Classical Force Fields Tailored for QM Applications: Is It Really a
Feasible Strategy?
Oliviero Andreussi,*
,†,‡,#Ingrid G. Prandi,
§,#Marco Campetella,
§Giacomo Prampolini,
∥and Benedetta Mennucci
§†Institute of Computational Science, Universitàdella Svizzera Italiana, Via Giuseppe Buffi 13, CH-6904 Lugano, Switzerland
‡Theory and Simulations of Materials (THEOS) and National Centre for Computational Design and Discovery of Novel Materials
(MARVEL), École Polytechnique Fedé rale de Lausanne, Station 12, CH-1015 Lausanne, Switzerland́
§Department of Chemistry, University of Pisa, Via Giuseppe Moruzzi 3, I-56124 Pisa, Italy
∥CNR, UOS Pisa, Istituto di Chimica dei Composti OrganoMetallici ICCOM CNR, Area della Ricerca, Via Giuseppe Moruzzi 1,
I-56124 Pisa, Italy
*
S Supporting InformationABSTRACT: Classical molecular dynamics is more and more often coupled to quantum mechanical based techniques as a statistical tool to sample configurations of molecular systems embedded in complex environments. Nonetheless, the classical potentials describing the molecular systems are seldom parametrized to reproduce electronic processes, such as electronic excitations, which are instead very sensitive to the underlining description of the molecular structure. Here, we analyze the challenging case of the peridinin molecule, a natural apocarotenoid responsible for the light-harvesting
process in the PCP antenna protein of dinoflagellates. Ground-state structural and vibrational properties, as well as electronic transitions of the pigment are studied by means of quantum-mechanical static and dynamic calculations. Thereafter, classical molecular dynamics simulations are performed with a number of different force-fields, ranging from a popular, general purpose
one to refined potentials of increasing level of complexity. From the comparison of classical results with their quantum
mechanical counterparts, it appears that, while very poor results are obtained from standard transferrable force-fields, specifically tuned potentials are able to correctly characterize most of the structural and vibrational features of the pigment. Nonetheless, only an advanced parametrization technique is able to give a semiquantitative description of the coupling between vibrations and electronic excitations, thus suggesting that the use of classical MD in combination of QM calculations for the study of photoinduced processes, albeit possible, should be considered with care.
1. INTRODUCTION
In the past years, hybrid computational methods, which combine quantum mechanical (QM) descriptions with classical models, have become standard techniques to study properties and processes of molecular systems embedded in different environ-ments. While for homogeneous solvents, continuum models are successfully used,1,2 for more complex environments where anisotropies and heterogeneities become important, atomistic models, such as those based on Molecular Mechanics (MM), are
to be preferred.3−6 The resulting QM/MM formulations,
however, need to be used in combination with statistical tools, allowing for a proper sampling of the configurational space of the full system.
In most cases, a sequential classical-QM strategy7,8 is used, where sampling is performed by means of Molecular Dynamics (MD) at a classical level because of the large dimensions of the system of interest or the long time window that has to be explored. As a result, the quality of thefinal QM description strongly relies on the accuracy through which the initial classical
MD describes both intra- and intermolecular interactions. The QM/MM calculations of the property of interest are in fact performed on configurations extracted from the classical MD trajectory, generally without any further refinement. If the internal structure of the QM subsystem and the relative position and orientations of the QM and the MM subsystems are not accurately predicted, the QM/MM calculations will lead to artifacts.9Even in the absence of an embeddingmedium, the most delicate aspect in a sequential MD-QM strategy is to achieve a reliable description of the intramolecular degrees of freedom of the investigated molecule.
As a matter of fact, a not accurate description of the geometry of the molecule and its vibrational modes is a real risk when photoinduced processes are investigated, since the QM calculations will be largely affected by the way the classically described nuclear degrees of freedom couple with the electronic
Received: July 19, 2017 Published: September 14, 2017
excitations. An effective way of quantifying these couplings is through the frequency dependent spectral density (SD) function.10 If we assume a linear coupling of the electronic degrees of freedom and the nuclear modes and treat them as harmonic oscillators, the spectral density can be computed using a Fourier transform of the excitation energy autocorrelation function, which can be in turn computed straightforwardly through the aforementioned sequential classical-QM ap-proach.11−15In such application, the nuclear degrees of freedom are treated using the classical MD whereas the excitation energies are calculated quantum mechanically along the classical trajectory. It is evident that the quality of the results will be almost completely determined by the quality of the forcefield
(FF) used in the MD.9 The simulation of the SD can thus
become an extremely important validation property for assessing the quality of MM FF to be used in quantum chemical based approaches for photoinduced processes.
In this work, a very challenging molecule, the apocarotenoid peridinin (PID), is used to investigate limits and potentials of this strategy. Peridinin represents the absorbing unit used in the peripheral water-soluble light-harvesting (LH) complex of most photosynthetic dinoflagellates, the peridinin-chlorophyll-com-plex (PCP), which constitute the main part of oceanic plankton. The uniqueness of PCP as LH antenna, is that carotenoid molecules (the PIDs) are preponderant over the chlorophylls, and they act as primary absorbing units. The key structural features of PID are a lactone and an allene group conjugated to the polyene chain, conferring special spectroscopic properties to the molecule.16−25 These unique properties are exactly the reason for our choice. Indeed the presence of a polar group (the lactone) within the polyene chain makes the coupling of the vibrational modes with the electronic degrees of freedom extremely challenging for classical FF based simulations.
Therefore, classical MD simulations are performed with three different FFs, ranging from a popular, general purpose, one to
more refined models, derived according to two different
strategies from Density Functional Theory (DFT) data, purposely computed on the target chromophore. These two
newly developed FFs, which represent extremely refined
formulations specifically developed for PID, are here used as
“optimal” FF descriptions. The results obtained from QM
calculations over trajectories obtained with such refined FFs, when compared with fully QM descriptions, should give us an insight about the limits and potentials of the sequential classical-QM strategy. Here two fully classical-QM descriptions are used as benchmarks, namely a static model based on a single QM optimized structure and a trajectory from an ab initio Born− Oppenheimer MD simulation. As the scope of this study is the analysis of the coupling between electronic and internal nuclear degrees of freedom arising form a classical description of the internal degrees of freedom, the whole investigation has been limited to peridinin in vacuum.
From the comparison of classical results with their quantum mechanical counterparts, it appears that, while very poor results are obtained from standard transferrable FF, specifically tuned potentials are able to correctly characterize most of the structural and vibrational features of the pigment. Nonetheless, only an advanced parametrization technique is able to give a semi-quantitative description of the coupling between vibrations and electronic excitations.
The article is structured as follows: insection 2we summarize
the different approaches exploited to generate and sample
configurations of PID, followed by a detailed description of
methods and parametrizations of the different classical Force Fields adopted; insection 3we report the results obtained for the different simulations in terms of the coupling between the lowest bright excitation of PID and its vibrational modes; this analysis is accompanied by an investigation of structural and vibrational properties.
2. METHODS AND PARAMETERIZATIONS
2.1. Structures and Trajectories.The following structures
or ensembles of structures were considered in this work: (1) Structures obtained by geometry optimization of the eight different molecules present in the crystallographic structure of the PCP monomer (PID611, PID612, PID613, PID614, PID621, PID622, PID623, PID624, as extracted from the 1PPR structure16found in the Protein Data Bank). Calculations were performed at the DFT level with the hybrid B3LYP functional and the Gaussian 6-311g(d,p) basis set, exploiting the Gaussian 09 program.26In the following sections, these results are reported with the label GAU-OPT.
(2) Single structure obtained by geometry optimization of the
first peridinin molecule (PID611) in the crystallographic
structure of the PCP monomer. Also in this case, calculations were performed at the DFT B3LYP level of theory, but exploiting the CP2K package,27using a mixed basis set composed by plane-waves and localized functions, consisting of plane-plane-waves within a cutoff of 300 Ry and a double-ζ plus polarization basis set (DZVP) with GTH pseudopotentials to handle core electrons. A simulation cell of 30 Å with the removal of periodic boundary artifacts was adopted. In the following these results are reported with the label CP2K-OPT.
(3) Structures sampled from an ab initio Born−Oppenheimer
MD (BOMD) simulation performed with the CP2K package,27
starting from the crystallographic structure of PID611 and using the same parameters as reported above. Simulations in the canonical NVT ensemble were performed using the thermostat from Bussi and co-workers,28with a target temperature of 300 K and a thermostat time constant of 1 ps. A simulation time step of 0.5 fs was adopted, for a total simulation time of 32.5 ps. For the collection of ensemble averages, only configurations from the second half of the trajectory were considered, whereas thefirst half of the simulation was used for equilibration. For the calculation of the IR spectrum and of the spectral density, configurations were sampled every 2 fs, for a total of about 8000 structures. In the following sections, these results are reported with the label CP2K-BOMD.
(4) Structures sampled from classical MD simulations performed with the Amber suite of programs and the general
Amber force-field (GAFF).29Atomic charges were computed
using the RESP procedure with a HF/6-31G(d) level of theory,
averaged over the first four peridinin structures
the following sections, these results are reported with the label GAFF-CLMD.
(5) Structures sampled from classical MD simulations performed with the Amber suite of programs30and a specifically tailored force-field, developed starting from first-principles
simulations performed on the first four peridinin molecules
(PID611-PID614) at the B3LYP/6-311G(d,p) level of theory. The Amber functional form of the force-field was adopted, and an atom-specific parametrization was used, similar to the strategy adopted by Prandi et al.31Details of the simulations are the same as in the previous GAFF-CLMD case. In the following sections, these results are reported with the label STAFF-CLMD.
(6) Structures sampled from classical MD simulations performed with the Gromacs software (version 4.6.1)32and a
tailored force-field generated with the help of the JOYCE
program,33,34applied tofirst-principles simulations performed at the B3LYP/6-311G(d,p) level of theory on the minimal energy configuration obtained among the different peridinin molecules (corresponding to PID613). Simulations were performed in the canonical NVT ensemble, with a target temperature of 300 K enforced by the velocity rescale thermostat of Bussi et al.35with 0.1 ps time constant. A simulation time step of 1 fs and no periodic boundary conditions were adopted. The hydrogen bonds were constrained with LINCS algorithm.36Following 5 ns of equilibration, a correlated 32 ps MD was performed and 8000 snapshots were extracted every 4 fs for the IR spectrum and spectral density analyses. For the uncorrelated MD we have continued the correlated simulation for other 5 ns and extracted
Figure 1.Unique atomic labels for the atoms of the peridinin molecule and corresponding atom types for the three different classical interaction potentials considered in this work. Labels are reported in black, red, and green to identify carbon, hydrogen, and oxygen atoms, respectively.
1000 frames from the MD trajectory (1 frame every 5 ps). In the following sections, these results are reported with the label JOYFF-CLMD.
For all the above structures, electronic excitation energies were
computed through the Gaussian 09 program using the Tamm−
Dancoff(TDA) formulation of the TD-DFT approach, coupled
with the hybrid B3LYP density functional and a 6-311G(d,p) basis set. IR vibrational spectra were computed either using linear response calculations on optimized structures (GAU-OPT and CP2K-OPT) or from the autocorrelation of QM electric dipoles, as computed along the trajectories by means of DFT calculations at the B3LYP/6-311G(d,p) level of theory, still exploiting the Gaussian 09 program.
2.2. Force-Field Parameterization.In the standard Amber
formulation, the generalized force-field (GAFF) is partitioned into bonded and nonbonded terms. The former are described through sums of bond stretching, angle bending, and two types of dihedral torsions (proper and improper) with the expression
∑
∑
∑ ∑
∑
θ θ φ γ φ φ = − + − + + − + −V k r r k
V
n
k
( ) ( )
2 [1 cos( )]
( ) b a n d n d n i bonded bonds eq 2 angles eq 2 dihedrals , , impropers eq 2 (1)
withkb,ka,Vd,n, andkibeing the force constants;randθare the bond length and bond angle respectively; req and θeq are the
corresponding bond and angle equilibrium values; φ
corre-sponds to the dihedral angle,nis its periodicity number,γd,nis its phase, andφeqis the equilibrium value of the improper dihedral angle.45
Nonbonded terms are instead modeled as a sum of pairwise interactions, namely long-range electrostatic Coulomb and short-range Lennard-Jones (LJ) interactions expressed as
∑
∑
ε=
ϵ + −
< < ⎡ ⎣ ⎢ ⎢ ⎛ ⎝ ⎜⎜ ⎞ ⎠ ⎟⎟ ⎛ ⎝ ⎜⎜ ⎞ ⎠ ⎟⎟⎤ ⎦ ⎥ ⎥ V q q R R R R R 4 1 4 1 2 i j i j
ij i j ij ij ij ij ij nonbonded 0
0 12 0 6
(2)
whereqi/jare atomic point charges,εijandRij0are the depth and
position of the LJ potential well, whileRijis the distance between atomsiandj. It might be worth noticing that GAFF uses the same nonbonded parameter set (i.e., LJ’sεand σ, plus charges) to describe either interaction with sites belonging to the same molecule (intramolecular FF) and pertaining to other molecules of the simulated systems (intermolecular FF). If on the one hand this facilitates the transferring process from one target molecule to another, on the other hand a highly accurate description of the nonbonded interaction should not be expected for a specific target. Indeed, bonded and nonbonded FF parameters are univocally determined in GAFF once each atom is assigned to one of the predefined atom types. While bonded and LJ terms are usually left unaltered from such initial assignment, it is common practice to derive atomic partial charges from ad hoc fi rst-principles calculations.
Following this idea, to derive the potential for peridinin, the crystallographic structures of thefirst four peridinin residues in the PCP structure from the Protein Data Bank (label 1PPR) were hydrogenated and structurally optimized using the Gaussian program at the standard B3LYP/6-31G(d) level of theory. GAFF atom types were automatically assigned by the Antechamber
package and are reported inFigure 1a. For each of the four optimized structures, atomic partial charges were then generated through the two-step restrained electrostatic potential (RESP) method also implemented in the Antechamber package. For the calculation of partial charges, the standard HF/6-31G(d) was used. Missing interaction parameters for specific groups of atom types where inferred from similar groups in the GAFF database using the parmchk tool,37(all thefinal parameters are included in theSupporting Information).
Specifically Tuned Amber Force-Field (STAFF).To overcome
some of the limitations of a general FF and to gain a description of the structure and dynamics of PID closer to the one obtained from QM calculations, a more advanced parametrization strategy was followed, similarly to what was recently proposed for other carotenoids.31,38−40 In particular, the functional form of the Amber FF as reported ineqs 1and2was fully retained, but each atom of the molecule was assigned to a new specific atom-type. Similarly to the GAFF potential, atomic partial charges were generated through the two-step RESP method, averaged between to thefirst four peridinin residues in the PCP structure. For the geometry optimization and the calculation of partial charges, we resort to DFT, making use of the B3LYP hybrid functional with 6-311G(d,p) basis set. Bonded terms, in particular all stretching and bending, plus all the possible proper dihedral torsions within the conjugated chain, were specifically tuned using the following multistep procedure:
(1) Bonds and angles equilibrium values and force constants
are first computed in the harmonic approximation from the
knowledge of the QM Hessian matrix of the molecule computed in the minimum-energy geometry. Similar to our previous application,31 in this work, we used the eigenvalue analysis
scheme available as a plugin named Paratool46 for the VMD
software,50already correcting for the electrostatic contributions, naturally included in the QM Hessians. For the calculations of Hessian matrix and normal modes, the same B3LYP/6-311G(d,p) level of theory used for the optimization and the determination of partial charges was adopted and the results were
averaged over the first four peridinin residues of the PCP
structure.
(2) A symmetrization procedure was adopted to reduce the number of independent atom types: atom types in similar chemical groups are assigned the same label and their corresponding bonds and angles parameters are averaged out, provided that thefinal values have less than a 5% difference with respect to the original unsymmetrized ones. This eventually corresponds in assigning equal labels to all hydrogen atoms connected to the same heavy atom, to assign the same labels to atoms in different methyl groups, and to assign the same labels to carbon atoms in similar positions in the side rings. Thefinal atom types resulting from this procedure are reported inFigure 1b, while all the values of the parameters are reported in theSI.
(3) Ad hoc scaling of bond and angle force-constants is performed to match the QM normal-modes distribution as computed on the equilibrium structure. The scaling procedure is
first performed for highly localized normal modes, namely C−H and O−H bond stretching, and the bond stretching of the atoms in the allene group and in the carboxyl groups. Eventually, the other force constants are tuned until a significant overlap between the classical andfirst-principles distributions of normal modes is obtained.
simplified (left and right head cores) as reported inFigure 2. Then, the above steps werefirst performed on the simplified
fragments and the resulting parameters were used as starting point for the determination of the bonded terms of the more complex fragments. Eventually, values from different fragments were combined to give the starting point for the parameters of the full molecule. In this way, the tuning procedure wasfirst applied to systems with a significantly reduced number of degrees of freedom, allowing a better determination of specific scaling factors. Similarly to what is done by default by the Paratool plugin, force constants were appropriately scaled in order to take into account of the constant factor betweenfirst-principles and experimental vibrational frequencies, as reported in the Minnesota Database of Frequency Scale Factors for Electronic Model Chemistries.50,51
Dihedral torsions involving the atoms of the conjugated chain not embedded in the lactone ring were parametrized individually on QM scans of the potential energy surface, computed at the B3LYP/6-31+G(d) level of theory. The reduced basis set was adopted to reduce the computational burden, since its effects on the determination of the dihedral torsional barriers was found to be negligible. The comparison between QM and MM scans of dihedral angles is reported inFigure 3, together with a graph summarizing the values of the first dihedral force coefficients (Vd,n=2ineq 1) for all the bonds of the chain and their GAFF
counterparts. It is clear from the results reported inFigure 3c that the chosen approach is able to overcome one of the limitations of GAFF, which is not able to describe correctly the connection between the lactone ring and the chain (failing to identify bond 10 as more double-bond-like in character). Moreover, it allows capturing the differences between different regions of the chain,
with different parameters when passing from the innermost,
more conjugated, parts of the chain toward the outermost parts, where the single/double nature of the bonds is stronger.
It has to be noted that to enforce the right rigidity on the connection between the chain and the left head ring afive-body interaction term would be required, able to include all the atoms of the allene group and theirfirst neighbors. To overcome this limitation, afictitious bond between the two external carbon atoms of the allene (C22 and C24, labeled as bond 0 inFigure 3a) was introduced and the dihedral torsion along this bond was
specifically parametrized with a single dihedral term of
Figure 2.Overlapping fragments (a, c, d) and simplified structures (b, e) used to derive bond and angle parameters fromfitting of normal modes distributions. The two cyclohexane-based head groups are first simplified into “core”structures (b and e), which are then used as starting guesses for the determination of the bond and angle parameters of the real fragments (a and d, respectively). The results obtained for the left head (a), chain (c), and right head (d) overlapping fragments are then combined to provide the initial guess for the parametrization of the
full peridinin molecule. Figure 3.(a) Labeling of PID bonds belonging to the polyene chain and (b) comparison of QM (black circles) dihedral scans (energy vs dihedral torsion) and corresponding classical curves (red lines) obtained with STAFF. Curves obtained with JOYFF are not reported for sake of clarity, as they would be virtually on top of STAFF results. (c) Comparison of thefirst dihedral force coefficients for the chain bonds as automatically assigned by GAFF (blue diamonds) and specifically optimized in STAFF (red squares).
periodicityn= 2, with a resulting force coefficient of about 2.8 kcal/mol. All other bonded interactions involving thisfictitious bond were set to zero.
The remaining bonded terms, namely dihedral torsions of atoms in the lactone ring, in the two side rings, and improper torsions, where assigned the values of the GAFF potential. Similarly, LJ parameters were assigned according to the atom types of the corresponding GAFF potential.
JOYCEForce-Field (JOYFF).A further step toward FF specificity
and hence, hopefully, accuracy, can be made by abandoning the strict Amber partition into bonded and nonbonded terms and the whole idea of transferability. In other words, a new FF can be completely reparameterized based on QM information only, purposely carried out on the target molecule. Indeed, this is the main aim of the JOYCEprocedure, proposed33some years ago and
successfully employed in several different systems.34,43−46 To fully exploit JOYCE’s procedure, thefirst step is to resort to a more flexible MD program, which should allow for the implementation of a larger variety of user-defined parameters. The GROMACS
engine was adopted for this reason, both because JOYCEdirectly
operates with GROMACSfile formats for FF parameters and in
consideration of the following features granted by this MD engine.
(1) Nonbonded LJ parameters and point charges of an atom can be assigned different values, depending if they concur to the computation of the intra- or intermolecular interaction energy.
(2)“Stiff” dihedrals, as those governing the planarity of the aromatic rings or ruling the (forbidden) rotation around a double bond, can be described through harmonic (improper) dihedral potentials of given force constants and equilibrium values.
(3) Highly redundant internal coordinates can be removed from the FF description and their parameters not assigned.
(4) In principle, an unlimited number of atom types can be defined within the FF.
Concretely, the standard expression for the FF does not differ
much from the Amber one reported ineq 1, being the bonded
and nonbonded terms respectively defined as
∑
∑
∑
∑ ∑
θ θ φ φ δ γ = − + − + − + + − θ μ °μ μ μ
μ °
μ μ μ
μ °
μ μ μ
μ ° = ° μ μ μ μ μ
V k r r k
k
A n
1
2 ( )
1
2 ( )
1
2 ( )
[1 cos( )]
N S N N N j N
j j j
BOND 0 2 0 2
ID 0 2
1 PD I P bonds angles dihedrals dihedrals cos (3) and
∑ ∑
∑ ∑
ε σ σ
π = − + ϵ ‐ ° < ° ° < ° ⎡ ⎣ ⎢ ⎢ ⎛ ⎝ ⎜⎜ ⎞⎠⎟⎟ ⎛⎝⎜⎜ ⎞⎠⎟⎟⎤ ⎦ ⎥ ⎥ V r r q q r 4 4 i N i j N ij ij ij ij ij i N i j N i j ij NON BOND 12 6 0 LJ LJ Coul. Coul. (4)
Ineq 3,kμS,k
μ θ, andk
μ
IDare, respectively, the stretching, bending
and improper dihedral force constant for harmonic potentials,rμ,
θμ, and ϕμ are the bond length, the angle and the improper dihedral angle,rμ0,θ
μ
0, andϕ
μ
0are their equilibrium positions. For
proper dihedrals, for each cosine term j, Ajμ
PD is the torsional
barrier;njμis the multiplicity,δμis the dihedral angle andγjμis a
phase factor. Conversely in eq 4, rij represents the distance between atomsiandj, whereasεijandσijare the LJ parameters, which are obtained for every atom pairs by the standard
Lorentz−Berthelot mixing rules, applied to the atomic
parametersεk andσk, (k = i orj). As already mentioned, the latter can be given a different value depending on the kind of interaction the defining atom is involved in, thus defining for each atomkthe two pairs of parameters ϵkinter/σkinterandϵkintra/
σkintra, for inter- and intramolecular interactions, respectively. The JOYCEparametrization of the peridinin FF was carried out
following the protocol outlined hereafter. Further details can be found in the original papers.33,34
(1) The parametrization was based on a starting geometry optimization of PID, again performed at DFT level, with the B3LYP functional and the 6-311G(d,p) basis set. Besides the fully optimized peridinin coordinates, the Hessian matrix (HQM) in the ground-state minimum geometry was also stored as JOYCE’s
input data. Furthermore, for all flexible dihedrals within the
conjugated chain (shown inFigure S1), QM torsional energy
profiles (VQM) were also computed and stored for JOYCE. Each
relaxed scan was carried out stepwise, varying the investigated dihedral every 30 degrees, and optimizing all coordinates but the scanned angle.
(2) As far as the atom types choice is concerned, the different employed labels are displayed in Figure 1d. To increase the specificity of the description, the greatest variety of possible atom types was chosen, with no restriction but those dictated by symmetry and chemical equivalence. Yet, to reduce the number of parameters to be determined, without significantly loosing on
specificity, some exceptions were made. Methyl and O−H
groups were considered as equivalent. Nonetheless, it should be pointed out that the regions embedding such groups are still
differentiated. For instance, each methyl group is also
characterized by the angle X−C−H, being X the atom,
specifically labeled, bearing the methyl group.
(3) At difference with the former FF parametrization
strategies, not all the possible redundant internal coordinates and potential functions were employed in JOYFF. First, the planarity of the aromatic cycles and the torsions around double bonds were described through improper harmonic potentials, which are more suited for such stiffdihedrals. In second place, intramolecular point charges were set to zero, describing the nonbonded interactions only through intramolecular LJ interactions. Finally, not all the possible atom pairs within PID were set to interact through nonbonded potentials, but only those necessary to prevent unphysical curling of the flexible moieties of the target molecule and avoid contacts along the chain. A complete list of the employed potential functions and the resulting parameter is given in theSupporting Information.
(4) Once the internal coordinates subtending the JOYFF and the connected potential functions have been chosen, the FF parametrization was carried out by the Joyce automated procedure based on the QM computed data, by minimizing JOYCE’s standard objective function
∑ ∑
∑
= − + ′ − = − = − =I w H H
N
w V V
( ) 1
( )
k M
l M
kl kl kl
i N
i i i
1
3 6
1
3 6
QM FF 2
geom
1
QM FF 2
geom
whereHQMandHFFare the Hessian matrices computed at QM
molecule,VQMandVFFthe torsional energies of theith geometry
sampled during the scan, Ngeom the number of scanned
geometries, and w and w′ user defined weights. It should be noted that, to reduce the computational burden, the second term of the sum is only exploited for the chainflexible dihedrals, for which the QM torsional profiles were computed, whereas only the Hessian matrix was employed to parametrize the dihedrals of the left and right end ring. Despite this clearly implies a less accurate description of the conformational behavior of these cycles, their influence on the electronic properties of interest should be negligible.
Although harmonic force constants were directly derived from
the QM Hessian and do reflect the DFT optimized structure,
some further improvements could be made to increase the accuracy of some key vibrational modes. More precisely, all of the mentioned terms ineq 3are expressed as sums of contributions, each one depending on a single internal coordinate, and such description is often termed as “diagonal”. A more accurate description should explicitly take into account the coupling of two (or more) internal coordinates. As a matter of fact, in the original FF functional form implemented in Joyce, several couplings between different internal coordinates (as bending-stretching terms) can be included. Unfortunately, most MD programs are able to take into account only uncoupled molecular motions like stretching or angular bending, without taking into account hybrid terms, or including them only in a partial fashion. To remedy for these missing coupling terms, we have slightly
tuned the force constants extracted from the Hessian accordingly to atomic displacements in backbone normal modes. The tuning is a way to recover some of the coupled terms and it is essential to bring back the conjugated normal modes of the backbone. To avoid a too rigid potential, in the tuning we have multiplied stretching constants by the factors reported in section SI5 of the
Supporting Information. The only changes in angular constant
were CB2−CA1−CA2 and CB2−CA1 = CA3 that were
multiplied by a 150% factor to be coherent with the others angular backbone constants and to avoid a break of the conjugation.
3. RESULTS AND DISCUSSION
3.1. Structural Properties. Values of the main bonds,
angles, and dihedrals of peridinin, obtained on the different structures and ensemble of structures considered in this work, are summarized in section SI7 of theSupporting Information. The time scale explored in the ab initio BO-MD does not allow seeing
significant changes in conformation with respect to the
optimized structure, when simulating peridinin at 300 K. On average, bonds are found to be elongated with respect to the optimized values, the two cyclohexane rings are rotated and, similarly, some of the dihedral torsions around the single bonds in the conjugated chain show largefluctuations.
Extensive classical MD simulations show larger structural changes. Similarly to what was observed for other carotenoids,31
simulations with the GAFF potential show significant
Figure 4.Conformational changes in left (left panels) and right (right panels) rings of PID, as identified by the behaviors of some dihedral angles as a function of time, along classical MD simulations performed with GAFF/STAFF (top panels) and JOYFF (bottom panels) interaction potentials.
modifications with respect to the optimized structure, with several isomerization events along the single bonds of the conjugated chain, in particular involving the dihedral angles of bonds 5, 9, 10, and 13 (see labels inFigure 3a). Note that, as the order of the bond between the lactone ring and the left side of the chain (bond number 10) is assigned a single-bond-like dihedral torsion in GAFF, isomerization along this bond is also observed in the simulations exploiting this forcefield. Similarly, torsions along the allene group are very easily observed in the GAFF-CLMD simulation, reflecting the unphysical lack of a proper interaction term. These changes in conformations are not seen in the MD performed with the specifically tuned forcefields, where only minorfluctuations are observed. In particular, as reported in
Figure 4, changes in the conformations of the cyclohexane rings are readily observed in both the STAFF-CLMD and JOYFF-CLMD simulations, and are reflected by the large spread in the values of dihedral angles along the dynamics (see tables in section
SI7 of Supporting Information). In agreement with fi
rst-principles results, also in classical MD simulations conformations of the right epoxy-containing ring are exchanged more often than the ones of the left ring. Simulations performed with the STAFF potential, using for these portions of the molecule the same dihedral parameters of GAFF, show one substantially dominant configuration for the left ring, while the right ring has a balanced bimodal distribution of probabilities between the two
con-formers (see section SI3 in Supporting Information). The
JOYFF results show similar trends, but with a significantly larger
flexibility in both rings, characterized by a faster exchange of configurations. As briefly discussed in the previous section, this last feature may be an artifact, due to the absence of the torsional barriers between ring conformers among the DFT database employed in Joyce parametrization. However, the rings internal coordinates are expected to be rather decoupled from the
conjugated part, and a negligible effect on the electronic
properties should be expected. Conversely, the degree of conjugation of the carotenoid backbone is known to have a strong influence on its electronic properties as well as in the electronic excitations.47,24Thus, a key geometric analysis is the measure of the length of the conjugated bonds in the peridinin backbone.Figure 5compares the bond lengths in the different structures considered in this work, while inTable 1the computed values of the bond length alternation (BLA) are reported. The bond length alternation is defined as the average difference between single and double bonds. For the case of peridinin, following previous works on this system,20,25the bonds entering
the definition of the BLA are the ones labeled from 1 to 14 in
Figure 3a.
Good agreement is found between the two sets of QM optimizations (GAU-Opt and CP2K-OPT) even if some differences are found possibly because of the difference in the description of the core electrons that are modeled through pseudopotentials in CP2K. In particular, all the bonds are found to be slightly more elongated in the CP2K optimization, but while for double bonds, this elongation appears to be homogeneous along the chain, single bonds show larger differences at the left side of the chain, while smaller differences are found in the center and in the lactone part of the conjugated system. As a consequence, the overall BLA value of the two optimized structures is slightly smaller for the CP2K-OPT structure. Comparing static and MD simulations at QM level, it appears that thermal effects are responsible for an overall elongation of both single and double bonds, but also in this case the effect is not completely homogeneous along the chain: more conjugated single bonds at the center and lactone sides of the chain show larger elongations due tofinite temperature effects. The overall BLA of the CP2K-BOMD average structure is larger than both the CP2K-OPT and the GAU-OPT ones.
When comparing QM and classical results, it is clear that the very general formulation of GAFF does not allow capturing the electronic delocalization in this system. The standard atom types
defined in GAFF for conjugated linear and aromatic systems
correspond to four possible different values of the equilibrium bond length of single and double bonds of the PID chain. The linear single-bond value is a good match of the QM result only for the longest (least conjugated) bond of the system (bond number 1), while the linear double-bond value is a good match only for the shortest (least conjugated) double bond of the system (bond number 14). The rest of linear single or double bonds are thus described by GAFF as not enough conjugated (too elongated or shortened, respectively) when compared to the corresponding QM results. On the contrary, the GAFF procedure assigns aromatic conjugation to the bonds (11−12) belonging to the lactone ring and to its connection to the left of the chain (10), which correspond to a slight overestimate of the conjugation with respect to QM results: despite an overall better agreement, double bonds in these region are larger, while the single bond is shorter, than their first-principles counterparts. The resulting BLA is mostly affected by the large differences in the linear part of the chain and is substantially larger than anyfirst-principle result. The above limitations of GAFF are clearly overcome by both
flavors of the specifically tuned FFs, which show bond lengths qualitatively in line with the QM structures used to parametrize them. Nonetheless, a significant difference is observed between STAFF and JOYFF results: while the former shows a visible elongation of all the bonds, more marked for some of the single bonds, the latter produces average bond lengths substantially on top of the GAU-OPT results. This difference, which is clearly
reflected in the computed BLAs, is due to the different
approaches adopted in the parametrization of the two FF and
in the definition of nonbonded terms. As a matter a fact,
nonbonded terms of STAFF were set to be equal to the transferable and general values of GAFF and were not tuned for
each specific atom-type of the system, thus acting in an
uncontrolled manner on top of the equilibrium geometry set by the bonded terms. The result is that the minimal energy
configuration of STAFF (see section SI6 in Supporting
Information) shows some minor, but observable, distortions with respect to the optimized QM configuration used for its
Figure 5.Lengths of the bonds of the conjugated polyene chain, bond numbers refer to labels inFigure 3a. Error bars correspond to one standard deviation, as computed from the MD trajectories.
parametrization: these distortions account for most of the
deviations observed in bond lengths and BLA inFigure 5and
Table 1.
Contrary to STAFF, the JOYCE procedure takes explicitly into account the effect of intramolecular short-range interactions in the equilibrium geometry, thus producing a resulting potential in better structural agreement with the QM data. Still, it seems that thermal effects on the structural features of the system are
differently described by the QM and classical MD (see also
section SI6 in Supporting Information). In particular, while a large relaxation of the most conjugated single bonds is observed in the CP2K-BOMD simulation, leading to a substantial increase in the BLA atfinite temperature, only a minor effect is captured in the JOYFF-CLMD results. This is not to be ascribed to the stiffness of the potential, as the two simulations show similar standard deviations of bond lengths, while it may be a reflection of stronger nontrivial anharmonic effects in the conjugated chain of carotenoids.
3.2. Electronic Excitations and Spectral Density. As
described in the Introduction, PID is a very complex molecule that combines the characteristic of a carotenoid with those of a polar molecule. As a result, the selection of the appropriate QM method to describe the correct nature of its excited states is not
trivial. The Tamm−Dancoff(TDA) formulation of the TD-DFT
approach was here used, being aware that it cannot describes the dark lowest state because of its double-excitation character, which is not accounted for in the selected approach.48As a result, the bright state of interest is here the lowest one, while experimentally it is the second one.
Results on excitation energies distributions and oscillatory strengths are summarized inFigure 6andTable 2. Calculations
on the GAU-OPT structuresfind thefirst excited state at energy of about 2.6 eV, with a significant oscillatory strength. Minor
deviations are observed among the different optimized
structures, with the minimum energy structure showing the highest excitation energy and the strongest oscillatory strength
(see section SI1 in Supporting Information). The same
calculation carried over the CP2K-OPT structure predicts slightly smaller excitation energy and oscillatory strength, compatibles with the small differences in the structural properties of the two systems.
Finite temperature effects, included in the BOMD simulation, show a red shift of the average excitation energy of the system of about 0.08 eV, together with a substantial broadening of the distribution, with a standard deviation of about 0.07 eV. By observing the behavior of thefirst three excited states along the trajectory (as reported in section SI4 of Supporting Informa-tion), we note that the different states have reasonably separated features, with only minor overlaps: while thefirst excited state is usually the brightest, some configurations show a second excited state very close in energy and with a substantial oscillatory strength.
Simulations performed on trajectories generated with standard GAFF show dramatically poor results, with a significant average red shift of the first excited state and several instances of unphysically low excitation energies (see inset in the bottom panel ofFigure 6). These correspond to distorted conformations of the conjugated chain, in particular featuring large rotations along double bonds or the allene group. The unphysical description of peridinin structure is well mirrored in the behavior of thefirst three excited states along the MD trajectory, where mixing of different states and drifts in excited state energies are observed (see results in section SI4 ofSupporting Information). The problems observed in the GAFF-CLMD system are overcome by bothflavors of specifically tuned FF. Both STAFF and JOYFF show excitation energy distributions in agreement with QM results, with a red-shift with respect to the minimal
energy configurations of about 0.03 (0.06) eV for STAFF
(JOYFF) and a standard deviation of about 0.06 (0.07) eV. The behaviors of the excited states along the MD trajectories are in line to what was observed for the CP2K-BOMD simulation, showing well-defined states with only minor overlaps and no significant drifts.
To have a more quantitative evaluation of the coupling between electronic and nuclear degrees of freedom, we have calculated the spectral densityJ(ω). The spectral density can be estimated by computing the time-dependentfluctuations of the excitation energy of the selected pigment along a molecular dynamics trajectory, either classical of fromfirst-principles. In particular, the spectral density is expressed as the Fourier transform of the autocorrelation function of the excitation energiesfluctuations,CΔE(t), of the pigment, namely
∫
ω βω
π ω
=
∞ Δ
J( ) C E( )cost t td
0 (7)
where β = 1/(kBT) is the Boltzmann factor, ΔE(t) are the
excitation energyfluctuations, and the autocorrelation function
Table 1. Bond Length Alternation (BLA) for the Different Structures Considered in This Work, As Computed from the Average Difference between Single-like and Double-like Bond Lengths Reported inFigure 5
structure GAU-OPT CP2K-OPT CP2K-BOMD GAFF-CLMD STAFF-CLMD JOYFF-CLMD BLA (Å) 0.0758 0.0724 0.0770 0.1030 0.0829 0.0764
Figure 6.Excitation energy distributions for the different structures considered in this work.
= ⟨Δ Δ ⟩ Δ
C E( )t E t( ) E(0) (8)
is averaged over a moving window of the correlated MD simulations.49
Alternatively, spectral densities can be evaluated without the need to perform an MD simulation, by means of a purely QM approach based on the Displaced Harmonic Oscillator (DHO)
model. Within this framework, the differences between the
ground and excited PES are assumed to be only in the position and vertical offset. At the ground-state equilibrium, the gradient of the excited state PES equals the gradient of the energy gap and, in this position, the excited-state gradient is named vertical gradient (VG). As a result, the spectral density can be calculated as a sum of discrete contributions, one for each normal mode. The following expression in terms of the Huang−Rhys factors (Sa) corresponding to each GS normal modeaof frequencyωa, is used41
∑
ω ω ωγ ω ω γ
=
− +
J ( ) S
( )
VG
a a a
a 2 2 (9)
whereγis a damping factor andλa=Saωais the reorganization energy of modea.42In the application of the VG approach, the Huang−Rhys factor of each mode is computed by projecting the excited state gradient (calculated at the GS optimized geometry) along the normal mode. In particular, we have considered the GAU-OPT structure, with energy gradients computed at the TDA B3LYP/6-311G(d,p) level of theory and using a damping constant of 10 cm−1.
Results of the calculation of spectral densities using the VG method on the GAU-OPT structure and using the evaluation of autocorrelation functions in MD simulations, both QM and classical, are displayed in Figure 7. Consistently with the computed shifts in the normal modes of the pigment found in the CP2K-BOMD and the GAU-OPT structures, it appears that both the position of the peaks (notwithstanding few slight displacements) and their relative intensities result in very similar
values when obtained by the VG approximation and througheq
7, applied over the CP2K-BOMD trajectory. Conversely, only a qualitative agreement is found for the whole spectral density, as the VG peaks are generally more intense, sometimes twice as much, than the CP2K-BOMD ones.
When trying to evaluate spectral densities with a MD performed with a classical interaction potential, the quality of the result depends crucially on the ability of the FF to reproduce structural and vibrational properties of the pigment. This is
clearly reflected in the computed results, where GAFF-CLMD
simulations perform very poorly, due to the lack of a proper specific parametrization. Also the simulation using STAFF is not able to capture the main features of the vibronic coupling of peridinin, missing the most important features in the conjugated chain region around 1600 cm−1. This is related to the inability of
the FF to capture the correct composition of the lowest-energy higher symmetry chain mode.
Eventually, with respect to the previous classical descriptions, JOYFF-CLMD results are in better agreement with the QM calculations, identifying most of the important peaks and their relative intensities. Nonetheless, two equally intense features are predicted by this classical potential in the conjugated chain region, still reflecting the limitation of a classical approach in modeling the complexity of such a moiety.
From the practical point of view, one key parameter that can be extracted from the above results is the reorganization energy, which can be computed from the frequency-weighted integral of the spectral density. In Figure 8, we report the cumulative integrals of the spectral densities computed for the different approaches considered in this work. The VG approach applied to the GAU-OPT structure shows a cumulative reorganization energy that is in perfect agreement with the one computed on the CP2K-BOMD trajectory, but for the modes in the low frequency
range (200−1000 cm−1), where the VG is known to
under-estimate the coupling of normal modes with electronic excitations.
As for the classical simulations, GAFF-CLMD results are clearly offfrom thefirst-principles ones, mostly due to the poor description of the spectral density in the low-frequency region, where some intense unphysical peaks are observed. The two
Table 2. Averages and Standard Deviations of Energy and Oscillatory Strength of the First PID Excited State, Computed at the TDA/B3LYP 6-311g(d,p) Level of Theory
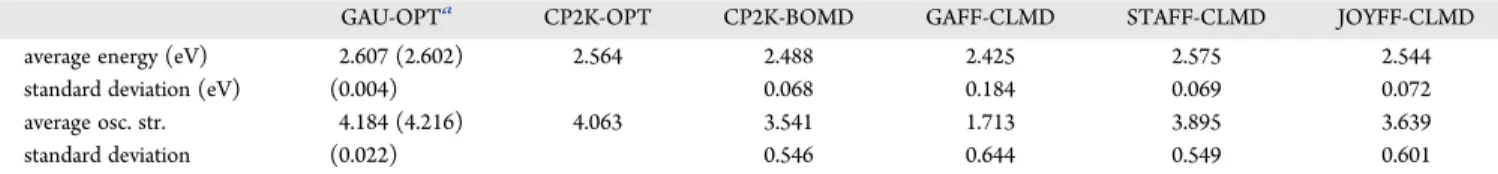
GAU-OPTa CP2K-OPT CP2K-BOMD GAFF-CLMD STAFF-CLMD JOYFF-CLMD average energy (eV) 2.607 (2.602) 2.564 2.488 2.425 2.575 2.544 standard deviation (eV) (0.004) 0.068 0.184 0.069 0.072 average osc. str. 4.184 (4.216) 4.063 3.541 1.713 3.895 3.639 standard deviation (0.022) 0.546 0.644 0.549 0.601 aThe value computed on the minimum energy structure is reportedfirst, while in parentheses the averages and standard deviations computed over
the eight different initial structures are reported.
Figure 7. Spectral densities (in cm−1) computed using the vertical gradient approach on the GAU-OPT structure (top panel) or the MD-based approach on the CP2K-BOMD ab initio trajectory (middle panel) and on the classical simulations (bottom panel).
specifically tuned FFs, instead, give rise to reorganization energies substantially in line with thefirst-principles results. In particular, STAFF appears to describe correctly the low-frequency range, which closely follows the CP2K-BOMD curve, while showing stronger deviations in the high-frequency range. Overall, the computed reorganization energy, which is very close to the BO one, is the result of a cancellation of errors,
where the presence of a very strong peak at 1200 cm−1 is
compensated by the missed peak at 1600 cm−1. On the other
hand, JOYFF shows significant deviations with respect to QM and STAFF results in the low-frequency range, possibly because of the less optimized set of parameters used for the non-conjugated parts of the molecule.
To better understand the differences found in the SD with the VG and the MD-based approaches, vibrational modes and the corresponding IR intensities have been also calculated. A particular important region of the vibrational spectrum of peridinin lies above 1550 cm−1, where highly localized normal modes are found, which can be easily assigned to specific bonds of the system. In particular, the two stretching modes of the
carbonyl CO bonds are observed at about 1700 cm−1,
stretching of the conjugated allene bond is found at about 2000 cm−1, while seven modes are observed around 1600 cm−1and correspond to different combinations of the double bonds of the conjugated chain. At higher frequencies, stretching modes of C− H and O−H bonds are observed. This is in compliance with the results obtained by Bovi and co-workers.52 The strongest IR intensities are not surprisingly found in the two carbonyl stretching modes, while a strong signal is also observed for the lowest energy, highest-symmetry, mode of the conjugated chain. Good agreement is found between the IR spectra computed onfirst-principles optimized structures and extracted from the BOMD simulation: for the sake of clarity the comparison between these results are reported in section SI2 of the
Supporting Information.
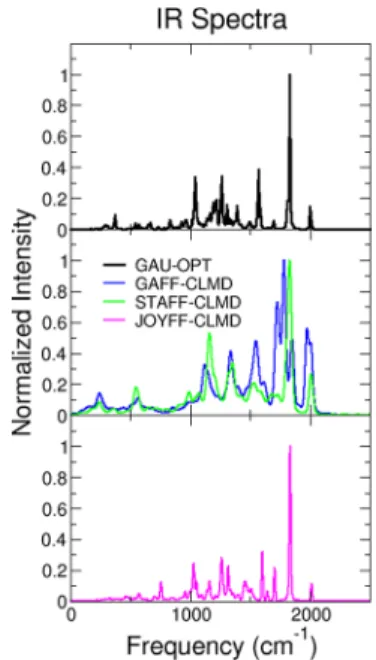
Simulations performed with GAFF (results inFigure 9) show significant deviations with respect to the QM results, consistently with the poor description of peridinin structure and with the lack of a specificfit to reproduce vibrational properties. In particular, several peaks of high intensity are observed in the important frequency region between 1600 and 2000 cm−1, where instead very specific and localized stretching modes should be present. Simulations with STAFF show, instead, a normal modes distribution in good qualitative agreement with the QM data used to parametrize the force-field. This is reflected in the correct position of the more localized peaks and in an overall fair
description of the IR spectrum of the carotenoid. Nonetheless, the main drawback of STAFF seems to be the lack of a proper description of the conjugated chain modes, with the quenching of the intense highly symmetric chain mode. An analysis of the composition of the normal modes in terms of atomic displacements shows that the presence of the lactone ring breaks the delocalization of normal modes on the two sides of the chain, producing vibrations that are more localized and have significantly reduced IR intensities.
On the other hand, the more flexible parametrization of
JOYFF produces results in very close agreement with the static QM results used for the parametrization, with similar IR peaks positions and intensities. Similarly to what was observed for the structural properties, results of JOYFF show small deviations due tofinite-temperature and anharmonic effects. For the specific case of vibrational properties, it is possible that the minor broadening observed in the JOYFF-CLDM simulations is related to the use of constraints to block hydrogen stretching, thus hindering a possible source of anharmonic coupling between normal modes. In particular, the intensity of the most important IR peaks seems to strictly mirror the original spectrum computed on equilibrium configurations, with the carbonyl features still predominant over the chain modes. In particular, also in this case the classical interaction potential may not be able to correctly describe modes delocalized over the whole conjugated chain, thus distributing the IR intensity of the lowest energy (most symmetric) mode over two more localized modes.
4. CONCLUSIONS
The application of quantum chemical methods to structures resulting from force field based molecular dynamics is a very common procedure in computational chemistry but no detailed investigations on the drawbacks of such a sequential approach have been presented so far, nor clear strategies have been developed to correct them. In this Article, we have tried to give an insight on both of these open issues. To do that, we have selected
Figure 8.Cumulative sums of the spectral densities reported inFigure 7.
Figure 9.Infrared spectra computed using a linear-response approach on the GAU-OPT minimum energy structure (top panel) and via Fourier transform of the QM dipole autocorrelation function along classical MD trajectories using the GAFF, STAFF (middle panel), and JOYFF (bottom panel) potentials.
a challenging test case, which is the simulation of electronic excitations of peridinin, a natural pigment combining the large conjugation typical of carotenoids with the presence of a polar group within the conjugation. To have a complete and detailed analysis, we have performed a benchmark QM-MD and compared that with classical MD using three different force
fields: the standard GAFF, and two newly developed FFs tailored on QM properties of the pigment. The two pigment-specific FFs,
which differentiate in their functional form and the
para-metrization technique, can be seen as two alternatives of a strategy which, by releasing the requirement of being trans-ferable, can achieve the“best”molecular mechanics description of the selected molecule.
From the comparison of classical results with their quantum mechanical counterparts, it appears clearly that poor results are obtained when using standard transferrable FFs. On the contrary,
specifically tuned potentials are indeed able to correctly
characterize most of the structural and vibrational features of the pigment. However, if a correct description of the coupling between structural fluctuations and electronic excitations is needed, an advanced parametrization technique must be used and also in that case, the description that can be achieved is only semiquantitative. This suggests that the use of force-field based MD in combination of QM calculations for the study of photoinduced processes, albeit possible, should be considered with care.
■
ASSOCIATED CONTENT*
S Supporting InformationThe Supporting Information is available free of charge on the
ACS Publications websiteat DOI:10.1021/acs.jctc.7b00777. All thefiles with the force-field parameters required to perform simulations with the force-fields developed and described in the article (Amber for GAFF and STAFF, Gromacs for JOYFF) (ZIP)
Additional commentedfigures and tables with analysis of the results of the different simulations and average and standard deviation of bond lengths, angles, and dihedrals of the peridinin molecule for all the different structures considered in the work (PDF)
■
AUTHOR INFORMATIONORCID
Oliviero Andreussi:0000-0002-1869-5678 Giacomo Prampolini:0000-0002-0547-8893 Benedetta Mennucci:0000-0002-4394-0129
Author Contributions
#O.A. and I.G.P. contributed equally to this work.
Notes
The authors declare no competingfinancial interest.
■
ACKNOWLEDGMENTSWe would like to acknowledge Lucas Viani for useful discussions. O.A., M.C., and B.M. acknowledge the European Research
Council (ERC) for financial support in the framework of the
Starting Grant (EnLight 277755). I.G.-P. acknowledges CNPq− Brazil for scholarship 236693/2012-3.
■
REFERENCES(1) Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models.Chem. Rev.2005,105, 2999−3093.
(2) Cramer, C. J.; Truhlar, D. G. Implicit Solvation Models: Equilibria, Structure, Spectra, and Dynamics.Chem. Rev.1999,99, 2161−2200.
(3) Gao, J. Hybrid Quantum and Molecular Mechanical Simulations: An Alternative Avenue to Solvent Effects in Organic Chemistry.Acc. Chem. Res.1996,29, 298−305.
(4) Senn, H. M.; Thiel, W. QM/MM Methods for Biomolecular Systems.Angew. Chem., Int. Ed.2009,48, 1198−1229.
(5) Brunk, E.; Rothlisberger, U. Mixed Quantum Mechanical/ Molecular Mechanical Molecular Dynamics Simulations of Biological Systems in Ground and Electronically Excited States.Chem. Rev.2015,
115, 6217−6263.
(6) Combining Quantum Mechanics and Molecular Mechanics. Some Recent Progresses in QM/MM Methods, Advances in Quantum Chemistry Vol.59; Sabin, J. R., Brändas, E., Eds.; Elsevier, 2010.
(7) Bergsma, J. P.; Berens, P. H.; Wilson, K. R.; et al. Electronic Spectra from Molecular Dynamics: A Simple Approach.J. Phys. Chem.1984,88, 612−619.
(8) Canuto, S., Ed.Solvation Effects on Molecules and Biomolecules. Computational Methods and Applications; Springer, 2008.
(9) Cerezo, J.; Santoro, F.; Prampolini, G. Comparing Classical Approaches with Empirical or Quantum-Mechanically Derived Force Fields for the Simulation Electronic Lineshapes: Application to Coumarin Dyes.Theor. Chem. Acc.2016,135, 143.
(10) Mukamel, S.Principles of Nonlinear Optical Spectroscopy; Oxford University Press, 1995.
(11) Chandrasekaran, S.; Aghtar, M.; Valleau, S.; Aspuru-Guzik, A.; Kleinekathöfer, U. Influence of Force Fields and Quantum Chemistry Approach on Spectral Densities of BChl a in Solution and in FMO Proteins.J. Phys. Chem. B2015,119, 9995−10004.
(12) Valleau, S.; Eisfeld, A.; Aspuru-Guzik, A. On the Alternatives for Bath Correlators and Spectral Densities from Mixed Quantum-Classical Simulations.J. Chem. Phys.2012,137, 224103.
(13) Wang, X.; Ritschel, G.; Wuster, S.; Eisfeld, A. Open Quantum System Parameters for Light Harvesting Complexes from Molecular Dynamics.Phys. Chem. Chem. Phys.2015,17, 25629−25641.
(14) Shim, S.; Rebentrost, P.; Valleau, S.; Aspuru-Guzik, A. Atomistic Study of the Long-Lived Quantum Coherences in the Fenna-Matthews-Olson Complex.Biophys. J.2012,102, 649−660.
(15) Viani, L.; Corbella, M.; Curutchet, C.; O’Reilly, E. J.; Olaya-Castro, A.; Mennucci, B. Molecular Basis of the Exciton-Phonon Interactions in the PE545 Light-Harvesting Complex. Phys. Chem. Chem. Phys.2014,16, 16302−16311.
(16) Hofmann, E.; Wrench, P. M.; Sharples, F. P.; Hiller, R. G.; Welte, W.; Diederichs, K. Structural Basis of Light Harvesting by Carotenoids: Peridinin-Chlorophyll-Protein from Amphidinium Carterae. Science
1996,272, 1788−1791.
(17) Krueger, B. P.; Lampoura, S. S.; van Stokkum, I. H.; Papagiannakis, E.; Salverda, J. M.; Gradinaru, C. C.; Rutkauskas, D.; Hiller, R. G.; van Grondelle, R. Energy Transfer in the Peridinin Chlorophyll-a Protein of Amphidinium Carterae Studied by Polarized Transient Absorption and Target Analysis.Biophys. J.2001,80, 2843− 2855.
(18) Damjanović, A.; Ritz, T.; Schulten, K. Excitation Transfer in the Peridinin-Chlorophyll-Protein of Amphidinium Carterae. Biophys. J.
2000,79, 1695−1705.
(19) Spezia, R.; Zazza, C.; Palma, A.; Amadei, A.; Aschi, M. A DFT Study of the Low-Lying Singlet Excited States of the All-Trans Peridinin in Vacuo.J. Phys. Chem. A2004,108, 6763−6770.
(20) Knecht, S.; Marian, C. M.; Kongsted, J.; Mennucci, B. On the Photophysics of Carotenoids: A Multireference DFT Study of Peridinin.
J. Phys. Chem. B2013,117, 13808−13815.
(21) Enriquez, M. M.; Hananoki, S.; Hasegawa, S.; Kajikawa, T.; Katsumura, S.; Wagner, N. L.; Birge, R. R.; Frank, H. A. Effect of Molecular Symmetry on the Spectra and Dynamics of the Intra-molecular Charge Transfer (ICT) State of Peridinin.J. Phys. Chem. B
2012,116, 10748−10756.
(22) Wagner, N. L.; Greco, J. A.; Enriquez, M. M.; Frank, H. A.; Birge, R. R. The Nature of the Intramolecular Charge Transfer State in Peridinin.Biophys. J.2013,104, 1314−1325.
(23) Kish, E.; Mendes Pinto, M. M.; Bovi, D.; Basire, M.; Guidoni, L.; Vuilleumier, R.; Robert, B.; Spezia, R.; Mezzetti, A. Fermi Resonance as a Tool for Probing Peridinin Environment.J. Phys. Chem. B2014,118
(22), 5873−5881.
(24) Coccia, E.; Varsano, D.; Guidoni, L. Ab Initio Geometry and Bright Excitation of Carotenoids: Quantum Monte Carlo and Many Body Green’s Function Theory Calculations on Peridinin. J. Chem. Theory Comput.2014,10, 501−506.
(25) Andreussi, O.; Knecht, S.; Marian, C. M.; Kongsted, J.; Mennucci, B. Carotenoids and Light-Harvesting: From DFT/MRCI to the Tamm-Dancoff Approximation.J. Chem. Theory Comput.2015,11, 655−666.
(26) Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; et al. Gaussian09, revision A.1; Gaussian, Inc.: Wallingford, CT, 2009.
(27) VandeVondele, J.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter, J. Quickstep: Fast and Accurate Density Functional Calculations Using a Mixed Gaussian and Plane Waves Approach.Comput. Phys. Commun.2005,167, 103−128.
(28) Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling.J. Chem. Phys.2007,126, 014101.
(29) Wang, J.; Wolf, R. M.; Caldwell, J. W.; Kollman, P. a; Case, D. a. Development and Testing of a General Amber Force Field.J. Comput. Chem.2004,25, 1157−1174.
(30) Case, D. A.; Cheatham, T. E.; Darden, T. A.; Simmerling, C. L.; Roitberg, A.; Wang, J.; Duke, R. E.; Luo, R.; Roe, D. R.; Walker, R. C.; et al.Amber14; University of California: San Francisco, CA, 2014.
(31) Prandi, I. G.; Viani, L.; Andreussi, O.; Mennucci, B. Combining Classical Molecular Dynamics and Quantum Mechanical Methods for the Description of Electronic Excitations: The Case of Carotenoids.J. Comput. Chem.2016,37, 981−991.
(32) Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A. E.; Berendsen, H. J. C. GROMACS: Fast, Flexible, and Free.J. Comput. Chem.2005,26, 1701−1718.
(33) Cacelli, I.; Prampolini, G. Parametrization and Validation of Intramolecular Force Fields Derived from DFT Calculations.J. Chem. Theory Comput.2007,3, 1803−1817.
(34) Barone, V.; Cacelli, I.; De Mitri, N.; Licari, D.; Monti, S.; Prampolini, G. Joyce and Ulysses: Integrated and User-Friendly Tools for the Parameterization of Intramolecular Force Fields from Quantum Mechanical Data.Phys. Chem. Chem. Phys.2013,15, 3736−3751.
(35) Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling.J. Chem. Phys.2007,126, 014101.
(36) Hess, B.; Bekker, H.; Berendsen, H. J. C.; Fraaije, J. G. E. M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem.1997,18, 1463−1472.
(37) Case, D. A.; Darden, T. A.; Cheatham, T. E.; Simmerling, C. L.; Wang, J.; Duke, R. E.; Luo, R.; Walker, R. C.; Zhang, W.; Merz, K. M.; et al. AmberTools13 Reference Manual; University of California: San Francisco, CA, 2012.
(38) Cerezo, J.; Zúñiga, J.; Bastida, A.; Requena, A.; Pedro Ceró n-Carrasco, J. Conformational Changes ofβ-Carotene and Zeaxanthin Immersed in a Model Membrane through Atomistic Molecular Dynamics Simulations.Phys. Chem. Chem. Phys.2013,15, 6527.
(39) Mayne, C. G.; Saam, J.; Schulten, K.; Tajkhorshid, E.; Gumbart, J. C. Rapid Parameterization of Small Molecules Using the Force Field Toolkit.J. Comput. Chem.2013,34, 2757−2770.
(40) Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics.J. Mol. Graphics1996,14, 33−38.
(41) Minnesota Database of Frequency Scale Factors for Electronic Model Chemistries, Version 3 Beta 2.https://comp.chem.umn.edu/ freqscale/version3b2.htm(accessed on 15 October 2014).
(42) Alecu, I. M.; Zheng, J.; Zhao, Y.; Truhlar, D. G. Computational Thermochemistry: Scale Factor Databases and Scale Factors for Vibrational Frequencies Obtained from Electronic Model Chemistries.
J. Chem. Theory Comput.2010,6, 2872−2887.
(43) Barone, V.; Bloino, J.; Monti, S.; Pedone, A.; Prampolini, G. Fluorescence Spectra of Organic Dyes in Solution: A Time Dependent Multilevel Approach.Phys. Chem. Chem. Phys.2011,13, 2160−2166.
(44) De Mitri, N.; Monti, S.; Prampolini, G.; Barone, V. Absorption and Emission Spectra of a Flexible Dye in Solution: A Computational Time-Dependent Approach.J. Chem. Theory Comput.2013,9, 4507− 4516.
(45) Prampolini, G.; Yu, P.; Pizzanelli, S.; Cacelli, I.; Yang, F.; Zhao, J.; Wang, J. Structure and Dynamics of Ferrocyanide and Ferricyanide Anions in Water and Heavy Water: An Insight by MD Simulations and 2D IR Spectroscopy.J. Phys. Chem. B2014,118, 14899−14912.
(46) Cacelli, I.; Ferretti, A.; Prampolini, G. Predicting Light Absorption Properties of Anthocyanidins in Solution: A Multi-Level Computational Approach.Theor. Chem. Acc.2016,135, 156.
(47) Knecht, S.; Marian, C. M.; Kongsted, J.; Mennucci, B. On the Photophysics of Carotenoids: A Multireference DFT Study of Peridinin.
J. Phys. Chem. B2013,117, 13808−13815.
(48) Andreussi, O.; Knecht, S.; Marian, C. M.; Kongsted, J.; Mennucci, B. Carotenoids and Light-Harvesting: From DFT/MRCI to the Tamm−Dancoff Approximation. J. Chem. Theory Comput. 2015,11, 655−666.
(49) Curutchet, C.; Mennucci, B. Quantum Chemical Studies of Light Harvesting.Chem. Rev.2017,117, 294.
(50) Lee, M. K.; Coker, D. F. Modeling Electronic-Nuclear Interactions for Excitation Energy Transfer Processes in Light-Harvesting Complexes.J. Phys. Chem. Lett.2016,7, 3171−3178.
(51) Lee, M. K.; Huo, P.; Coker, D. F. Semiclassical Path Integral Dynamics: Photosynthetic Energy Transfer with Realistic Environment Interactions.Annu. Rev. Phys. Chem.2016,67, 639−668.
(52) Bovi, D.; Mezzetti, A.; Vuilleumier, R.; Gaigeot, M.-P.; Chazallon, B.; Spezia, R.; Guidoni, L. Environmental Effects on Vibrational Properties of Carotenoids: Experiments and Calculations on Peridinin.
Phys. Chem. Chem. Phys.2011,13, 20954−20964.