pre-transition state, the Natom of Lys111forms a salt bridge with the O␥1 atom of ATP (2.9 Å), which also forms a strong electrostatic interaction with the catalytic Mg2⫹ion (2.4
Å). This interaction presumably is important for stabilizing the developing charge on the PPileaving group released after tryp-tophan activation. Additionally, in the pre-transition state,
Lys111is in position to act as a hydrogen bond donor to the one water molecule coordinated to the Mg2⫹ion (Fig. 5). The subtle
opening of this loop in the inhibited state weakens the salt bridge between the Lys111Natom and the O␥1 atom of ATP (3.4 Å) from that observed in the preTS. Now the closest inter-actions are with three water molecules (2.6, 2.8, and 2.8 Å), none of which are coordinated directly to the catalytic Mg2⫹ ion so they are not shown in Fig. 5.
Replacement of tryptophan(amide) with indolmycin in the active site alters the coordination and placement of the Mg2⫹
ion used during the activation step of the aminoacylation reac-tion. Presumably because its orientation is fixed by the hydro-gen bonding network described above, the oxazolinone forms hydrogen bonds with two water molecules. Introduction of these two water molecules is associated with the movement of the Mg2⫹ ion into a hexavalent coordination that closely
resembles stable configurations generated in quantum mechanical simulations of Mg2⫹䡠ATP.4
As is also true in the PreTS structure, the Mg2⫹ion in the
inhibited BsTrpRS structure coordinates with a non-bridging oxygen from each phosphate group and three water molecules (Figs. 3B and 4B), two of which are further stabilized by the presence of indolmycin. In addition to the three electrostatic interactions with ATP, only one water molecule was seen to
4S. Liu, unpublished data.
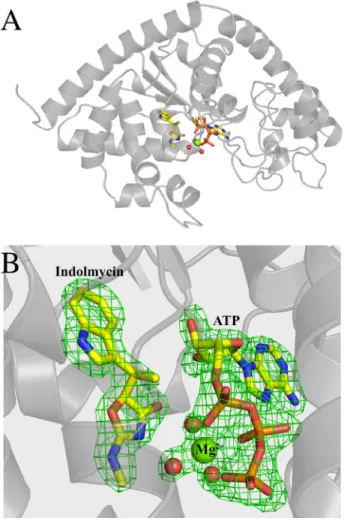
FIGURE 2. BsTrpRS forms a ternary complex with indolmycin and Mg2ⴙ䡠ATP in a closed conformation.A, functionally dimeric BsTrpRS
crys-tallizes with one monomer in the asymmetric unit.B, in addition to indolmy-cin and ATP, the active site contains a Mg2⫹ion and three stable water mol-ecules. TheFo⫺Fcomit map, derived by omitting indolmycin, ATP, Mg
2⫹, and three water molecules from a final round of refinement, contoured to 4.0
is depicted ingreen.
FIGURE 3.Hydrogen bonding and electrostatic interactions promote indolmycin binding and Mg2ⴙcoordination within the active site.A,
BsTrpRS residues His43, Asp132, and Gln147make stabilizing contacts with IND
via side chain atoms. The ATP and indolmycin binding subsites are linked via a hydrogen bond (2.7 Å) between Asp146and Gln147of the conserved GXDQ
coordinate with the Mg2⫹ion in the PreTS structure (Protein Data Bank code 1MAU) (Figs. 4Aand 5). The side chain resi-dues that accept and donate hydrogen bonds to this water mol-ecule differ between these two states (Fig. 5). Due to the differ-ent Gln107 rotamer and slight opening of the mobile loop around Lys111, these two residues no longer interact with this water molecule in the inhibited state.
The Mg2⫹ion is closer and more central to the triphosphate
moiety in the inhibited structure than in the PreTS structure
(Fig. 4C) and makes equivalent interactions with the phosphate oxygen atoms. The significance of the differences in metal posi-tions is evident from several measurements. (i) Metal to oxygen distances are significantly shorter (0.2 Å; p⫽ 0.002) in the inhibited complexes. (ii) Movement of the divalent metal ion into closer contact with the ATP phosphate oxygen atoms is associated with a subtle but statistically significant opening of the active site crevice among the N-terminal helix of the second crossover connection (the GXDQ motif), the KMSKS signa-ture, and the mobile loop containing Lys111.
Ligand-induced Stability Changes Imply Cooperative Sources of High Indolmycin Affinity—The nature of the ligands within the active site has a significant effect on the conformation and thermal stability of an enzyme. For small perturbations, the fractional change in melting temperature (⌬Tm/Tm) induced by ligand binding is proportional to the free energy change in sta-bility, the proportionality constant being the enthalpy change (⌬H) (28). We use this implicit relationship to assess the stabi-lizing or destabistabi-lizing effects of various ligands on the BsTrpRS enzyme. Binding of ATP, tryptophan, or tryptophanamide sta-bilizes the thermal transition of molten globule formation by 3, 7, and 7%, respectively (Table 3). Indolmycin enhances the ther-mal stability of BsTrpRS by 20%, increasingTmby 13.5 °C. The enhanced affinity for indolmycin over tryptophan results in a shift by 8 °C to higher temperature in the thermal transition due to molten globule formation in the presence of indolmycin compared with tryptophan.
The linkage between protein stability and ligand binding (29 –31) implies that we can attribute differences in stability changes to binding affinity. Two of the stabilizing interactions formed between indolmycin and Mg2⫹-coordinated water molecules are associated with a change in the metal position relative to the ATP phosphate oxygen atoms. A key implication of the structural observations in Figs. 3Aand 4Cis that binding of indolmycin to BsTrpRS should be potentiated by the pres-ence of Mg2⫹䡠ATP.
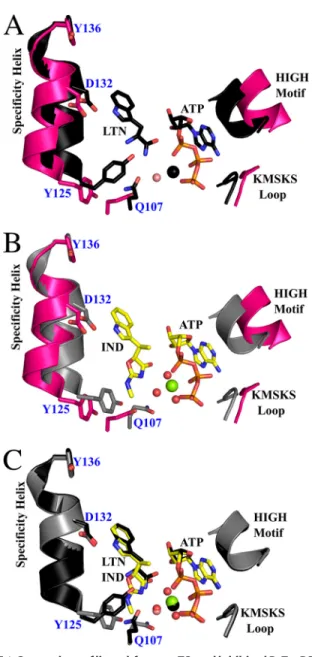
FIGURE 4.Comparison of ligand-free, preTS, and inhibited BsTrpRS struc-tures.A, compared with the apo form (pink; Protein Data Bank code 1D2R), the fully occupied PreTS structure (black; Protein Data Bank code 1MAU) assumes a closed conformation. The C␣of Tyr125is shifted inward by 2.4 Å,
and the side chain is flipped⬃45° (measured from OH-Ca-OH). A Mg2⫹ion (black sphere) forms electrostatic interactions with ATP and one water mole-cule (salmon sphere).B, binding of indolmycin and ATP causes similar shifts in the backbone (gray) as the enzyme adopts a closed conformation. However, due to the addition of the methylamino-substituted oxazolinone ring, this movement to the closed conformation is not accompanied by a rotamer change of Tyr125in the inhibited structure.C, consequently, Gln107is
con-strained and is rotated 106° around Caway from the specificity helix in the inhibited state compared with the pre-transition state. Finally, the Mg2⫹ (green sphere) is shifted toward the␣PO4and has hexavalent coordination to
ATP and three water molecules (red spheres) as compared with the preTS structure.
FIGURE 5.Differential BsTrpRS side chain interactions with the water molecules electrostatically coordinated with Mg2ⴙin the PreTS and
inhibited structures. Gln9, Gln107, and Lys111 are in position to accept
(Gln107) or donate (Gln9and Lys111) hydrogen bonds (black dashed lines) to
the water molecule (salmon sphere) coordinated with Mg2⫹(black sphere) in the PreTS (black sticks). As indolmycin binding leads to opening of the mobile loop containing Gln107and Lys111, these residues are too far (red dashed lines)
The presence/position of Mg2⫹in the active site is strictly dependent on ATP because the protein makes no contacts with the metal. As no direct ATP-indolmycin interactions are observed, we therefore expected that ATP would enhance the thermal stability of the BsTrpRS䡠indolmycin complex by a larger amount in the presence compared with the absence of Mg2⫹. Additionally, we did not expect Mg2⫹to contribute to thermal stability in the absence of ATP. As expected, the differ-ential scanning fluorimetry measurements show that the BsTrpRS in complex with indolmycin and Mg2⫹䡠ATP has a 27% increase in melting temperature compared with ligand-free enzyme with Mg2⫹䡠ATP contributing an additional 5 °C of thermal stability on top of the 13.5 °C provided by indolmycin binding. By contrast, binding of indolmycin, indolmycin ⫹ Mg2⫹, or indolmycin⫹ATP all elicit far smaller changes of ⬃20% in thermal stability, demonstrating that both Mg2⫹and
ATP are required to confer additional thermal stability to the BsTrpRS䡠indolmycin (IND) complex.
The conclusion that the metal is essential to the enhanced affinity of indolmycin to the pre-transition state complex can also be derived using the three-dimensional thermodynamic cycle of contributions to stability from ATP, the methylamino-substituted oxazolinone ring (OXA), and the presence/absence of Mg2⫹(Fig. 6). Differences in binding and thermal stability
between tryptophanamide and indolmycin were attributed to the methylamino-substituted oxazolinone ring as this is the major structural difference between these two ligands.
Stabilizing and destabilizing interactions are distinguished by positive and negative non-additivity, respectively. If thermal stability were unaffected by interactions between the ligands, then we expect the effects of binding multiple ligands,e.g.ATP and LTN, to be additive and the effects of binding one ligand not to be affected by the presence of a second ligand, thus giving Equations 5 and 6.
⌬Tm(ATP)⫹ ⌬Tm(LTN)⫽ ⌬Tm共ATP⫹LTN) (Eq. 5) ⌬Tm(LTN)⫽Tm(LTN)⫺Tm(LF)
⫽Tm(ATP⫹LTN)⫺Tm(ATP) (Eq. 6)
An interaction between the ligands would introduce a term,
⌬Tm,int,to describe the non-additivity (32), giving Equation 7. ⌬Tm(ATP)⫹ ⌬Tm(LTN)⫹ ⌬Tm,int⫽ ⌬Tm(ATP⫹LTN)
(Eq. 7)
In the absence of Mg2⫹, there is no significant ATP-OXA
inter-action, and binding either IND or LTN reduces the effect of ATP onTmby⬃1.5 °C. The ATP-LTN and ATP-IND interactions are both destabilizing;i.e.the doubly liganded complexes melt at lower temperatures. In contrast, addition of Mg2⫹stabilizes the interac-tions of ATP with LTN and IND to varying degrees. In the case of LTN-ATP, addition of Mg2⫹compensates for the destabilizing
ATP-LTN interaction such that the interaction is no longer signif-icant. The metal compensates for the⫺1.3 °C destabilizing ATP-IND interaction and allows for an additional stabilizing interaction of 2.6 °C. Thus, the⌬Tm(ATP-OXA) interaction is comparable with that of the⌬Tm(ATP-IND) interaction. The crystal structure suggests that the stabilizing effect of Mg2⫹ on the interaction FIGURE 6.Contributions of Mg2ⴙ, ATP, indolmycin, and
tryptophana-mide to the thermal stability of BsTrpRS.TrpRS enzymes require a Mg2⫹ ion for tryptophan activation. Although Mg2⫹does not change the thermal stability of BsTrpRS on its own, its presence or absence significantly impacts the interaction of ATP with both LTN and IND. In the absence of Mg2⫹(top row), both the ATP-LTN and ATP-IND interactions lower the fractional change inTmby 2% from its expected value. In the presence of Mg2⫹(bottom row), the interaction between ATP and LTN is insignificant, whereas the ATP-IND inter-action raises theTm4% higher than expected. As expected from the crystal structure, the Mg2⫹-dependent ATP-IND interaction is mediated through the oxazolinone moiety of indolmycin.LF, ligand-free.
TABLE 3
Thermofluor analysis of ligand-dependent stability
Differential scanning fluorimetric analysis of ligand-dependent thermal stability of BsTrpRS revealed a stabilizing interaction between indolmycin and ATP via Mg2⫹. Substrates (ATP and tryptophan) and substrate analogs (LTN and IND) induce conformational changes in BsTrpRS. These changes vary with ligand type and confer varying degrees of thermal stability in the transition to molten globule formation. ATP, tryptophan, tryptophanamide, and indolmycin separately enhance the thermal stability of BsTrpRS. Mg2⫹is required for additional stabilization by ATP of both LTN-bound and IND-bound BsTrpRS. The inhibited complex (BsTrpRS bound with IND and Mg2⫹䡠ATP) is the most thermally stable with aTm18.4 and 10.7 °C higher than that of ligand-free (LF) enzyme and PreTS complex, respectively. n/a, not applicable.
Ligand Mg2ⴙ T
m ⌬Tm (⌬Tm/Tm)ⴛ100
°C °C %
LF ⫺ 69.0⫾0.2 n/a n/a
ATP ⫺ 71.1⫾0.1 2.1⫾0.2 3.0⫾0.2
TRP ⫺ 74.7⫾0.1 5.7⫾0.3 8.2⫾0.4
LTN ⫺ 74.1⫾0.1 5.1⫾0.3 7.4⫾0.4
LTN⫹ATP ⫺ 74.7⫾0.2 5.7⫾0.2 8.3⫾0.3
IND ⫺ 82.5⫾0.2 13.5⫾0.3 19.6⫾0.5
IND⫹ATP ⫺ 83.3⫾0.3 14.3⫾0.4 20.7⫾0.6
LF ⫹ 69.0⫾0.1 n/a n/a
ATP ⫹ 71.2⫾0.03 2.2⫾0.1 3.1⫾0.2
TRP ⫹ 74.1⫾0.1 5.1⫾0.1 7.4⫾0.1
LTN ⫹ 74.2⫾0.2 5.1⫾0.2 7.4⫾0.3
LTN⫹ATP ⫹ 76.8⫾0.2 7.7⫾0.1 11.2⫾0.2
IND ⫹ 82.7⫾0.04 13.6⫾0.2 19.7⫾0.3
between ATP and indolmycin is mediated through the oxazolin-one ring, whose orientation is, in turn, stabilized by hydrogen bonds to His43and Gln147as discussed above.
Discussion
An array of crystal structures of both BsTrpRS and HcTrpRS provide snapshots of the enzymes along their catalytic paths and demonstrate the conformational changes that result from binding of various ligands (12, 13, 24). From these structures, it is evident that HcTrpRS uses a greater number of binding deter-minants for tryptophan recognition and that binding of trypto-phan causes an induced fit rearrangement of the active site in HcTrpRS but not BsTrpRS. Here we discuss possible structural and mechanistic reasons for the tight binding of indolmycin to BsTrpRS and the inability of indolmycin to inhibit eukaryotic TrpRSs.
Why Is Indolmycin a High Affinity Inhibitor of Bacterial TrpRS?—There are no drastic global changes between struc-ture 1MAU and the inhibited BsTrpRS strucstruc-ture (Protein Data Bank code 5DK4). We propose that subtle, mechanistically rel-evant differences in the active site metal configuration account for the ability of indolmycin to inhibit BsTrpRS as tightly as it does. We observe stronger Mg2⫹-ATP and weaker
BsTrpRS-ATP interactions (Fig. 6) as well as altered Mg2⫹coordination and placement in the inhibited state (indolmycin⫹Mg2⫹䡠ATP;
Figs. 3Band 4) compared with the pre-transition state (trypto-phanamide⫹Mg2⫹䡠ATP) structure. We attribute these differ-ences to the replacement of tryptophanamide with indolmycin that varies mainly at the methylamino-substituted oxazolinone ring of indolmycin. Interactions among His43, Gln147, and indolmycin restrict the oxazolinone ring orientation, thereby reducing the entropy of the␣-carbon mimic in the inhibited complex compared with the pre-transition state complex. This unfavorable entropy change is compensated by the enthalpy from additional hydrogen bonds formed among the Mg2⫹ -co-ordinated water molecules and the oxazolinone nitrogen and carbonyl oxygen atoms as well as the interaction with His43.
These hydrogen bonds stabilize the water molecules that are also tightly coordinated to the catalytic Mg2⫹ion. Functional
groups of the␣-carbon atoms of tryptophan and tryptophana-mide can adopt alternative conformations that are similar in energy, none of which allow for completion of the Mg2⫹ coor-dination sphere. We conclude from these observations that completion of that coordination sphere allows the metal to form significantly tighter interactions with all three phosphate oxygen atoms and hence that indolmycin stabilizes a ground state Mg2⫹䡠ATP configuration, opposing the tendency of the
PreTS state to promote the metal to a high energy state that assists in transition state stabilization.
Furthermore, the oxazolinone ring of indolmycin, stabilized by hydrogen bonds with His43and Gln147, prevents the rotamer switch of Tyr125in the specificity helix that is part of the struc-tural transition from the open to the closed state. To avoid a steric clash with the constrained Tyr125residue, Gln107likewise does not switch rotamers in the presence of indolmycin. Gln107 is part of a highly mobile loop that shows a subtle but significant opening in the inhibited state compared with the pre-transition state. This opening results in the weakening of ATP-BsTrpRS
interactions, specifically those between Lys111and the␥ -phos-phate group.
In the catalytically competent PreTS configuration, coordi-nation by lysine residues of the phosphate oxygen atoms pro-motes the metal to an activated, less stable state with weaker interactions to the three phosphate oxygen atoms and prevents the Mg2⫹ ion from assuming a lower energy position with
stronger contacts to ATP. The positively charged Natom of Lys111competes with the Mg2⫹ion for stabilization of a nega-tively charged oxygen atom (O␥) of the␥-phosphate group. In the PreTS state, the Mg2⫹-O␥and Lys111-O␥distances are 2.4 and 2.9 Å, respectively.
Substitution by indolmycin for tryptophanamide simultane-ously weakens the Lys111-O␥(3.4 Å) interaction and strength-ens that between that oxygen atom and the Mg2⫹ion (2.2 Å). Additionally, the 0.4-Å shift in Mg2⫹placement along with the opening of the mobile loop around Lys111 allows for tight, hexavalent Mg2⫹coordination accompanied by stronger, more
nearly equivalent interactions between Mg2⫹ and the three
ATP phosphate groups.
Mutation of His43Results in Indolmycin Resistance—When both tryptophanamide and ATP are bound, the His43side chain switches from one rotamer to another in the transition from open to closed PreTS state and back again in the closed product state. This rotamer switch does not occur when the active site is bound to AMP, PPi, and tryptophan (33). In the PreTS (Mg2⫹䡠ATP⫹LTN) and inhibited (Mg2⫹䡠ATP⫹IND) states, N⑀2 of His43interacts with O␦2 of Asp132. In all other observed states, N⑀2 of His43forms an interaction with the carbonyl oxy-gen of Tyr125. His43also contributes to indolmycin binding via N␦1. This reorientation of His43appears to be correlated with the succession of ligands most similar to the putative catalytic reaction path, and it may thus also be functional.
Several groups have identified mutations that confer high level indolmycin resistance (34 –36). One of the mutant sites, His43, is of direct interest in the context of the present inhibited structure, which furnishes a semiquantitative explanation for the mutational effects at position 43. We have implicated His43 in a hydrogen bond network that requires hydrogen bonds to both indole nitrogen atoms that stabilize the orientation of the plane of the oxazolinone ring of indolmycin. Fixing the orien-tation of the ring consequently allows formation of a full hexa-coordinated environment for the catalytic Mg2⫹ion, which we have shown accounts for the additional stabilization of the indolmycin䡠Mg2⫹䡠ATP complex.
Modeling Reveals Why Indolmycin Is a Weak Inhibitor of Eukaryotic Cytosolic TrpRS Enzymes—The selectivity ratio of indolmycin for cytosolicB. taurus(Bt)TrpRSversusBsTrpRS is 106-fold in favor of BsTrpRS binding. This selectivity factor far surpasses those of most therapeutic drugs, including trim-ethoprim (selectivity ratio rat/Toxoplasma gondii dihydrofo-late reductase, 49) and metoprolol (selectivity ratio 2/1 -adrenergic receptor, 6.0), which treat toxoplasmosis and cardiovascular disease, respectively (38, 39). This dramatic selectivity arises by enhancing indolmycin binding recognition by BsTrpRS as we have just shown while reducing eukaryotic cytosolic TrpRS affinity by similar magnitudes. HcTrpRS shares 93% sequence identity with BtTrpRS and is therefore highly likely to have a comparably weak millimolar affinity for indol-mycin. Interest in developing indolmycin as a lead compound for anti-infective therapy as well as the extensive kinetic and structural studies conducted on HcTrpRS led us to examine the repertoire of deposited HcTrpRS structures with the purpose of identifying potential means by which eukaryotic cytosolic TrpRS enzymes evade inhibition by indolmycin (13, 14, 40, 41). Whereas BsTrpRS uses an induced fit mechanism for ATP binding, HcTrpRS uses induced fit for tryptophan binding. Binding of tryptophan to BsTrpRS is stabilized by one hydrogen bond between the indole nitrogen of tryptophan and O␦2 of Asp132and-interactions with Phe5. Meanwhile, H
cTrpRS makes seven direct and water-mediated contacts to its trypto-phan substrate. The determinants for tryptotrypto-phan binding to HcTrpRS include Glu
199, which has a direct and
water-medi-ated interaction with the␣-amino group of tryptophan (Fig. 7A). Modeling of indolmycin into the amino acid binding site introduces steric clashes between indolmycin and Glu199(Fig. 7B). Furthermore, Glu199cannot adopt an alternative rotamer conformation without introducing additional clashes between Glu199 and Thr196, Trp203, or Phe280. Besides clashing with Glu199, this indolmycin conformer would not form any of the hydrogen bonds observed when tryptophan is bound aside from the bifurcated hydrogen bond with the indole nitrogen, Tyr159, and Gln194. These interactions are preserved because indolmycin was modeled into the active site by overlaying its indole moiety with that of tryptophan (Protein Data Bank code 2QUH).
Indolmycin can be modeled into the active site of HcTrpRS by rotating the oxazolinone ring away from Glu199to an orien-tation perpendicular to the indole moiety (Fig. 7B). Although this indolmycin conformer does not clash with active site resi-dues, it does disrupt the hydrogen bonding pattern used by the cytosolic enzyme to identify tryptophan as the bound substrate. The␣-amino group of tryptophan, which is protonated at phys-iological pH, is recognized by Glu199, Gln284, and Gln313(Fig. 7A). Each of these residues acts a hydrogen bond acceptor, and the negatively charged carboxylate of Glu199forms additional electrostatic interactions with the amino group. According to our model, the side chain amide group of Gln284would act as a hydrogen bond donor for the cyclic oxygen atom in indolmycin that is in the equivalent position of the␣-amino group (Fig. 7B). Although Glu199cannot form a salt bridge with indolmycin, it can instead share a bifurcated hydrogen bond from the
meth-ylamino group nitrogen with Gln313. This nitrogen can accept a hydrogen bond from the hydroxyl group of Tyr316.
Finally, Lys200cannot form a salt bridge with indolmycin as it does with the tryptophan carboxylate. This electrostatic inter-action is also missing when tryptophanamide is bound in place of tryptophan and appears to be critical for progression from the pre-transition state to transition state as this is the only interaction used for tryptophan substrate recognition and binding that cannot form when tryptophanamide occupies the active site. Indolmycin differs from tryptophan by a greater degree than does tryptophanamide. The inability of indolmycin to fully retain the tryptophan-HcTrpRS side chain interactions, including the salt bridge with Lys200, allows HcTrpRS to dis-criminate between tryptophan and the inhibitor. For these rea-sons, a stable HcTrpRS䡠ATP䡠indolmycin complex is⬃1000 less likely to form than that of HcTrpRS䡠ATP䡠tryptophan. Contrast-ingly, BsTrpRS, which has a 103-fold higher affinity for indol-mycin over tryptophan, is⬃1500 more likely to form a stable, inhibited complex than a catalytically competent tryptophan-bound complex.
FIGURE 7.Steric hindrance and altered hydrogen bonding pattern allow HcTrpRS to discriminate between tryptophan and indolmycin.A, the
elec-trostatic and hydrogen bonding interactions that facilitate specific recogni-tion and binding of Trp to HcTrpRS (Protein Data Bank code 2QUH) are shown
inyellow dashes.B, IND (yellowandpink sticks) from the BsTrpRS complex was modeled into the tryptophan binding site of HcTrpRS (Protein Data Bank code
2QUH) so the indole moieties superimposed. This maintained the H-bonding interactions between the indole nitrogen and Tyr159and Gln194. Rigid body
modeling shows that none of the interactions important for tryptophan sub-strate recognition/binding can form between active site residues and indol-mycin (yellow sticks). Additionally, rotating the oxazolinone ring away from Glu199eliminates a prominent steric clash between the methylamino group
of indolmycin and the side chain carboxylate group of Glu199. This alternate
In this work, we determined the structural basis for high affinity inhibition of BsTrpRS by indolmycin. The simultaneous binding of indolmycin and Mg2⫹䡠ATP results in (i) movement of the Tyr125and Gln107side chains, (ii) opening of the mobile loop containing Lys111, (iii) displacement of the Mg2⫹ion by 0.4 Å, (iv) hexavalent metal coordination, (v) stronger, nearly equivalent electrostatic interactions of Mg2⫹with an oxygen
from each phosphate group of ATP, and (vi) weaker coordina-tion of phosphate group oxygen atoms by active site lysine res-idues. These changes are reinforced by the hydrogen bonding interactions of Gln147, His43, and Mg2⫹-coordinated water molecules with indolmycin. We propose that weaker coordina-tion by lysine residues of the phosphate oxygen atoms and stronger Mg2⫹-ATP interactions induced by indolmycin
bind-ing allow the Mg2⫹ ion to settle into a lower energy state,
thereby significantly increasing affinity by preventing activa-tion of the metal required for use in amino acid activaactiva-tion.
Author Contributions—C. W. C. and T. L. W. conceived the experi-mental plan based on previous work by Y. W. Y., T. L. W. carried out all experimental work, analyzed the data, and wrote the paper in consultation with C. W. C. and Y. W. Y.
Acknowledgments—We are grateful to D. So¨ll and to J. Sello for discussions and unpublished data on indolmycin-resistant mu-tants of E. coli and indolmycin-resistant TrpRS variants in S. gri-seus. Shubin Liu kindly performed quantum mechanical simula-tions of Mg2⫹䡠ATP.
References
1. Werner, R. G., Thorpe, L. F., Reuter, W., and Nierhaus, K. H. (1976) In-dolmycin inhibits prokaryotic tryptophanyl-tRNA ligase.Eur. J. Biochem. 68,1–3
2. Ogilvie, A., Wiebauer, K., and Kersten, W. (1975) Inhibition of leucyl-transfer ribonucleic acid synthetasymol.Biochem. J.152,511–515 3. Nakama, T., Nureki, O., and Yokoyama, S. (2001) Structural basis for the
recognition of isoleucyl-adenylate and an antibiotic, mupirocin, by isoleu-cyl-tRNA synthetase.J. Biol. Chem.276,47387– 47393
4. Konrad, I., and , R. (1977) Inhibition of phenylalanine tRNA synthetase fromBacillus subtilisby ochratoxin A.FEBS Lett.83,341–347 5. Sutherland, R., Boon, R. J., Griffin, K. E., Masters, P. J., Slocombe, B., and
White, A. R. (1985) Antibacterial activity of mupirocin (pseudomonic acid), a new antibiotic for topical use.Antimicrob. Agents Chemother.27, 495– 498
6. Kanamaru, T., (2001)In vitroandin vivoantibacterial activities of TAK-083, an agent for treatment ofHelicobacter pyloriinfection.Antimicrob. Agents Chemother.45,2455–2459
7. Werner, R. G., and Reuter, W. (1979) Interaction of indolmycin in the metabolism of tryptophan in rat liver.Arzneimittelforschung29,59 – 63 8. Weinreb, V., Li, L., Campbell, C. L., Kaguni, L. S., and Carter, C. W., Jr.
(2009) Mg2⫹-assisted catalysis byB. stearothermophilus TrpRS is
pro-moted by allosteric effects.Structure17, 952–964
9. Retailleau, P., Weinreb, V., Hu, M., and Carter, C. W., Jr. (2007) Crystal structure of tryptophanyl-tRNA synthetase complexed with adenosine-5⬘ tetraphosphate: evidence for distributed use of catalytic binding energy in amino acid activation by class I aminoacyl-tRNA synthetases.J. Mol. Biol. 369, 108 –128
10. Weinreb, V., Li, L., and Carter, C. W., Jr. (2012) A master switch couples Mg2⫹-assisted catalysis to domain motion inB. stearothermophilus
tryp-tophanyl-tRNA Synthetase.Structure20, 128 –138
11. Zhou, M., Dong, X., Shen, N., Zhong, C., and Ding, J. (2010) Crystal struc-tures ofSaccharomyces cerevisiaetryptophanyl-tRNA synthetase: new in-sights into the mechanism of tryptophan activation and implications for
anti-fungal drug design.Nucleic Acids Res.38,3399 –3413
12. Retailleau, P., Vonrhein, C., Bricogne, G., Roversi, P., Ilyin, V., and Carter, C. W., Jr. (2003) Interconversion of ATP binding and conformational free energies by tryptophanyl-tRNA synthetase: structures of ATP bound to open and closed, pre-transition-state conformations.J. Mol. Biol.325, 39 – 63
13. Shen, N., Zhou, M., Yu, Y., Dong, X., and Ding, J. (2008) Catalytic mech-anism of the tryptophan activation reaction revealed by crystal structures of human tryptophanyl-tRNA synthetase in different enzymatic states. Nucleic Acids Res.36,1288 –1299
14. Yang, X. L., McRee, D. E., and Schimmel, P. (2007) Functional and crystal structure analysis of active site adaptations of a potent anti-angiogenic human tRNA synthetase.Structure15,793– 805
15. Studier, F. W. (2005) Protein production by auto-induction in high density shaking cultures.Protein Expr. Purif.41,207–234
16. Fersht, A. R., Ashford, J. S., Bruton, C. J., Jakes, R., Koch, G. L., and Hartley, B. S. (1975) Active site titration and aminoacyl adenylate binding stoichi-ometry of aminoacyl-tRNA synthetases.Biochemistry14,1– 4
17. Francklyn, C. S., First, E. A., Perona, J. J., and Hou, Y. M. (2008) Methods for kinetic and thermodynamic analysis of aminoacyl-tRNA synthetases. Methods44,100 –118
18. Schneider, C. A., Rasband, W. S., and Eliceiri, K. W. (2012) NIH Image to ImageJ: 25 years of image analysis.Nat. Methods9,671– 675
19. SAS Institute (2013)JMP: The Statistical Discovery Software, SAS Insti-tute, Cary, NC
20. Kabsch, W. (2010) XDS.Acta Crystallogr. D Biol. Crystallogr.66,125–132 21. Adams, P. D., Afonine, P. V., Bunko´czi, G., Chen, V. B., Davis, I. W., Echols, N., Headd, J. J., Hung, L. W., Kapral, G. J., Grosse-Kunstleve, R. W., Mc-Coy, A. J., Moriarty, N. W., Oeffner, R., Read, R. J., Richardson, D. C., Richardson, J. S., Terwilliger, T. C., and Zwart, P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solu-tion.Acta Crystallogr. D Biol. Crystallogr.66,213–221
22. Emsley, P., Lohkamp, B., Scott, W. G., and Cowtan, K. (2010) Features and development of Coot.Acta Crystallogr. D Biol. Crystallogr.66,486 –501 23. Cleland, W. W., and Northrop, D. B. (1999) Energetics of substrate
bind-ing, catalysis, and product release.Methods Enzymol.308,3–27 24. Ilyin, V. A., Temple, B., Hu, M., Li, G., Yin, Y., Vachette, P., and Carter,
C. W., Jr. (2000) 2.9Å crystal structure of ligand-free tryptophanyl-tRNA synthetase: domain movements fragment the adenine nucleotide binding site.Protein Sci.9,218 –231
25. Doublie´, S. (1993)2.9 Å Crystal Structure of Bacillus stearothermophi-lus Tryptophanyl-tRNA Synthetase Complexed to its Adenylate, Tryp-tophanyl-5⬘AMP. Ph.D. thesis, University of North Carolina at Chapel Hill
26. Retailleau, P., Yin, Y., Hu, M., Roach, J., Bricogne, G., Vonrhein, C., Rov-ersi, P., Blanc, E., Sweet, R. M., and Carter, C. W., Jr. (2001) High resolution experimental phases for tryptophanyl-tRNA synthetase (TrpRS) com-plexed with tryptophanyl-5⬘AMP.Acta Crystallogr. D Biol. Crystallogr. 57,1595–1608
27. Yin, Y. (1995)Crystallographic Study of Bacillus stearothermophilus tryp-tophanyl-tRNA synthetase in its catalytic reaction. Ph.D. thesis, University of North Carolina at Chapel Hill
28. Calvin, M., Hermans, J., Jr., and Scheraga, H. A. (1959) Effect of deuterium on the strength of hydrogen bonds.J. Am. Chem. Soc.81,5048 –5050 29. Matulis, D., Kranz, J. K., Salemme, F. R., and Todd, M. J. (2005)
Thermo-dynamic stability of carbonic anhydrase: measurements of binding affinity and stoichiometry using thermofluor.Biochemistry44,5258 –5266 30. Niesen, F. H., Berglund, H., and Vedadi, M. (2007) The use of differential
scanning fluorimetry to detect ligand interactions that promote protein stability.Nat. Protoc.2,2212–2221
31. Weber, P. C., Wendoloski, J. J., Pantoliano, M. W., and Salemme, F. R. (1992) Crystallographic and thermodynamic comparison of natural and synthetic ligands bound to streptavidin.J. Am. Chem. Soc.114,3197–3200 32. Jencks, W. P. (1981) On the attribution and additivity of binding energies.
Proc. Natl. Acad. Sci. U.S.A.78,4046 – 4050
in molecular simulations.Proc. Natl. Acad. Sci. U.S.A.106,1790 –1795 34. Kitabatake, M., Ali, K., Demain, A., Sakamoto, K., Yokoyama, S., and So¨ll, D. (2002) Indolmycin resistance ofStreptomyces coelicolorA3(2) by in-duced expression of one of its two tryptophanyl-tRNA synthetases.J. Biol. Chem.277,23882–23887
35. Hurdle, J. G., O’Neill, A. J., and Chopra, I. (2004) Anti-staphylococcal activity of indolmycin, a potential topical agent for control of staphylococ-cal infections.J. Antimicrob. Chemother.54,549 –552
36. Vecchione, J. J., and Sello, J. K. (2009) A novel tryptophanyl-tRNA synthe-tase gene confers high-level resistance to indolmycin.Antimicrob. Agents Chemother.53,3972–3980
37. Ali, K. S. (2002)Bacillus stearothermophilus Tryptophanyl-tRNA Synthe-tase: Mutations Leading to Indolmycin Resistance. Ph.D. thesis, Yale University
38. Gangjee, A., Vasudevan, A., Queener, S. F., and Kisliuk, R. L. (1996) 2,4-Diamino-5-deaza-6-substituted pyrido[2,3-d]pyrimidine antifolates as potent and selective nonclassical inhibitors of dihydrofolate reductases. J. Med. Chem.39,1438 –1446
39. Smith, C., and Teitler, M. (1999)-Blocker selectivity at cloned human 1- and2-adrenergic receptors.Cardiovasc. Drugs Ther.13,123–126 40. Yang, X. L., Otero, F. J., Skene, R. J., McRee, D. E., Schimmel, P., and Ribas
de Pouplana, L. (2003) Crystal structures that suggest late development of genetic code components for differentiating aromatic side chains.Proc. Natl. Acad. Sci. U.S.A.100,15376 –15380