organic papers
o1504
Quet al. C6H5FN2O4 doi:10.1107/S1600536806009676 Acta Cryst.(2006). E62, o1504–o1506
Acta Crystallographica Section E Structure Reports Online
ISSN 1600-5368
5-Fluorouracil-1-acetic acid
Jian-Qiang Qu,a* Ling Quband Xiao-Fei Jiaa
a
Department of Chemistry, Tianjin University, Tianjin 300072, People’s Republic of China, andbNingxia Academy of Agriculture and Forestry Sciences, Yinchuan 750002, People’s Republic of China
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study
T= 294 K
Mean(C–C) = 0.003 A˚
Rfactor = 0.041
wRfactor = 0.121
Data-to-parameter ratio = 12.0
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
Received 4 March 2006 Accepted 15 March 2006
#2006 International Union of Crystallography
All rights reserved
In the title compound, C6H5FN2O4, the acetic acid group lies
out of the pyrimidine plane. In the crystal structure, molecules are connected by intermolecular N—H O, O—H O and C—H O hydrogen bonds, forming a three-dimensional network.
Comment
The cycle-specific schedule-dependent antimetabolite 5-fluorouracil has been used in clinics for 40 years and has evolved as an important agent in the treatment of a large spectrum of tumours, including breast cancer, gastric carci-noma and bladder cancer (Duschinsky et al., 1957; Heidel-bergeret al., 1957; Correale et al., 2005). However, its slight harmfulness to the liver, kidney and digestive system limits its wider applicability (Wasterack & Bettina, 1987). For these reasons, many derivatives of 5-fluorouracil have been synthesized and some compounds have better biological activity. 5-Fluorouracil-1-acetic acid, (I), is a member of the family (Tada, 1975). Its metal complexes have been reported to have biological activity (Wanget al., 1993; Quet al., 2001; Huanget al., 2005).
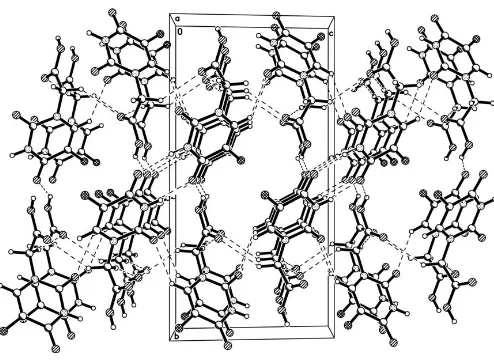
The acetic acid group lies out of the pyrimidine plane (Fig. 1). The C—F, C—O and C—N bond distances are given in Table 1. In (I), there are intermolecular N—H O, O— H O and C—H O hydrogen bonds (Table 2), forming a three-dimensional network (Fig. 2).
Experimental
Crystal data
C6H5FN2O4
Mr= 188.12
Monoclinic,P21=n
a= 4.9730 (10) A˚
b= 17.093 (3) A˚
c= 8.7485 (17) A˚
= 97.424 (3)
V= 737.4 (2) A˚3
Z= 4
Dx= 1.694 Mg m
3 MoKradiation Cell parameters from 1757
reflections
= 2.6–26.2
= 0.16 mm1
T= 294 (2) K Block, colourless 0.240.200.18 mm
Data collection
Bruker SMART CCD area-detector diffractometer
’and!scans
Absorption correction: multi-scan (SADABS; Sheldrick, 1996)
Tmin= 0.963,Tmax= 0.972 4104 measured reflections
1507 independent reflections 1139 reflections withI> 2(I)
Rint= 0.023
max= 26.4
h=6!6
k=11!21
l=10!10
Refinement
Refinement onF2
R[F2> 2(F2)] = 0.041
wR(F2) = 0.121
S= 1.03 1507 reflections 126 parameters
H atoms treated by a mixture of independent and constrained refinement
w= 1/[2(F
o2) + (0.0616P)2 + 0.3397P]
whereP= (Fo2+ 2Fc2)/3 (/)max< 0.001
max= 0.38 e A˚
3
min=0.19 e A˚
3
Table 1
Selected bond lengths (A˚ ).
F1—C3 1.422 (2)
O1—C2 1.232 (2)
O2—C1 1.213 (2)
O3—C6 1.200 (2)
O4—C6 1.322 (2)
N1—C2 1.370 (3)
N1—C1 1.385 (3)
N2—C1 1.374 (2)
N2—C4 1.376 (3)
[image:2.610.316.563.341.523.2]N2—C5 1.461 (2)
Table 2
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
O4—H4 O1i 0.98 (3) 1.81 (3) 2.696 (2) 148 (3) N1—H1 O1ii
0.91 (3) 2.00 (3) 2.904 (2) 171 (2) C4—H4A O2iii
0.93 2.53 3.284 (3) 139
C5—H5A O3iv 0.97 2.56 3.368 (2) 141 C5—H5B O3v
0.97 2.50 3.459 (3) 172
Symmetry codes: (i) xþ1 2;y
1 2;zþ
1
2; (ii) xþ1;yþ1;z; (iii)
x1 2;yþ
1 2;zþ
1 2; (iv)xþ
1 2;yþ
1 2;zþ
1
2; (v)xþ1;y;z.
The H atoms attached to O and N atoms were located in a difference map and refined freely. Other H atoms were placed in geometrically calculated positions, with C—H = 0.93 or 0.97 A˚ , and refined as riding atoms, withUiso(H) = 1.2Ueq(C).
Data collection:SMART(Bruker, 1997); cell refinement:SAINT
(Bruker, 1997); data reduction: SAINT; program(s) used to solve structure: SHELXS97(Sheldrick, 1997); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics:
SHELXTL (Bruker, 1997); software used to prepare material for publication:SHELXTL.
This work was supported by the Scientific Research Foun-dation of Tianjin University
References
Bruker (1997). SMART (Version 5.06a), SAINT (Version 5.501) and
SHELXTL(Version 5.10). Bruker AXS Inc., Madison, Wiscosin, USA. Correale, P., Fulfaro, F., Marsili, S., Cicero, G., Bajardi, E., Intrivici, C., Vuolo,
G., Carli, A. F., Caraglia, M., Prete, S. D., Greco, E., Gebbia, N. & Francini, G. (2005).Cancer Chemother. Pharmacol.56, 563–568.
Duschinsky, R., Pleven, E. & Heidelberger, C. (1957).J. Am. Chem. Soc.79, 4559–4560.
Heidelberger, C., Chaudhuri, M. S., Danneberg, P., Mooren, D., Duschinsky, R., Schnitzer, R. J., Pleven, E. & Scheiner, J. (1957).Nature (London),179, 663–666.
Huang, J., Qu, J.-Q., Wang, L.-F., Liu, Y.-Q., Wang, Y.-Y., Song, Y.-M., Zhang, C.-J. & Zhan, R. (2005).Chem. Pap.59, 267–270.
Qu, J.-Q., Huang, J., Wang, L.-F. & Sun, G.-C. (2001).Chem. Pap.55, 319–322.
organic papers
Acta Cryst.(2006). E62, o1504–o1506 Quet al. C
6H5FN2O4
o1505
Figure 1
The molecular structure of (I), showing the atom-labelling scheme. Displacement ellipsolids are drawn at the 30% probability level.
Figure 2
Sheldrick, G. M. (1996).SADABS. University of Go¨ttingen, Germany. Sheldrick, G. M. (1997). SHELXS97 and SHELXL97. University of
Go¨ttingen, Germany.
Tada, M. (1975).Bull. Chem. Soc. Jpn,48, 3427–3428.
Wang, L.-F., Yang, Z.-Y., Peng, Z.-R., Cheng, G.-Q., Guo, H.-Y., Sun, A.-L., Wang, Q. & He, F.-Y. (1993).J. Coord. Chem.28, 167–172.
Wasterack, C. & Bettina, H. (1987).Pharmazie,12, 73–75.
organic papers
o1506
Quet al. Csupporting information
sup-1 Acta Cryst. (2006). E62, o1504–o1506
supporting information
Acta Cryst. (2006). E62, o1504–o1506 [https://doi.org/10.1107/S1600536806009676]
5-Fluorouracil-1-acetic acid
Jian-Qiang Qu, Ling Qu and Xiao-Fei Jia
5-Fluorouracil-1-acetic acid
Crystal data
C6H5FN2O4 Mr = 188.12 Monoclinic, P21/n a = 4.973 (1) Å
b = 17.093 (3) Å
c = 8.7485 (17) Å
β = 97.424 (3)°
V = 737.4 (2) Å3 Z = 4
F(000) = 384
Dx = 1.694 Mg m−3
Melting point: 549 K
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 1757 reflections
θ = 2.6–26.2°
µ = 0.16 mm−1 T = 294 K Block, colorless 0.24 × 0.20 × 0.18 mm
Data collection
Bruker SMART CCD area-detector diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
φ and ω scans
Absorption correction: multi-scan (SADABS; Sheldrick, 1996)
Tmin = 0.963, Tmax = 0.972
4104 measured reflections 1507 independent reflections 1139 reflections with I > 2σ(I)
Rint = 0.023
θmax = 26.4°, θmin = 2.4°
h = −6→6
k = −11→21
l = −10→10
Refinement
Refinement on F2
Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.041 wR(F2) = 0.121 S = 1.03 1507 reflections 126 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H atoms treated by a mixture of independent and constrained refinement
w = 1/[σ2(Fo2) + (0.0616P)2 + 0.3397P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001
Δρmax = 0.38 e Å−3
Δρmin = −0.19 e Å−3
Special details
Experimental. IR (KBr, ν cm-1): 3191, 1705, 1668, 1242; 1H NMR (d
6DMSO): δ 11.85 (s, 1H), 11.80 (s, 1H), 8.18 (d,
1H, J = 7.0 Hz), 4.50 (s, 2H); analysis calculated for C6H5FN2O4: C 38.29, H 2.68, N 14.90%; found: C 38.20, H 2.75, N
supporting information
sup-2 Acta Cryst. (2006). E62, o1504–o1506
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full
covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
F1 0.1657 (3) 0.44882 (8) 0.43020 (17) 0.0553 (4)
O1 0.2805 (3) 0.50870 (9) 0.14724 (18) 0.0439 (4)
O2 0.8668 (3) 0.31106 (9) 0.08864 (18) 0.0462 (4)
O3 0.3581 (3) 0.18579 (9) 0.20437 (19) 0.0467 (4)
O4 0.6516 (3) 0.10093 (9) 0.3280 (2) 0.0434 (4)
H4 0.504 (7) 0.064 (2) 0.298 (4) 0.084 (10)*
N1 0.5887 (4) 0.41255 (10) 0.1279 (2) 0.0340 (4)
H1 0.625 (5) 0.4325 (16) 0.036 (3) 0.056 (7)*
N2 0.6630 (3) 0.31179 (9) 0.30709 (18) 0.0304 (4)
C1 0.7176 (4) 0.34248 (12) 0.1693 (2) 0.0306 (5)
C2 0.3980 (4) 0.44955 (11) 0.2018 (2) 0.0319 (5)
C3 0.3542 (4) 0.41234 (12) 0.3438 (2) 0.0373 (5)
C4 0.4805 (4) 0.34640 (12) 0.3916 (2) 0.0355 (5)
H4A 0.4449 0.3234 0.4832 0.043*
C5 0.7651 (4) 0.23365 (11) 0.3501 (2) 0.0317 (5)
H5A 0.7944 0.2291 0.4615 0.038*
H5B 0.9377 0.2257 0.3119 0.038*
C6 0.5671 (4) 0.17181 (12) 0.2844 (2) 0.0310 (5)
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
F1 0.0655 (9) 0.0500 (9) 0.0564 (9) 0.0226 (7) 0.0302 (7) 0.0137 (6)
O1 0.0514 (9) 0.0316 (8) 0.0502 (9) 0.0127 (7) 0.0126 (7) 0.0095 (7)
O2 0.0558 (10) 0.0443 (10) 0.0415 (9) 0.0172 (8) 0.0182 (8) 0.0074 (7)
O3 0.0431 (9) 0.0425 (10) 0.0500 (10) 0.0013 (7) −0.0113 (7) 0.0041 (7)
O4 0.0417 (9) 0.0251 (8) 0.0620 (11) 0.0023 (7) 0.0010 (7) 0.0061 (7)
N1 0.0424 (10) 0.0284 (9) 0.0321 (9) 0.0055 (7) 0.0084 (8) 0.0076 (7)
N2 0.0348 (9) 0.0259 (9) 0.0307 (9) 0.0015 (7) 0.0050 (7) 0.0048 (6)
C1 0.0327 (10) 0.0281 (10) 0.0308 (10) 0.0012 (8) 0.0033 (8) 0.0004 (8)
C2 0.0341 (10) 0.0242 (10) 0.0373 (11) 0.0015 (8) 0.0042 (8) 0.0002 (8)
C3 0.0431 (12) 0.0344 (12) 0.0366 (11) 0.0056 (9) 0.0131 (9) −0.0006 (9)
C4 0.0418 (12) 0.0353 (12) 0.0306 (11) 0.0010 (9) 0.0095 (9) 0.0049 (9)
C5 0.0322 (10) 0.0280 (11) 0.0345 (11) 0.0028 (8) 0.0028 (8) 0.0072 (8)
supporting information
sup-3 Acta Cryst. (2006). E62, o1504–o1506
Geometric parameters (Å, º)
F1—C3 1.422 (2) N2—C1 1.374 (2)
O1—C2 1.232 (2) N2—C4 1.376 (3)
O2—C1 1.213 (2) N2—C5 1.461 (2)
O3—C6 1.200 (2) C2—C3 1.437 (3)
O4—C6 1.322 (2) C3—C4 1.331 (3)
O4—H4 0.98 (3) C4—H4A 0.9300
N1—C2 1.370 (3) C5—C6 1.507 (3)
N1—C1 1.385 (3) C5—H5A 0.9700
N1—H1 0.91 (3) C5—H5B 0.9700
C6—O4—H4 109 (2) C4—C3—C2 121.91 (19)
C2—N1—C1 126.97 (18) F1—C3—C2 116.70 (17)
C2—N1—H1 118.0 (17) C3—C4—N2 120.81 (19)
C1—N1—H1 114.7 (17) C3—C4—H4A 119.6
C1—N2—C4 121.75 (17) N2—C4—H4A 119.6
C1—N2—C5 118.27 (16) N2—C5—C6 110.77 (16)
C4—N2—C5 119.09 (16) N2—C5—H5A 109.5
O2—C1—N2 123.12 (19) C6—C5—H5A 109.5
O2—C1—N1 121.89 (19) N2—C5—H5B 109.5
N2—C1—N1 115.00 (17) C6—C5—H5B 109.5
O1—C2—N1 121.22 (19) H5A—C5—H5B 108.1
O1—C2—C3 125.51 (19) O3—C6—O4 124.76 (19)
N1—C2—C3 113.27 (18) O3—C6—C5 123.85 (18)
C4—C3—F1 121.38 (19) O4—C6—C5 111.38 (17)
C4—N2—C1—O2 −176.1 (2) O1—C2—C3—F1 −2.2 (3)
C5—N2—C1—O2 −7.0 (3) N1—C2—C3—F1 177.90 (17)
C4—N2—C1—N1 3.8 (3) F1—C3—C4—N2 −179.83 (18)
C5—N2—C1—N1 172.85 (16) C2—C3—C4—N2 1.1 (3)
C2—N1—C1—O2 173.5 (2) C1—N2—C4—C3 −1.5 (3)
C2—N1—C1—N2 −6.3 (3) C5—N2—C4—C3 −170.5 (2)
C1—N1—C2—O1 −174.0 (2) C1—N2—C5—C6 −87.1 (2)
C1—N1—C2—C3 5.9 (3) C4—N2—C5—C6 82.3 (2)
O1—C2—C3—C4 176.9 (2) N2—C5—C6—O3 1.8 (3)
N1—C2—C3—C4 −3.0 (3) N2—C5—C6—O4 −177.26 (16)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
O4—H4···O1i 0.98 (3) 1.81 (3) 2.696 (2) 148 (3)
N1—H1···O1ii 0.91 (3) 2.00 (3) 2.904 (2) 171 (2)
C4—H4A···O2iii 0.93 2.53 3.284 (3) 139
C5—H5A···O3iv 0.97 2.56 3.368 (2) 141
C5—H5B···O3v 0.97 2.50 3.459 (3) 172