Aymetrix Miroarrays
Dissertation
zur Erlangung des Doktorgrades
der Naturwissenshaften
vorgelegt beim Fahbereih Informatik und Mathematik
der Johann Wolfgang Goethe-Universität
in Frankfurt am Main
von
Frau Dipl.-Biol. Claudia Döring
aus Frankfurt am Main
Frankfurt (2009)
Dekan: Prof. Dr. Detlef Krömker
Gutahter: Prof. Dr. Dirk Metzler
Prof. Dr. Lars Hedrih
Prof. Dr. Martin-Leo Hansmann
1 Einleitung 1
1.1 Biologishe Grundlagen . . . 4
1.2 Lymphome . . . 5
1.3 Die Miorarray-TehnologievonAymetrix . . . 7
1.4 Das Miroarray-Chip-Design von Aymetrix . . . 9
1.5 Zieldieser Arbeit . . . 11
2 Material und Methoden 15 2.1 Verwendete Genexpressionsdatensätze . . . 15
2.1.1 Datensatz vonKüppers et al. . . 15
2.1.2 Datensatz vonPialuga etal. . . 16
2.1.3 Test-Datensatz . . . 17
2.1.4 Datensatz vonBrune etal. . . 18
2.2 Ablauf eines Miroarray-Experiments . . . 18
2.3 Tehnishe Vorraussetzungen . . . 19
2.4 Explorative Datenanalyse . . . 20
2.4.1 Boxplot . . . 20
2.4.2 Histogramm . . . 20
2.4.3 Q-Q-Plot . . . 21
2.5 PräprozessierungsalgorithmenfürdieNormalisierungvon Aymetrix-Miroarray-Rohdaten . . . 21
2.5.1 Ziel der Präprozessierung. . . 21
2.5.2 Das ModellvonAymetrix . . . 23
2.5.3 ModellvonKlein etal. . . 28
2.5.4 Das VSN-ModellvonHuber etal. . . 29
2.5.8 Das sVSN-Modell . . . 40
2.6 Simulation von Daten . . . 42
2.6.1 Kontinuierlihe Verteilungen . . . 42
2.6.2 Simulieren der Daten . . . 47
2.6.3 Lineare gemishteModelle . . . 48
2.7 Cluster-Analysen . . . 51
2.8 Heatmap . . . 55
2.9 Denition der dierentiellenGenexpression . . . 55
2.9.1 Denition des Fold-Changes . . . 57
2.9.2 Das multipleTesten-Problem . . . 58
2.10 Signalweg- und Genfunktionsgruppen-Analysen . . . 59
2.10.1 Signalweg-Software und Datenbanken . . . 59
2.10.2 Überrepräsentationsanalysen mitGen-Ontologien . . . 60
2.11 Qualitätskriterienzur Beurteilung der Miroarray-Ergebnisse . . . . 63
3 Ergebnisse 65 3.1 Auswertung des Datensatzes von Küppers etal. . . 65
3.1.1 Beshreibung der statistishen Analyse-Strategie . . . 65
3.1.2 Betrahtung der dierentiell-exprimiertenGene . . . 66
3.1.3 Betrahtung der GO-Analyse . . . 71
3.2 Auswertung des Datensatzes von Pialugaet al. . . 73
3.2.1 Beshreibung der statistishen Analyse-Strategie . . . 73
3.2.2 Clusteranalyse. . . 74
3.2.3 Betrahtung der dierentiell-exprimiertenGene . . . 76
3.2.4 Betrahtung der GO-Analyse . . . 77
3.3 Zusammenfassung . . . 80
3.4 Vergleih der Normalisierungsmethodenmitsimulierter Expressions-daten . . . 81
3.4.1 Wahlder kontinuierlihen Verteilungsfunktion . . . 81
3.4.2 SimulationderDaten aufBasiseinerangepassten Gammaver-teilung . . . 94
al. . . 111
3.5.1 DeskriptiveBetrahtung der Expressionsdaten . . . 112
3.5.2 Betrahtung der dierentiell-exprimiertenGene . . . 114
3.6 Zusammenfassung . . . 117
4 Diskussion 119 4.1 Reanalyse vonpublizierten Datensätzen. . . 119
4.1.1 Reanalyse des Datensatzes vonKüppers etal. . . 119
4.1.2 Reanalyse des Datensatzvon Pialugaetal.. . . 120
4.2 Simulation von Expressionsdaten . . . 121
4.2.1 Simulation der Expressionsdaten basierend auf einer kon-tinuierlihen Verteilung. . . 121
4.2.2 Simulationder Expressionsdaten basierendauf einer ange-passten Gammaverteilung . . . 122
4.2.3 Simulation von Expressionsdaten basierend auf einem ge-mishten linearenModell . . . 122
4.3 Vergleih der vershiedenen Präprozessierungsmethoden bei einem ausgewählten Vergleih aus dem Datensatz von Brune et al. . . 123
4.3.1 Dierentielle Expression von HL im Vergleih zu normalen B-Zellen . . . 124
4.4 Ausblik . . . 124
5 Zusammenfassung 127 6 Appendix 129 6.1 R-Skript sVSN . . . 129
6.2 R-Skripte für dieSimulationvonExpressionsdaten . . . 133
6.3 Reanalyse der Miroarray-Daten vonKüppers et al. . . 149
6.4 GO-AnalysederGesamt-GenlistenahReanalysederDatenvon Küp-pers etal. . . 161
6.5 GO-Analyseder299Probesets,dienurnahReanalysederDatenvon Küppers etal. gefundenwurden . . . 162
6.6 Der Zytokine-Zytokine-Interaktionssignalweg . . . 163
and Shimodaira,2006℄ . . . 167
6.10 GO-Analyse der gesamten Genliste (599 Probesets),die nah Reana-lyse der Daten von Pialugaet al.gefundenwurde . . . 168
6.11 GO-Analyseder gesamten Genliste(363Probesets),dienurnah Re-analyse der Daten vonPialugaet al.gefunden wurden . . . 169
6.12 Ausshlieÿlih nah sVSN-Methode gefundene Probesets an einem Teildatensatz vonBrune etal. . . 169
Literatur. . . 173 Abkürzungsverzeihnis . . . 184 Danksagung . . . 185 Publikationen . . . 188 Lebenslauf . . . 191 Erklärungen . . . 193
1.1 Aymetrix Chip . . . 2
1.2 Molekulare Struktur der DNA, inklusive Transkription und Bildung der mRNA (Quelle:National Human Genome Researh Institute) . . 4
1.3 Eine B-Zellewird durheine T-Helferzelleaktiviert. (Quelle:Charles A. Janeway jr. u. a.: Immunologie. 5. Auage. Spektrum Akademi-sher Verlag GmbH, Heidelberg,Berlin 2002). . . 6
1.4 Eine B-Zelle wird nah Antigenkontakt zur Antikörper produzieren-denPlasmazelle.(Quelle:Dr.med.MarioShubert,Heidelberg, Deutsh-land) . . . 6
1.5 Allgemeines Probeset-Design für alle 3'-Expressions-Miroarrys von Aymetrix . . . 8
2.1 Ablauf eines Miorarray-Experiments . . . 19
2.2 Darstellung eines horizontalen Boxplots . . . 20
2.3 Histogramm-Beispiel . . . 21
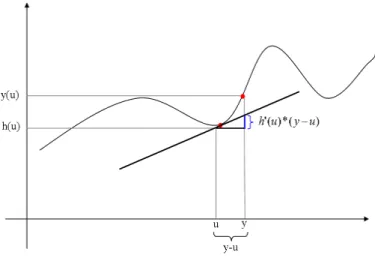
2.4 Shematishe Darstellungder Delta-Methode . . . 31
2.5 Funktionsverlaufvonf(x) und
h
s
(
x
)
[Huberet al.,2002℄. . . 332.6 Shematisher Aufbaueiner mRNA . . . 39
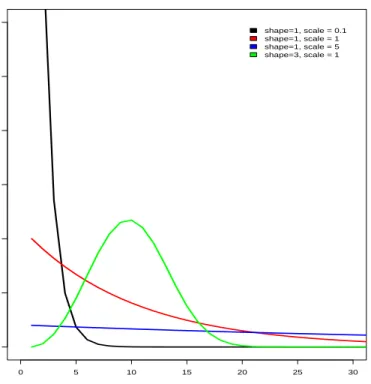
2.7 Dihtefunktion der Weibullverteilung bei vershiedenen Form- und Skalenparametern. . . 44
2.8 DihtefunktionderLognormalverteilungbeivershiedenen
σ
undµ
= 0
. 45 2.9 Dihtefunktion der Gammaverteilung bei vershiedenen Form- und Skalenparametern. . . 46entsprehen den Probesets/Genen. Die absoluten Expressionswerte
sind in einer logarithmishenSkalazur Basis 2farbkodiert. . . 69
3.2 Unsupervised Clustering der 291 Probesets mit
σ
≥
1
,
5
nah Reana-lyse der Daten von Pialuga et al.. Spalten repräsentierenindivi-duelle Patienten, Zeilen entsprehen den vershiedenen dargestellten
Genen.Die absoluten Expressionswerte sindineiner logarithmishen
SkalazurBasis2farbkodiert.DieUntersheidung derGruppenunter
dem Dendrogramm der Spaltenerfolgt durh zusätzlihen Farbode:
Rot fürPTZLs, Grün für AILTs und Blau für ALCLs.. . . 75
3.3 Heatmap der 30Probesets, dienurnah Reanalysegefunden wurden
und den gröÿten FC haben. Spalten repräsentieren die Stihprobe,
Zeilen entsprehen den Probsets/Genen. Die absoluten
Expressions-werte sind ineiner logarithmishen Skalazur Basis 2 farbkodiert. . . 78
3.4 Heatmap der 7 Gene, die nur nah Reanalyse gefunden wurden und
den gröÿten FChaben. Spaltenrepräsentieren dieStihprobe, Zeilen
entsprehen den Probsets/Genen. Die Expressionswerte sind ineiner
logarithmishen Skalazur Basis 2 farbodiert. . . 80
3.5 Histogrammder logarithmiertenrealen Expressionsdaten . . . 82
3.6 Histogramme zu den simulierten Expressionswerten und die
dazuge-hörigen Q-Q-Plots. . . 83
3.7 Histogramme zu den simulierten und gemessenen Mittelwerten und
Varianzen und den dazugehörigen Q-Q-Plots . . . 84
3.8 DieQ-Q-PlotsdersimuliertenundgemessenenExpressionswerte,
Mit-telwerteund Varianzen nahder Anpassungder empirishenVerteilung 85
3.9 Vergleih der Boxplots vonrealen und simuliertenExpressionsdaten . 86
3.10 Boxplotder Expressionsdaten miteinfaher IVT . . . 86
3.11 Vergleih der RNA-Degradation beieinfaher und doppelter
Invitro-transkription. Die einzelnen Linien repräsentieren jeweils einen
Mi-roarray-Chip.. . . 87
besets, die bei allen 10 Simulationen wiedergefunden wurden. Jedes
Feld entspriht einem anderen Quantilbereih. . . 90
3.14 Boxplots zum Wiederndender simuliertendierentiell-exprimierten
Probesets für die 10 Simulationen, d. h. die Expressionswerte sind
überalleExpressionsstärken fürdieeinzelnen Simulationendargestellt. 92
3.15 Sensitivität und Spezität der einzelnen Methoden für die 10
Simu-lationen . . . 93
3.16 SensitivitätundSpezität,dargestelltineiner ROC-Kurvebei
Simu-lation der Expressionsdaten miteiner empirishen Verteilung . . . 94
3.17 Histogramm der simulierten PM-Expressionswerte resultierend aus
einer angepassten Gammaverteilung und der QQ-Plot im Vergleih
zu den realen PM-Expressionsdaten . . . 96
3.18 Boxplotder Gamma-simuliertenExpressionsdaten . . . 97
3.19 RNA-Degradationsplot der Gammasimulierten Expressionsdaten . . 98
3.20 ProzentualerAnteilderrihtigPositivendierentiell-exprimierten
Pro-besets bei gamma-verteilten Daten. Jedes Feld entspriht einem
an-deren Quantilbereih. . . 99
3.21 Boxplotfür diePräprozessierungsmethoden für alle10Simulationen . 100
3.22 SensitivitätundSpezitätderPräprozessierungsmethodenfüralle10
Simulationen . . . 101
3.23 SensitivitätundSpezität,dargestelltineiner ROC-Kurvebei
Simu-lation der Expressionsdaten miteiner angepassten Gammaverteilung. 102
3.24 Histogramm der simulierten PM-Expressionswerte resultierend aus
einem linearengemishten Modell und der QQ-Plot im Vergleih zu
den realen PM-Expressionsdaten. . . 104
3.25 Boxplot der simuliertenExpressionsdaten basierend auf einem
li-nearen gemishten Modell . . . 105
3.26 RNA-DegradationsplotdersimuliertenExpressionsdatenresultierend
aus einemlinear gemishten Modell . . . 106
3.27 ProzentualerAnteilderrihtigpositivendierentiell-exprimierten
Pro-besets. Der Faktor gibt dieStärke der dierentiellen Expression an. . 108
3.30 Sensitivitätund Spezitätdargestellt ineiner ROC-Kurvebei
Simu-lation der Expressionsdaten miteines linear gemishten Modells . . . 111
3.31 Histogrammder logarithmiertenExpressionsdaten . . . 112
3.32 Boxplot der logarithmierten Expressionsdaten, der 12 HLs und 10
Keimzentrum-B-Zellen . . . 113
3.33 Degradationsgrad der PM-Werte in Abhängigkeit von ihrer Position
in den einzelnen Miroarrays . . . 114
6.1 GO-Analyse der Gesamt-Genlistenah der Reanalyse der Daten von
Küppers etal. . . 161
6.2 GO-Analyseder 299 Probesets dienurnahder Reanalyseder Daten
vonKüppers et al.gefunden wurden. . . 162
6.3 Signalweg Zytokine-ZytokineInteraktion . . . 163
6.4 Signalweg humaneB-Zellen Netzwerk . . . 164
6.5 Stabilitätsprüfung des Clusterverfahrens nah Suzuki et al.[Suzuki
and Shimodaira,2006℄. . . 167
6.6 GO-Analyse über die gesamte Genliste (599 Probesets) die nah der
Reanalyse der Daten von Pialugaetal. gefundenwurde . . . 168
6.7 GO-Analyse über die Genliste (363 Probesets) die nur nah der
2.1 Shematishe Darstellung der einzelnen Shritte der vershiedenen
Präprozessierungsalgorithmen bei Aymetrix-Miroarrays . . . 41
3.1 Die 299 dierentiell-exprimierten Probesets, die nur nah Reanalyse
identiziertwurden.DerShwerpunktder Regulationsstärkeliegtbei
einemFCzwishen2und3imVergleihvonHL-Linienundnormalen
B-Zellen. . . 67
3.2 Die 17 Gene, dienah Reanalyseidentiziert wurden (FDR
≤
0
.
05
), mitdemgröÿtenFCzwishen HL-LinienundnormalenB-Zellen.Ge-ne diemit* markiertsind, wurdenimVergleihzwishenHLsund
Keimzentrums B-Zellen alssignikant-reguliertgefunden. . . 68
3.3 Die fünfGenemitdem höhstenFCzwishenHL-Linienund
norma-len,reifenB-Zellen,dienurnahReanalyseidentiziertwerden
konn-ten. Diese Gene haben eine FDR
≤
0
.
05
und wurden als signikant-dierentiell exprimiert in primären HL-Patienten gefunden ([Bruneet al.,2008℄) . . . 70
3.4 Anzahl der Gene, die dierentiell sind, und nah Durhführung der
vershiedenen Normalisierungsmethoden amDatensatzvonBrune et
al. gefundenwurden . . . 115
3.5 Ein Teil der Gene, die ausshlieÿlih mit der sVSN-Methode im
Da-tensatz vonBrune et al.als dierentiellgefunden wurden . . . 116
6.1 Genlisteder zusätzlihgefundenen 299ProbesetsnahReanalysedes
Datensatzes von Küppers etal. [Küppers et al.,2003℄ . . . 149
6.2 Genliste der zusätzlih gefundenen 30 Probesets mit
F C
≥
10
oder≤ −
10
nahReanalysedesDatensatzesvonPialugaetal.[Pialuga et al.,2007℄ . . . 165Einleitung
DieMiroarray-Tehnologieermöglihtheute,simultanmehrals20000Genezu
mes-sen.Seit 1994produziertdieFirmaAymetrixkommerzielleMiroarrays.Zunähst
arbeiteten nur wenige Institute und Firmen mit diesen Miroarrays. Dies lag an
den hohen Kosten und der anfangs shlehten Reproduzierbarkeit der Ergebnisse.
InzwishenistAymetrixMarktführerfürdieMiroarray Tehnologiemitvielen
ver-shiedenen Miroarrays.Vershiedene Spezies werden abgedekt. Gemessen werden
kann sowohl auf mRNA- und DNA-Ebene. 3'Expressions-Miroarrays messen auf
der mRNA-Ebene. Auÿerdem gibt es sogenannte SNP-Miroarrays (DNA-Ebene)
und mikroRNA (regulatorish wirkende RNAs) Miroarrays.
Führt man das Experiment mit selbst hergestellten DNA-Miroarrays durh, gibt
esofttehnishe Fehler.Diesewerdendurhdiestandardisierte Produktionder
Mi-roarrays und das Vorhandensein standardisierter Protokolle zur experimentellen
Aufbereitung der Gewebeproben verhindert.
MitMiroarrays können neue Gene und Signalwege gefunden werden,die füreinen
Krankheitssubtyp spezish sind. So führen Miroarrays zu einem besseren
Ver-ständnis der Pathogenese und dadurh zu einem besseren Therapiekonzept. 2006
gelanges Hummel etal. [Hummel etal., 2006℄ zum ersten Mal mitAymetrix
Mi-roarrays einGensetzuidentizieren,daseindeutigzwishendemdiusgroÿzelligen
B-Zell-Lymphom (DLBCL) und den Burkitt-Lymphom (BL) untersheiden kann.
Aymetrix-Miroarrays sind einfester Bestandteil der neuen biologishen und
me-dizinishenForshunggeworden.Zukunftsversion ist,dassMiroarrays alsStandard
in Kliniken eingesetzt werden können und so jeder Patient von dieser Tehnologie
DieserShritt verlangt neben einer standardisierten Produktion und
experimentel-lenDurhführungauheine standardisierte Dokumentation der verwendeten
statis-tishen Analysen. Hierzu gibt es viele Arbeiten, die zeigen, dass vershiedene
sta-tistishe Methoden zu vershiedenen Ergebnissen führen können [Homann et al.,
2002;Parrish and Spener,2004℄.
DerMiroarray auh Chipgenannt istdabeider Träger auf dem biologishes
Material, in unserem Fall Nukleotide (Sonden) in hoher Dihte und Anzahl (10
6
-10
7
Kopien) und ineiner denierten Anordnung aufgetragen sind (Abb.1.1).
Abbildung1.1: Aymetrix Chip
Miroarrarys basieren auf dem Prinzip der Hybridisierung. Bei der Hybridisierung
bildet unter bestimmten Bedingungen eine RNA-Kette mit dem komplementären
DNA-Strang eine stabile Doppelhelix. Die Eigenshaften des so durh
Basenpaa-rung entstandenen Hybridmoleküls führten zur Entwiklung der Methode der
Nu-kleinsäurehybridisierung.
Um die Genexpression messen zu können, muss vor der Hybridisierung die mRNA
mit uoreszierenden Farbsto markiert werden. Messbar wird die
Fluoreszensin-tensität auf Grund des linearen Zusammenhangs, dass umso mehr mRNA an den
immobilisierten Sonden bindet, desto mehr Fluoreszens wird gemessen. Die
Fluo-reszenz kann nur mit einem speziellen Sanner aus den Miroarrays gemessen
werden.DieSignalintensitätenkönnendannje nahFragestellungmitweiteren
sta-tistishen Methodenausgewertet werden.Die Signalintensitäten sindRohdaten, die
normalisiert werden müssen, um systematishe und tehnishe Fehler (z.B. bei
un-tershiedliher mRNAQualität und Quantität) zu eliminieren.
Beiallen für diese Arbeitdurhgeführten Analysen werdenvershiedene
Generatio-nen humaner Miroarrays der Firma Aymetrix verwendet. Die statistishe
Aus-wertungder numerishen Rohdaten wird mit der frei verfügbaren Statistiksoftware
R[IhakaandGentleman,1996℄unddenimBioondutorexistierendenPaketenay
[Gautieretal.,2004℄, VSN [Huber etal.,2002℄, geneplotter,multest, usw.
durhge-führt. Ende2001 wurdedas Open-Soure-and-Development-Softwareprojekt
Bion-dutoramInstitutdes DanaFarberCaner Institutder HarvardUniveritätinitiiert
[Gentlemanetal.,2004℄.R undBioondutorsollen das statistishund grash
ad-äquate Auswerten immer gröÿerer Menge genomisher Daten ermöglihen.
InsbesondereBioondutoristfester BestandteilfürdiestatistisheAuswertungvon
Miroarrays geworden.Deshalbintegriertauhkommerziellentwikelte Miorarray
-Software(GeneSpring,Spotre,Partek, Aymetrix,usw.)entwederPakete aus
Bio-ondutor oder stellt eine direkte Shnittstelle zu Rund Bioondutor bereit.
In der Literatur gibt es bereits einige Evaluationen zu den Untershieden zwishen
denvershiedenenAuswertungsalgorithmen[Irizarryetal.,2006;Ploneretal.,2005;
Choeetal.,2005;Homannetal.,2002℄.Allerdingswurdendiesenurmit
standardi-siertenDatenentwikelt,dasheiÿtunterBerüksihtigungderAngabendes
Miroar-ray-Herstellers zur Ausgangsmenge an Gesamt-mRNA. Bei manhen
Erkrankun-gensind nurwenigeTumorzellenvorhanden.Deshalb isthierdas Ausgangsmaterial
sehr begrenzt. Zum Zeitpunkt der Datenerhebung gab es noh kein kommerzielles
Protokoll, das mit so einer geringen Ausgangsmenge zu Ergebnissen geführt
hät-te. Kommerzielle Protokolle benötigen immer noh die zehnfahe Menge an Zellen
(
≥
10000),diefürdieExperimentebereitgestelltwerdenkönnen.DadieverwendetenMiroarrays vonAymetrixnihtfüreine solhe Vorbehandlungentwikeltwurden,
sind spezielle Anpassungen an die weiterführenden statistishen Methoden nötig,
vor allembeider Normalisierung.Zu klären ist,obund im welhem Umfangsolhe
1.1 Biologishe Grundlagen
Die DNA liegt als doppelsträngige Helix in jeder eukaryotishen Zelle vor. Das
GrundgerüstbestehtausZukerundPhosphatmolekülen,dieumzwei
Nukleotidket-tenangeordnetsind.AndiesenKettensinddievierBasenAdenin,Guanin,Thymin
undCytosinangeordnet.DiebeidenDNA-SträngewerdendurhWasserstobrüken
zusammengehalten, die von den komplementären Basen Adenin und Thymin oder
CytosinundGuaningebildetwerden.Bei derTranskription müssenzuerstdie
Was-serstobrüken aufgebrohen werden, damit die DNA teilweise in zwei einzelnen
Strängen vorliegt. Nur einer der beiden Stränge, der sogenannte odogene Strang,
wirdverwendetummitHilfederRNA-PolymerasediemRNAzu bilden.Die
Gense-quenz auf der DNA wird währendder Transkription in mRNAübersetzt und dann
durhdieTranslationandenRibosomenzufunktionell-biologishwirkenden
Protei-nensynthetisiert.DieRNA-Polymeraseverknüpftdieangelagertenkomplementären
Basen.Dabeiwirdstatt ThyminUrailindiemRNAeingebaut. Beider Basenfolge
ist,abgesehenvonUrail, diemRNAeine komplementäre Kopie eines T
eilabshnit-tes des odogenen Stranges der DNA [Wehner and Gehring,1995℄ (Abb.1.2).
Abbildung1.2: Molekulare Strukturder DNA, inklusiveTranskription und Bildung
der mRNA(Quelle: National HumanGenome Researh Institute)
mR-NAundderdarausresultierenden Proteinmenge,aberdurhMiroarrays kannman
diemRNA-Menge imVergleihzu einem anderem Experiment ermitteln. Eine
Va-lidierung der Ergebnisse eines Miroarray-Experiments auf mRNA- und auf
Pro-teinebene mit einer unabhängigen anderen Methode ist unerlässlih, um Artefakte
auszushlieÿen.DieBestätigungdurheineunabhängigeandereMethodeistgängige
Laborpraxis.
1.2 Lymphome
AllgemeinsindLymphomeShwellungenderLymphknotenoderTumoreim
Lymph-systembeigutartigemoderbösartigemGewebe.Diebösartigen(malignen)
Lympho-me werden nah der WHO-Klassizierung in Hodgkin-Lymphome (HL) und
Non-Hodgkin-Lymphome (NHL) unterteilt [Jae et al., 1998℄. Alle malignen Lymhome
stammen von malignen Lymphozyten ab. Etwa 90 % der NHLs stammen von
B-Zellenab [Fisher, 2003℄.
Zuden Lymphozyten gehören dieB-Lymphozyten (B-Zellen) und T-Lymphozyten
(T-Zellen). B-Zellen und T-Zellen werden beim Menshen im Knohenmark
gebil-det. B-Zellen sind als einzige Lymphozyten in der Lage, Antikörper herzustellen.
Sie bilden zusammen mit den T-Zellen das adaptive Immunsystem. Die T-Zellen
übernehmendieRolleder zellvermitteltenImmunantwort.WennnaiveB-Zellen,die
inunserem Blutkreislaufund imLymphsystemzirkulieren,aufeinfremdes Antigen
stoÿen,werdendieB-Zellen durhihren B-ZellRezeptor gebunden,indieZelle
auf-genommenund dann miteinem MHC-Molekül wiederan der Membran-Oberähe
präsentiert (Abb.1.3).
BeiderT-ZellenabhängigenAktivierungbedarfesnuneinergeeignetenT-Helferzelle,
die durh ihren Rezeptor die T-Zelle an den Antigen-MHC-Komplex binden kann.
Erst jetzt wird die B-Zelle durh Ausshüttung von Zytokinen der gebundenen
T-Zelleaktiviert. Ausder aktivierten naiven B-Zelle(Abb.1.4)vermehrt und
dieren-ziert sih eine antikörperproduzierende Plasmazelle (PC). Werden B-Zellen durh
körperfremde Antigene aktiviert, können sih Plasmazellen oder Gedähtnis-Zellen
entwikeln. DiesenVorgangbezeihnet manalsKlassenwehsel derB-Zelle.Die
Ver-mehrung(klonaleExpansion)ndetimKeimzentrum(GC)desLymphknotens oder
Abbildung1.3:EineB-ZellewirddurheineT-Helferzelleaktiviert.(Quelle:Charles
A. Janeway jr. u. a.: Immunologie. 5. Auage. Spektrum Akademisher Verlag
GmbH,Heidelberg, Berlin2002)
bezeihnet [Alberts etal.,1995℄.
Abbildung1.4: Eine B-ZellewirdnahAntigenkontakt zurAntikörper
produzieren-den Plasmazelle.(Quelle: Dr. med. MarioShubert, Heidelberg, Deutshland)
Sind dieAntigene oft wiederholte Polysaharide (Unterklasse der Kohlenhydrate),
wie sie z. B. bei Bakterien vorkommen, ist eine zusätzlihe Aktivierung durh eine
T-Helferzellenihtnötig.DieB-Zelleistaktiviertalleinüberihren B-Zell-Rezeptor,
diesogenannteT-Zell-unabhängigeAktivierung,ohneeinenKlassenwehsel oderdie
Bildung von Gedähtnis-B-Zellen [Alberts et al.,1995℄.
Die starke Proliferation (Zellteilung) der GC-B-Zellen erhöhen das Risiko,
(Entstehung)vonB-Zell-LymphomeneineRolle[RalfKüppers,2003℄.Währendder
Keimzentrumsreaktion nden vershiedene Umbauprozesse statt, wie z.B. die
Ein-führungvonDNA-Strangbrühen [Brossetal.,2000;Goossensetal.,1998;
Papava-siliouand Shatz, 2000℄, diesomatishe Hypermutation (Einfügen von Mutationen
in die Antikörpergene einer reifenden B-Zelle) und den Klassenwehsel. Genau in
diesenRegionen ndetman oft TranslokationenvonOnkogenen [Klein etal.,1998;
Küppers andDalla-Favera,2001℄.Auh Punktmutationenkönneneine Folgeder
so-matishen Hypermutation sein [Küppers, 2005℄.
Keimzentrums-B-Zellen zeigen harakteristish somatish mutierte V-Gene (V =
Variablitätselement der V-Gene). Dieses Merkmal ermögliht Vergleihe zwishen
B-Zell-Lymphomen und normalen B-Zellen und kann damit zur Identizierung des
Entwiklungsstadiums der Vorläuferzellendienen.
T-Zellen werden im Knohenmark gebildet, aber im Thymus ausdierenziert.
Zel-len, die fremde Antigene auf ihrer Membranoberähe repräsentieren, werden von
zytotoxishenT-Zellen(CD4
−
CD8
+
),dieeinenMHCIerkennendenT-ZellRezeptor
besitzen,erkanntundgetötet.T-Zellen,dieeinenMHCIIerkennendenT-Zell
Rezep-torbesitzen, werden allgemeinalsT-Helfer-Zellen bezeihnet. Es gibt des Weiteren
nohregulatorishe T-Zellen(Treg), T-Gedähtniszellen und NK (Natürlihe Killer
T-Zellen). Da bei der Entwiklung und Ausdierenzierung der T-Zellen weder ein
Klassenwehsel noh eine somatishe Hypermutation stattndet, ist es shwer, die
Vorläuferzellen der T-Zell-Lymphomezu denieren [Alberts et al.,1995℄.
1.3 Die Miorarray-Tehnologie von Aymetrix
DieMiroarraysvonAymetrixverwendeneineKombinationausPhotolithographie
und einer kombinatorishen hemishen Synthese, um die einzelsträngigen
DNA-Fragmente (sogenannte Oligonukleotide) auf eine Glasoberähe zu synthetisieren
[Lipshutz et al.,1999℄.
Diese Fragmente haben eine Länge von 25 Basen (auh Probe genannt). Jedes zu
messendeGenwirddurheinebestimmteAnzahlvonProbes,abhängigvon
Miroar-ray-Typ und Gen, repräsentiert. Die Anzahl shwankt zwishen 11 und 20 Probes
proGen.DiesesogenanntenPerfetMathes (PM)repräsentierendie
einesGens.DasMM hatander13.BasenpositioneinenkomplementärenAustaush
derreellenBaseundstelltdamitdienihtspezishe Bindungunddas
Hintergrund-raushen dar.
In Abbildung 1.5 ist shematish der Aufbau eines Probesets dargestellt. Die PMs
eines Probesets sind niht an nur einer Stelle auf dem Miroarray, sondern
rando-misiertaufdem Chip verteilt, um möglihe lokaleräumlihe Eekte zu minimieren.
Auh wenn nur ein Teil der PMs eines Probesets detektiert werden konnte, ist das
Signal aus den verbliebenen PMs noh zu verwenden. Jedes Probe ist mit 10
6
bis
10
7
Kopien auf dem Chip vertreten und bildet die sogenannte Probe-Cell. Das
Se-quenzdesign der einzelnen Probes auf dem Chip istso gewählt, dass Sequenzen der
mRNA nahe 3'-Ende abgebildet werden, um Probleme von teilweise degradierter
mRNAoder durhdieInvitrotranskription(Verfahrenzur Vermehrungder mRNA)
verkürzter RNAszuverringern.Am3'-EndebindetdiePolymerase,diefürdas
Invi-trotranskriptionsverfahrenbenötigtwird.JeweiterdiePolymeraseRihtung5'-Ende
wandert, umso höher ist die Wahrsheinlihkeit, dass diese ihre Funktion abbriht.
DaherkommtesvorallembeilangenTranskriptenoderbeishlehterRNA-Qualität
zu einer shlehten Abdekung der Probes Rihtung 5'-Ende.
Abbildung1.5: Allgemeines Probeset-Design füralle 3'-Expressions-Miroarrys von
Aymetrix
FürdievorliegendeArbeitwurden vershiedene Chip-GenerationenvonAymetrix
verwendet (HGU95, HGU133A, HGU133Plus2.0), die sih im Probe-Design und in
derAnzahl der Gene untersheiden. BeimSannen wird dieFluoreszenz aller Probe
Cells gemessen und im sogenannten DAT-File gespeihert. Diesen kann man mit
Ayme-trixvisualisieren. Ausden einzelnenProbe-Cells wirddieFluoreszenzstärkeineinen
numerishen Expressionswert umgewandelt und im sogenannten CEL-File
gespei-hert. Das CEL-File repräsentiert also die Rohdaten in Form einer numerishen
Matrixund kann fürweitere statistishe Analysen verwendet werden.
Aymetrix selbst hat Algorithmen entwikelt, um aus den einzelnen
Expressions-wertenderProbes einenaggregiertenWert fürdas Probeset oderGen zuberehnen.
Allerdings wurde dieses Verfahren shon mehrfah in der Literatur kritisiert. Dort
wurdebelegt, dass diese Methode einigeShwähen aufweist [Irizarry etal., 2003b;
Lazaridisetal.,2002℄.Esgibteine Vielzahlkommerziellerund freierhältliher
Soft-ware zur statistishen Analyse von Miroarray-Daten. R ist die freie Software mit
einer Sammlung von Softwarepaketen mit neu implementierten Algorithmen. Alle
statistishenAnalysen dieserArbeitwurdenmitRund Bioondutordurhgeführt.
Bioondutor ist ein frei erhältlihes Softwareprojekt, aufbauend auf R zur
Erstel-lung von Werkzeugen, zur Analyse und zur Interpretation genetisher Daten.
DasvierstugeVerfahren(Hintergrundkorrektur,Normalisierung,PM-Adjustierung,
PM-Signal Zusammenfassung), gehört zum ay-Paket von Bioondutor. Es wird
imMaterial- und Methodenteil detailliertbeshrieben und aggregiert aus den
Pro-be-Signalendes CEL-Fileseinen Expressionswert pro Probeset oder Gen.
1.4 Das Miroarray-Chip-Design von Aymetrix
Gegeben ist ein Projekt mit n Chips, wobei jeweils ein Chip eine Gewebeprobe
repräsentiert. Für jeden Chip
i
= 1
, . . . , n
sind m Gene vorhanden, die durhl
j
(
j
= 1
, . . . , m
)
Probe-Pairs vertreten sind. Jedes Probe Pair besteht aus einemPM und einem MM (Siehe Abb.1.5). Gegeben ist also
P M
ijk
, wobeii
= 1
, . . . , n
der Chip,j
= 1
, . . . , m
das Gen undk
= 1
, . . . , l
j
das Probe ist. Abhängig vom Chiptyp und dem jeweiligen Gen kann dieAnzahl vonl
j
dierieren. Somitergeben sihn
·
P
m
j
=1
l
j
Werte für diePM-Werte, äquivalenterhält man dieMM-Werte mitMM
ijk
.Übersiht der Analyse im Bioondutor
Zunähst wird eine Hintergrundkorrektur vorgenommen. Dies ist nötig, da auh
Bio-ondutor stellt im ay-Paket drei vershiedene Möglihkeiten zur Auswahl. Da in
der Normalisierungmethode VSN von Huber et al. dieser Shritt shon enthalten
ist,wird aufdiese Korrekturverzihtet. Aufgrund untershiedliher mRNA-Mengen
kann eszu vershiedenen Fluoreszenzintensitätenkommen.Im zweitenShritt wird
deshalb eine Normalisierung durhgeführt. Bioondutor bietet dazu eine Vielzahl
Möglihkeiten. Alle Reanalysen wurden mit der Normalisierungsmethode VSN von
Huber et al.normalisiert. Im Material- und Methodenteil wird hierauf detaillierter
eingegangen.Bei diesem Shritt erhält man die normalisiertenWerte
P M
norm
ijk
undMM
norm
ijk
. Für den nähsten Shritt, die PM-Adjustierung, wird auf dieMM
norm
ijk
verzihtet, das heiÿt hier ndet keine Verrehnung der
P M
norm
ijk
mit denMM
norm
ijk
statt.DiesesVorgehenshlagenBolstadetal.[Bolstad etal.,2003℄ vor.
Berüksih-tigtwird damit,dass dieMM Signaleoft niht wie vonAymetrix angenommen
unspezishe Bindungen darstellen [Naef et al., 2002℄. Der Wert für das
Probe-Pair-k wird durh
P M
norm
ijk
repräsentiert. Dien
·
P
m
j
=1
l
j
Werte haben sih in diesem Shritt halbiert. Wie wird nun ausl
j
Wertendes Gensj
einWertfürdieExpressiondiesesGensj
berehnet? Bioondu-tor stellt einige Möglihkeiten zur Verfügung. Dieser abshlieÿende Shritt bei denReanalysenwirdmitder Methode vonTukeyet al.[Tukey, 1977℄ durhgeführt.Der
Material-undMethodenteilenthält eine detaillierteBeshreibung dieserMethoden.
Die verrehneten
P M
norm
ijk
, oder allgemeinery
ijk
, für jeden Chipi
und jedes Genj
werden imWeiteren mitx
ij
bezeihnet und bildenzusammen dieMatrixX
.X
=
x
11
x
12
· · ·
x
1
n
x
21
x
22
· · ·
x
2
n
. . . . . . . . . . . .x
m
1
x
m
2
· · ·
x
mn
DieseMatrixist dieessentielle Grundlagefür allestatistishen Analysen. Abhängig
von der Fragestellung gibt es vershiedene weitere statistishe Analysen-Methoden.
Eine wihtige Fragestellung ist z. B., welhe Gene dierentiell zwishen zwei
de-niertenGruppen exprimiertsind und in welhem Ausmaÿ.
Das Ausmaÿ, also die Stärke der dierentiellen Expression wird auh Fold Change
(FC)genannt.WerdendieGruppenimVorfelddeniert,sprihtmanauhvon
über-wahten (supervised) Analysen.Sowohl dieSignikanz eines Gens alsauh der FC
ha-ben, werden alsbiologishrelevantangesehen (z. B. FCmindestens2- oder 3-fah).
So können z. B. Subtypen einer Erkrankung, z. B. mit Hilfe von Clusteranalysen
entdektwerden.BeidiesenClustermethodenwirdimAllgemeinennihtüberwaht
(unsupervised) vorgegangen, das heiÿt die Gruppen werden niht vorher deniert.
Auh gibt es Methoden, die erlauben, einen Prädiktor zu erstellen, der
zukünfti-ge Patientengruppen eindeutig einer bestimmten Klasse zuordnet. Immer mehr an
Bedeutung gewinnt die Fragestellung nah involvierten Signalwegen und
Genfunk-tionsgruppen. Dabeiwird niht mehrauf der Einzel-Gen-Ebeneanalysiert, sondern
in zu einem Signalweg gehörenden Gengruppen oder einer Funktionsgruppe. Von
besonderembiologishenInteressesind Gengruppenoder Signalwege,dieeine
Über-repräsentierung oder Unterrepräsentierung zeigen.
1.5 Ziel dieser Arbeit
Dievorliegende Arbeit istin zweiTeile untergliedert:
1. Die Vorteile neuer statistisher Methoden bei der Miorarray Auswertung an
shon veröentlihten Datensätzen werden dargestellt:
•
Am Beispiel der Analyse von Klein et al. [Klein et al., 2001℄ werden Ansätze zur Verbesserung der Methode aufgezeigt.•
Ergebnisse derReanalyse sindzusätzlihe Gene,dienurnahder Reana-lyse gefunden werden.•
diebiologishe Relevanz der zusätzlihgefundenen Gene.2. Expressionsdaten, die aus geringen mRNA-Mengen stammen, werden
statis-tish analysiert:
•
Präprozessierungs-Algorithmen werden ansimuliertenRohdaten geprüft und bewertet.•
Welhe Methoden zeigen welhe Vorteile?•
ZeigtdieGewihtungder Probes inAbhängigkeitihrerPositionimT ran-skript die Vorteile der Analyse?•
Reanalyse eines Vergleihes mit allen angewendeten Präprozessierungs-Algorithmen.FürdieReanalyse wurden dieDatensätze vonKüppers etal.[Küppers etal.,2003℄
und Pialugaet al. [Pialuga et al., 2007℄ verwendet. Dr.Ulf Klein hat beide
Da-tensätze mitderselben Methode statistish analysiert.
Wie shon im Vorfeld erwähnt, gibt es bestimmte Erkrankungen, bei denen das
Ausgangsmaterial nur begrenzt zur Verfügung steht. Mit dem in unserem Institut
entwikeltemProtokollisteserstmalsmöglih,ausnur1000ZellengenügendmRNA
zuamplizieren,umeinMiorarray-Experimentdurhführenzukönnen.Dader
ver-wendeteMiroarray für dieseArt ampliziertermRNA niht konzipiertist,kommt
eszu systematishenFehlern.DerAnteilanfalshnegativenWerten isthoh.Inder
vorliegendenArbeitwirdnahderNormalisierungnoheineGewihtungder Probes
vorgenommen. Diese Methode berüksihtigt wesentlihe Teile des systematishen
Fehlers,der währendder mRNA-Prozessierung entsteht.
Zwar haben Cope et al. [Cope et al., 2006℄ gewihtete Probes in ihren
Algorith-musRMA(Robust MultihipAverage)implementiert,konntenaberkeinedeutlihen
Untershiede und Verbesserungen zum Standard RMA-Algorithmusfeststellen.Die
verwendete Normalisierungsmethode VSN von Huber etal. zeigte eine höhere
Spe-zität und Sensitivität bei der dierentiellen Expression. Damit war ein anderes
ErgebnisalsbeiCope etal.zu erwarten.
ZuerstwurdensimulierteDatenverwendet,umdievershiedenen
Präprozessierungs-algorithmenvergleihenzukönnen.AneinemdafürkonzipiertenTestdatensatz
wur-den dievershiedenen Methoden verglihen. Dazu wurden zwei vershiedene
Grup-penvonAusgangsgewebegewählt,dieeinhohesMaÿandierentiellerExpression
er-wartenlieÿen.AnhandvonQualitätskriteriensolltegeklärtwerden, obdie
vershie-denenMethoden einenEekt aufdieweitere statistishe Analyse haben.
Qualitäts-kriteriensinddabeiSensitivitätundSpezität,Anzahlderdierentiell-expremierten
Gene und der Einuss auf resultierende Gengruppen (z. B. Gen-Ontologien und
Signalweg-Analysen).LetztendlihsolltendievershiedenenMethodenaneinem
rea-lenTeildatensatzvonBrune etal.[Brune et al.,2008℄angewendet und miteinander
verglihen werden.
Im zweiten Kapitel werden die verwendeten Datensätze bezüglih ihrer
mRNA-Prozessierung,der verwendeten Materialienund Gewebebeshrieben. ImAnshluss
Daten-sätzeangewendet wurden. Esfolgteine Beshreibung der gewihtetenProbes durh
ein angepasstes lineares Modell. Im Ergebnisteil wird deren potentieller Vorteil
er-örtert.
Im dritten Kapitel werden zunähst die Ergebnisse der Reanalyse der beiden
Da-tensätze dargestellt. Anshlieÿendgeht es um dieSimulationvon Expressionsdaten
mit vershiedenen Modellen und die Anwendung der vershiedenen Methoden, die
zuletzt auf einen Teildatensatz angewendet und verglihen werden. Das Verhalten
im biologishen Kontext wird besonders herausgearbeitet und wenn möglih
eine Empfehlungzur weiteren statistishen Analyse gegeben.
Material und Methoden
2.1 Verwendete Genexpressionsdatensätze
2.1.1 Datensatz von Küppers et al.
Küppers etal.[Küppers etal.,2003℄haben dieerstegenomweite
Genexpressionsan-layse mit Aymetrix-Miroarrays an Hodgkin-Lymphom-Zelllinien (HL-Zelllinien)
imVergleih zu normalen B-Zellen durhgeführt. Bei den HL-Zelllinien, handelt es
sihum kontrollierte,etablierte und kultivierteZellliniendievonPatienten mitHL
stammen.
Thomas Hodgkin beshrieb 1832 zum ersten Mal das HL [Hodgkin, 1832℄. Fast 70
JahrespäterwurdeesvonSternberg [Sternberg,1898℄und Reed[Reed,1902℄weiter
harakterisiert. In der westlihen Weltist das HL mit 2- 4 Fällenpro 100.000eine
selteneErkrankung, abermit einemAnteilvon30% eines der häugsten malignen
ErkrankungendeslymphatishenSystems.DasHLlässtsihinzweiIdentitäten
un-terteilen:dasklassishe HL(HL)unddasnodulärelymphozyten-prädominanteHL
(NLPHL).95%aller HLssindder IdentitätHLzuzuordnen. Die Besonderheit,die
beideIdentitätenharakterisiert,sinddiewenigengroÿenneoplastishen
(neubilden-den)Zellen,diesihineinemzellulärenInltratnihtneoplastisherZellenbenden
[Jaeetal.,1998℄. Bei HLspriht man dannvonHRS-Zellen und beiNLPHL von
L&H-Zellen.Wenigerals1%des inltriertenGewebesmahendieHRS-Zellenoder
L&H-Zellenaus[Hansmannetal.,1999;Weissetal.,1999℄.Allerdingsuntersheiden
sihbeide Identitätendes HLsu.a.imImmunphänotyp,inder Epidemiologieund
Das HL wird vor allem in vier Subtypen unterteilt: nodulär sklerotisierend (NS;
60 - 80 % der HL), mishzellig (MC; 15 - 30 % der HL), lymphozytenarm (LD;
1%der HL) und lymphozytenreih (IrHL;6 %der HL) [Harris,1999℄. Moderne
Therapiekonzepte haben zu einem sehr guten Therapie- und Heilungserfolg beim
HLgeführt unabhängigvomSubtyp. Eine genetishe Disposition liegt nahe, daein
gehäuftesAuftretendesHLsineinigenFamilienundein99-fahhöheresRisikoeiner
ErkrankungbeieineiigenZwillingenvermutet wird [Gruerman and Delzell, 1984℄.
Epidemiologishen Studien zeigten eine Abhängigkeit des Erkrankungsrisikos von
Geshleht, sozialem Lebenssituation und ethnishen Hintergrund [Gutensohn and
Cole,1980℄.
DieHL-ZellliniendientenbeiKüppersetal.alsrepräsentativesModellzumprimären
HLdeslymphatishenGewebes. DieHL-ZelllinienL428undHDLM2stammenvon
PatientenmitHLmitdem SubtypNSund dieHL-ZelllinienKMH2undL1236vom
Subtyp MC. Die HDLM2 stammen von T-Zellen, die anderen drei stammen von
B-Zellenab. Allenormalen B-Zellender vershiedenen Subtypen (GC-B-Zellen,
Ge-dähtnis B-Zellen,NaiveB-Zellen und PC-Zellen) wurden aus menshlihen T
onsil-len(Mandeln) gewonnen.
Der Datensatz von Küppers et al. wurde mit dem Aymetrix Miroarray Typ
HGU95A erstellt. Auf diesem Miroarray benden sih 12626 Probesets.
Verwen-detwurdedas Standard-AymetrixProtokoll(Einsatzmengeungefähr
1
·
10
6
Zellen
pro Miroarray).
2.1.2 Datensatz von Pialuga et al.
Der zweite reanalysierte Datensatz ist von Pialuga et al. [Pialuga et al., 2007℄.
IndieserArbeitwurdeeine bestimmteGruppevonT-Zell-LymphomenimVergleih
zunormalenT-Zellenuntersuht.BeidenT-Zell-Lymphomenhandeltessihum
so-genannte periphere T-Zell-LymphomeTyp unspezish (PTZL/U). In diese
Grup-pefallenalleT-Zell-Lymphome,dienihtweiter harakterisiertwerden können.Die
PTZL/U-LymphomesinddieammeistenverbreitetenT-Zell-Lymphomeundzeigen
morphologishund phänotypisheine sehrgroÿeHeterogenität.Auÿerdemsprehen
diese Lymphome nur shwah auf die Therapie an und haben eine shlehte
Pro-gnose. 60 - 70 % der PTZLs fallen in die Gruppe der PTZL/U und können niht
mit der Morphologie, mit dem Phänotyp oder durh molekulare Untersuhungen
ag-gressivsteNHL. Insgesamt wurden 28PTZL/U und 20normleT-Zell-Populationen
(bestehend aus CD4 und CD8) verwendet. Auÿerdem wurden zusätzlih zwei
wei-tere Subtypen des T-Zell-Lymphomsmit jeweils 6 Miroarays verwendet (AITL =
angioimmunoblastishesT-Zell-Lymphom,ALCL=anaplastishesgroÿzelliges
Non-Hodgkin-Lymphom).DienormalenT-Zell-Populationenstammenentwederausdem
Blutoder aus Tonsillen gesunder Spender.
FürdieseStudiewurdederHGU133Plus2.0Miroarray-TypvonAymetrix
verwen-det.Dieserhat 54676 Probesets auf demMiroarray, wasungefähr20000 einzelnen
Genenentspriht. Verwendet wurde das Standardprotokollvon Aymetrix.
Pialugaetal.haben sogenannte Ganzshnitte fürdieeinzelnen Tumorprole
ver-wendet.HierwurdeeinganzerQuershnitt einesTumorarealsgewählt,derzum Teil
groÿeMengenanKontaminationenenthält.MitKontaminationsind hieralleZellen
gemeint,die nihtTumorzellensind.
2.1.3 Test-Datensatz
Bevor der Datensatz von Brune et al. [Brune et al., 2008℄ generiert wurde,
muss-te eine geeignete Methode etabliert werden, um aus dem zur Verfügung stehenden
MaterialüberhauptGenexpressionsanalysenmitMiroarrays vonAymetrix
durh-führen zu können.
Die Zellen des HLs wurden einzeln per Lasermikrodissektion aus dem
Gewebe-shnittisoliert. Deshalb konntenureine Mengevonmaximal 1000 Zellen pro
Pati-entisoliertwerden.DieLaser-MikrodissektionunddiePressure-Catapulting-Tehnik
(LMPC) mit UV-Laser der Firma P.A.L.M. mahten es erstmals möglih, einzelne
Zellen so aus dem Gewebe zu isolieren, dass die DNA- und RNA-Qualität
ausrei-hend gut war, um weitere Experimente durhzuführen. Zum Zeitpunkt der
Date-nerhebung gab es nur das Standardprotokoll von Aymetrix und anderen Firmen,
derenProtokolleaber mindestens
1
·
10
6
Zellen benötigen.
Um trotzdem Genexpressionsanalysen an nur 1 000 Zellen durhführen zu können,
musste eine neue mRNA-Isolierungs- und mRNA-Amplikationsmethode gefunden
werden. Nahdem eine geeignete Methode gefunden wurde [Brune etal., 2008℄
tes-tete man zunähst an einem kleinen Datensatz die Qualität des neuen Protokolls.
Hierfür wurden eine HL-Zelllinie und ein Burkitt-Lymphom-Probe verwendet. Da
inFolgedes neuenProtokollszweiVermehrungsshritte (Invitrotranskription)der
maskiertsind. Das Probe-Design von Aymetrix istauf dem verwendeten
Miroar-ray HGU133Plus2.0 niht für solhe vorbehandelten Zellproben vorgesehen. Zwar
konnten dieDaten erfolgreihmitder VSN-Methode vonHuberetal.[Huberetal.,
2002,2003℄analysiertundpubliziert[Bruneetal.,2008℄werden,dieFragenaheiner
geeigneten Anpassung der Daten an das Miroarray-Design blieb allerdings oen.
DerTestdatensatz eignetsih,um eine Methode zu entwikeln, diedas Miroarray
-Designmiteinieÿenlässt. Duplikateeiner HL-Zelllinieund einesBL-Falleswurden
mitdem neuen Protokollund dem Standardprotokoll vonAymetrix durhgeführt,
um beide zu vergleihen.
2.1.4 Datensatz von Brune et al.
Der Datensatz von Brune et al. [Brune et al., 2008℄ wurde mit etablierten
statis-tishen Verfahren ausgewertet, allerdings ohne auf das Miroarray-Design und die
zusätzlihe Problematikmitder geringen Ausgangsmengean mRNAeinzugehen.
Der Datensatz von Brune et al. enthält zwei Gruppen: Zum einen die
mikrodissi-zierten Lymphomzellen, die entweder in Zellgruppen oder als Einzelzellen isoliert
wurden,zumanderen durhFluoreszenz-aktivierterZellseperation(FACS) sortierte
normaleB-Zellen aus der Tonsillevongesunden Spendern. Inder ArbeitvonBrune
et al.wurden Gene identiziert, die bei der Pathogenese des NLPHL eine wihtige
Rollespielen.ZumVergleihwurden dieniht-malignenUrsprungszellen,die
GC-B-Zellen,verwendet.DerVergleihvonHLundGC-B-Zellenwirdindervorliegenden
Arbeitreanalysiert und mit den Standardverfahren verglihen.
2.2 Ablauf eines Miroarray-Experiments
InAbbildung2.1istshematishdargestellt,wieeinGenexpressionsprolausZellen
erstellt wird. Im ersten Shritt wird aus einem Zellpool aus Tumorgewebe oder
einer Zellkulturdie mRNA extrahiert. Diese wird im zweiten Shritt vermehrt und
in unserem Fall, mit Fluoreszenz markiert. Im dritten Shritt wird die markierte
mRNA auf dem Miroarray-Chip hybridisiert. Im letzten vierten Shritt wird die
FluoreszenzmitdemAymetrix-Sannergemessen.GenauereInformationenenthält
Abbildung2.1: Ablaufeines Miorarray-Experiments
2.3 Tehnishe Vorraussetzungen
Die statistishe Auswertung wurde mit der frei verfügbaren Statistiksoftware R,
Versionen 2.1und 2.8, durhgeführt [Ihakaand Gentleman,1996; Gentleman etal.,
2004℄.Auÿerdem wurden vielePakete aus dem Bioondutor-Projektbenutzt(ay,
vsn,geneplotter,mathprobes usw. [Gautieretal.,2004; Huberetal., 2002;
Gentle-manetal.,2004; Huberand Gentleman,2004℄).Bioondutorist fester Bestandteil
inder Analyse vershiedener, hohdimensionalergenomisher Daten geworden.Das
liegt an der groÿen Anzahl implementierter Algorithmen und der Tatsahe, dass
die Software frei verfügbar ist und von herausragenden Wissenshaftlern gepegt,
und durh neue Pakete erweitert wird. Die Software ist niht nurim akademishen
Bereihverbreitet, sondern bietet auh fürkommerzielle Software Shnittstellen zu
Bioondutor. FürdieDatenanalyse wurden einRehner mitIntelArhitektur(3,2
GHzCPU) und 4GBRAM mitBetriebssystem Windows XPund einLinux-Server
mit16GBRAMverwendet.VorallemderVSN-AlgorithmusvonHuberetal.[Huber
2.4 Explorative Datenanalyse
2.4.1 Boxplot
Zurgrashen Visualisierungeines Sets von n Miroarrays kann manBoxplots
ver-wenden. Durh die grashe Dartsellung bekommt man eine gute Übersiht über
dieVerteilungder Signalintensitäten der einzelnen Chips. Wihtige Streuungs- und
Lagemaÿe(Median,diezweiQuartileundExtremwerte)zeigtenshnell,inwelhem
Bereihdie Daten liegen und wie dieDaten überdiesen Bereih verteilt sind.
Die Box beinhaltet die mittleren 50 % der Daten. Sie wird durh das obere und
untereQuartilbegrenzt.Die Ausdehnung der Boxrepräsentiert die
Interquartilran-ge (IQR). Sie ist ein Maÿ für die Streuuung der Daten und wird aus der Dierenz
des oberen und unteren Quartilsberehnet. Whiskers sind die Datenpunkte
auÿer-halb der Box. Die Länge der Whiskers sind niht unbedingt festgelegt.John Tukey
[Tukey, 1977℄ legte fest, dass die Whiskers maximal die 1,5 fahe der IQR haben.
Allerdingsendet derWhisker niht genauandiesem Punkt,sondern andem jeweils
äuÿersten Datenwert, der noh innerhalb dieser Grenze liegt. Die Daten auÿerhalb
der Whiskers werden alsAusreiÿer deniert (Abb.2.2).
Abbildung 2.2: Darstellungeines horizontalenBoxplots
2.4.2 Histogramm
Ein Histogrammkann einen guten Überblik über die Verteilung der Daten geben
(Abb.2.3). Zu bedenken ist allerdings, dass das Aussehen von Histogrammen oft
starkvon der Klassenanzahlund beiwenigen Klassenauhvonder Platzierung der
Histogramm
Werte logarithmiert
Häufigkeiten
4
6
8
10
12
14
0e+00
1e+05
2e+05
3e+05
4e+05
Abbildung2.3: Histogramm-Beispiel 2.4.3 Q-Q-PlotDer Quantil-Quantil-Plot (Q-Q-Plot)stellt dieQuantilezweier statistishen V
aria-blengrashgegenüber. DabeiwerdendieBeobahtungsmerkmalezweierMerkmale
der Gröÿe nah sortiert, als Wertepaare zusammengefasst und grash in einem
Koordinatensystem dargestellt. Ergeben die Punkte annähernd eine Gerade
y
=
x
kannman davonausgehen,dass beideMerkmalegleihverteiltsind. BeimVergleihmit den Quantilen der Standard-Normalverteilung gilt zusätzlih, dass die Gerade
y
=
a
+
bx
der Normalverteilungmit(
µ, σ
2
)
entspriht.
2.5 Präprozessierungsalgori t hmen für die
Normali-sierung von Aymetrix-Miroarray-Rohdaten
2.5.1 Ziel der Präprozessierung
Aufgabenstellung:
AusdenBilddatenvonAymetrixsollenmitgeeignetenVerfahrenExpressionswerte
fürdieProbesets ermitteltwerden.Typisherweise lässtsihdiePräprozessierungin
vierShritteunterteilen,diesihjenahangewandterMethode starkuntersheiden.
Zunähstwirderläutert,auswelhemGrunddieeinzelnenPräprozessierungsshritte
1. Die Hintergrundkorrektur:
EinHintergrundsignalkanndurhEigenuoreszenzderReagenzien,Streuliht
oder sonstigetehnisher Problemeentstehen. Selbstnahdem Sannen eines
Arrays ohneRNA (z. B. mitdestilliertemWasser) wird man ein
Fluoreszenz-signalmessen können.Zielder Hintergrundkorrektur ist,dieStärke dieses
ar-tiziellenSignalszu shätzen und vonden gemessenen Werten zu eliminieren.
Ist dieses Verfahren erfolgreih, sollte ohne RNA auf dem Miroarray keine
Fluoreszenzsignalmehr meÿbarsein [Gautier etal.,2004℄.
2. Normalisierung:
Unabhängigdavon,welhedervielenvershiedenenNormalisierungsmethoden
verwendetwird,allehabendas Ziel,dien-ArrayseinesExperiments
vergleih-bar zu mahen. Besteht ein Experiment aus mehreren Arrays, so ist die
Va-riabilitätder Messwerte sehrgroÿ. Dabeimuss man zwishen der biologishen
(Untershied zwishen Tumorzellenundnormalen Zellen)undder tehnishen
Variabilität(BenutzungvoneinemodervershiedenenProtokollen)
untershei-den. Die biologishe Variabilität ist die für den Wissenshaftler interessante.
Dietehnishe Variabilitätisthiereher störendundkann dazuführendie
bio-logishe Variabilitätzu verändern. Ziel istdeshalb, durh die Normalisierung
dietehnishe Variabilitätzu minimieren.DieUrsahen fürdietehnishe
Va-riabilitätkönnen vielfältigsein. Einussreihste Quelle ist wahrsheinlih die
Durhführung des Experiments, aber auh die Herstellung der Miroarrays
und der Laborreagenziensind eine Fehlerquelle. Shon geringfügige
Änderun-gen des Protokolls (z. B. Ausgangsmenge mRNA) können dabei zu massiven
Untershieden des Fluoreszenzsignalsführen.
Mit Hilfe von Boxplots, QQ-Plots und Histogrammen kann man wesentlihe
Untershiede der vershiedenen Experimente erkennen. Ohne Normalisierung
käme eshier zu dierentiell exprimierten Genen.
Die Normalisierungsverfahren lassen sih in zwei Gruppen aufteilen. Es gibt
Verfahren, die einen Miroarray als Basis verwenden, mit dessen Werten die
anderen n - 1 Arrays normalisiert werden. Andere Methoden benötigen für
die Normalisierung alle Arrays. Es gibt bis heute keinen Goldstandard für
dieNormalisierungmethode. Allerdingsgeben Boes und Neuhäuser [Boesand
Neuhäuser, 2005℄ einen guten Überblik über vershiedene
3. Probe-spezishe Hintergrundkorrektur:
Nah der Normalisierung ist zu entsheiden, ob und wie die PM-Werte und
MM-Wertemiteinanderverrehnetwerden.ZielderPM-Adjustierungist,einen
Wert für das Probe Pair zu berrehnen. Bolstad et al. [Bolstad et al., 2003℄
haben alsErste vorgeshlagen,nurdiePM-Werte für diePM-Adjustierung zu
verwenden.
4. Aggregationsmethoden:
Nahder PM-Adjustierung hatmaneinenWert
y
ijk
fürjedes Probeset bereh-net,dabei isti
der Miroarray,j
das Gen undk
das Probe.Zielder Aggrega-tionsmethode ist es, aus den Einzelwerten eines Probesets einen Wertx
ij
zu berehnen.Die detaillierte Beshreibung der in dieser Arbeit verwendeten Modelle folgt
die-sem Kapitel. Es gibt auÿer den genannten eine Vielzahl weiterer Modelle für die
Präprozessierung.
2.5.2 Das Modell von Aymetrix
Aymetrix[Aymetrix, 2002℄ hat einen eigenenAlgorithmusentwikeltund diesen
ineine Software Namens Miroarray Suite (MAS)implementiert.Mit diesem
Algo-rithmus werden die vier Shritte der Präprozessierung durhgeführt. Die MAS gab
eszunähstindenVersionen4.0und5.0.BeiVersion4.0warennegativeSignalwerte
möglih.Dasistaus biologisherSiht nihtsinnvollund fürweitereAnalysen istes
problematish,mitnegativen Werten umzugehen. Bei Version 5.0wurde dies
geän-dert. Heute istdie Software von Aymetrix (Expression Console 1.1= EC1.1) frei
verfügbar und neben dem MAS 5.0-Algorithmus sind weitere Algorithmen (unter
anderem RMA) implementiert.
MAS-4.0-Algorithmus
DieMethode zurBerehnungder ExpressionbeiMAS4.0wirdauhAverage
Die-rene genannt.Diese mittlerweileveralteteMethode wurdebeidem Datensatzvon
Küppers et al. [Küppers et al.,2003℄ verwendet. Deshalb hier eine kurze
Beshrei-bung:
DurhshnittderDierenzenderPM-WerteundderdazugehörigenMM-Werteeines
Probeset ist.ZielvonAymetrixfürdieProbe-spezisheHintergrundkorrekturwar
das Einstellen von PM-MM. Bis November 2002 war dieser Algorithmus Standard
beiAymetrix.Umeinen robusten Durhshnittswert fürdieeinzelnen ProbePairs
zugewährleisten,werdenzunähstmehrere Werte ausder Durhshnittsberehnung
herrausgenommen.
Minimalen und Maximalen Wert der
y
ijk
über alle Probe Pairsk
eines Gensj
imi
-tenMiroarray werden imerstenShritt zunähst ignoriert,daessihumAusrei-ÿer handeln könnte. Aus den verbleibenden Werten werden der Mittelwert
y
ij
unddieStandardabweihung
s
(
y
ij
)
berehnet. Im letzten Shritt werden alleWertey
ijk
gelösht,diemehr alsdrei StandardabweihungenvomMittelwertzeigen. Wenn diegelöshtenWertenihtmehralsdreiStandardabweihungenvomMittelwertentfernt
abweihen,können siedabei wiederberüksihtigtwerden. DieMenge
A
ij
wird also aus den verbliebenden Werten der Probe-Pairs gebildet. Dieses wird alsAverage-Dierene,oder kurz AvgDi,bezeihnetund lässtsihfürjedesGen
j
einesChipsi
folgendermaÿen berehnen:AvgDif f
ij
=
1
N
(
A
ij
)
X
k
∈
A
ij
y
ijk
(2.1)Dabei ist
N
(
A
ij
)
die Anzahl der Werte inder MengeA
ij
.FürdieAnalysemitMAS5.0werdenzunähstdieWertederProbe-CellseinesChips
benötigt.AlsNähsteswirdeineHintergrundkorrekturdurhgeführt.Fürdie
Hinter-grundkorrektur wird der Miroarray in K gleih groÿe Segmente
Z
k
(
k
= 1
, . . . , K,
Voreinstellung K=16) unterteilt. Kontroll-Probe-Cells und maskierte Probe Cellsbleiben bei der Kalkulation unberüksihtigt. Die Probe-Cell Intensitäten werden
nah Gröÿe sortiert. Die kleinsten 2 % repräsentieren das Hintergrundsignal b für
das Segment (
bZ
k
). Die Standardabweihung von den kleinsten 2 % Probe Cell -Intensitäten wird berehnet und als Shätzer für die Hintergrundvariabilitätn fürjedes Segment (
nZ
k
)verwendet.Um einen nahtlosen Übergang zwishen den Segmenten zu gewährleisten, wird der
räumlihe Abstandvon jedemProbeCell aufdem Miroarray zu denvershiedenen
K
-Segmentshwerpunkten mitd
k
(
x, y
) (
k
= 1
,
· · ·
, L
)
berehnet. Als Gewiht für dieBerehnung des Hintergrunds jeder Probe Cell wird der reziproke Abstand derjeweiligen Probe Cell zu den
K
-Segmentshwerpunkten berehnet. Deshalb hat ein SegmentmitnaheliegendemShwerpunkteingröÿeres GewihtbeiderBestimmungdes Hintergrundsignals als ein von der Probe-Cell weit entferntes Segment. Die
er-mitteltenGewihte werden mit
w
k
(
x, y
)
bezeihnet. Siekönnen berehnet werden:w
k
(
x, y
) =
1
d
2
k
(
x, y
) +
i
(2.2)
i is dabei eine Konstante (Voreinstellung = 100), die verhindert, dass der Nenner
Null wird. Mit dem berehneten Segmenthintergrundsignal
b
Z
und den Gewihtenw
k
(
x, y
)
kann man das Hintergrundsignal jeder einzelnen Probe-Cell berehnen:b
(
x, y
) =
P
K
1
k
=1
w
k
(
x, y
)
K
X
k
=1
w
k
(
x, y
)
bZ
k
(2.3)UmhintergrundbereinigteWerte für jedes Probe-Cell zu erhalten, subtrahiert man
diein(2.3)geshätztenHintergrundwertevondenjeweilsgemessenenProbe-Intensitäten.
DieShätzungdesHintergrundsignalskanngröÿeralsdiegemesseneProbe-Intensität
sein.UmkeinenegativenWertezuerhalten,werdendiehintergrundbereinigten
Wer-tefolgendermaÿenberehnet:
A
(
x, y
) =
max
(
I
′
(
x, y
)
−
b
(
x, y
)
,
0
.
5
·
(
x, y
))
,
(2.4)
wobei
I
′
(
x, y
) =
max
(
I
′
(
x, y
)
,
0
.
5)
ist.I(x,y)repräsentiertdie
P M
ijk
bzw.dieMM
ijk
mitden jeweiligen Koordinaten x und y.BeiAymetrixndeteine Normalisierungaufeineranderen Ebene stattalsbeiden
anderen aufgeführten Verfahren. Die Normalisierung wird niht auf Probe-Ebene
durhgeführt, sondern mitden shon aggregiertenWerten des Probesets.
Zielist es, jedem Miroarray mit einem konstanten Wert zu multiplizieren, sodass
alle Mittelwerte gleih dem Mittelwert des Basis-Chips sind. Der erste Miroarray
dient dabei alsBasis-Array. Die übrigen n-1-Arrays werden dann normalisiert:
An-genommen
z
base
derMittelwert derWerte von
z
i
(
z
i
..
=
1
P
m
j
=1
l
j
P
m
j
=1
P
l
j
k
=1
z
ijk
)
.Dannberehnetmanβ
i
wie folgt:β
i
=
z
base
jk
z
i
..
(2.5)
DieExpressionswerte der normalisiertenChips erhält man folgendermaÿen:
z
norm
ijk
=
β
i
z
ijk
(2.6)Das beshriebene Skalieren ist äquivalent zu einer linearen Anpassung des
Basis-Arraysanjeden anderen Miroarray miteinemAhsenabshnitt0.Die MM-Signale
repräsentieren das Ausmaÿ der unspezishen Bindung und damit die
Hybridisie-rung von Niht-Zielsequenzen auf dem Miroarray. Verwendet man diese Methode
kommt es zu negativen Signalwerten. Biologish ist dies Unsinn, weil ein Gen
ent-weder ausgeprägt ist oder niht, negativ ausgeprägt kann es niht sein. Aymetrix
änderte deshalb ihren Algorithmus[Aymetrix, 2001℄. Dieser Algorithmus auh
Miroarray Suite 5 (MAS5) genannt verhindert negative Werte nah der
Sub-traktionder PM-Werte vonden MM-Werten.
HateinMM einengröÿerenSignalwert alsdasdazugehörigePMund kommtes
des-halb zum negativem Signalwert, ist einidealer MM (IM) zu wählen, der kleiner als
das PM ist. Deshalb berehnet man zuerst für jedes Probeset einen Probe
spezi-shen Hintergrund, der das durhshnittlihe Verhältnis zwishen PM und MM in
einemProbeset angibt:
SB
ij
=
T
bi
(log
2
(
P M
ijk
)
−
log
2
(
MM
ijk
))
, k
= 1
,
· · ·
, l
j
=
T
bi
log
2
P M
ijk
MM
ijk
, k
= 1;
· · ·
, l
j
,
(2.7)dabei ist mit
T
bi
der Tukey's Biweight[Aymetrix, 2002℄ gemeint.Der Tukey's Bi-weightisteinrobusterShätzerfürdenLageparametereinerFunktion.DieFunktiong(z) und ihre Ableitungsind gegeben durh:
g
(
z
) =
a
2
6
·
[1
−
(1
−
z
2
a
2
)
3
]
für|
z
| ≤
a
a
2
6
für|
z
|
> a
(2.8)g
′
(
z
) =
z
(1
−
z
2
a
2
)
2
für|
z
| ≤
a
0
für|
z
|
> a
(2.9)DerShätzwert für den Tukey's Biweightminimiert
X
i
g
(
z
i
−
m
)
(2.10)füreine gegebene Stihprobe
z
i
, i
= 1
,
· · ·
, n
.M ist der gesuhte Lageparameter. Im Paket ay wird m zunähst als Median
geshätzt und dann für die
z
i
−
m
wie folgtfortgefahren:Da h dieAbleitungvong ist,giltX
i
h
(
z
i
) = 0
(2.11)und dieGewihtefunktion von
w
(
z
) =
h
(
z
)
/z
für Tukey's Biweightergibt:w
(
z
) =
(1
−
z
2
a
2
)
2
,
für|
z
| ≤
a
0
,
für|
z
|
> a
(2.12)Datenabhängig von der Anzahl der Ausreiÿer wird der Parameter a gewählt. Der
IM berehnet sih deshalb:
IM
ijk
MM
ijk
fürMM
ijk
< P M
ijk
P M
ijk
2
S
B
ij
fürMM
ijk
≥
P M
ijk
undSB
ij
> τ
P M
ijk
2
τ /
(1+((
τ
−
SBij
)
/ν
))
fürMM
ijk
≥
P M
ijk
undSB
ij
≤
τ
Aymetrix shlägt Standardeinstellungen für
τ
undν
vor, die bei 0.03 bzw. 10 lie-gen.NahdemmandenIMberehnethat,lassensihPMundMMzusammenfassen:y
ijk
=
max(
P M
ijk
−
IM
ijk
, κ
)
,
(2.14)wobei Aymerix für
κ
eine Standardeinstellung vorgibt(
κ
= 2
−
20
)
. Nah der
Pro-be-spezishen Hintergrundkorrektur der Probe-Pair-Werte werden diese zunähst
logarithmiert
(
P V
ijk
=
log
2
(
V
ijk
))
, um die Varianz zu stabilisieren. Mit Tukey Bi-weight [Aymetrix, 2002℄ werden diese logarithmierten Werte zu einemExpressi-onswert pro Probeset berehnet:
MAS5Signal
ij
=
T
bi
(
P V
ij
1
,
· · ·
, P V
ijl
j
)
(2.15)DasobenberehneteMAS5SignalwirddannabshlieÿendzurBasiszweiexponiert:
x
ij
= 2
MAS5Signal
ij
(2.16)
2.5.3 Modell von Klein et al.
Klein et al. [Klein et al., 2001℄ haben zur Präprozessierung des Datensatzes von
Küppersetal.dieMethode vonAymetrix(MAS4.0)miteinerglobalenSkalierung
verwendet. Alle negativen Expressionswerte und sehr kleine positive Werte wurden
durh den Wert 20 ersetzt. Eine Denition ab welher Grenze ein Wert als klein
positivklassiziertwirdwird,gebenKleinetal.nihtan.Pialugaetal.verwenden
2.5.4 Das VSN-Modell von Huber et al.
Erst dadurh, dass niht alle Gene nah einem Experiment dierentiell exprimiert
sind, wird eine Normalisierung möglih. Denn nur Probesets, die keine
dierentiel-le Expression zeigen, lassen tehnishe Variabilität erkennen. Bei den
nahfolgen-denPräprozessierungsmodellenwerden ausshlieÿlihdiePM-Werte verwendet.Die
MM-Werte werden falls nötig (so bei Irizarry et al.[Irizarry et al., 2003a℄)
nur für die Hintergrundkorrektur berüksihtigt. Vor der Normalisierung mit VSN
erfolgt keine Hintergrundkorrektur. VSN selbst shätzt und subtrahiert einen
Ge-samthintergrundshätzer(pro Miroarray),sodass eine zusätzlihe
Hintergrundkor-rekturnurbeilokalerVariabilitätübereinenMiroarray, z.B.räumliher Gradient,
sinnvoll wäre.
DerVSN-AlgorithmuslässtsihinzweiKomponentenaufteilen:Beiderersten
han-delt es sih um eine ane Transformation, um die systematishen experimentellen
Faktoren, wie die Markierungsezienz und die Detektionssensivität, zu kalibrieren.
Bei der zweiten handelt es sih um eine
g
log
2
-Transformation, um die Varianz zu stabilisieren[Huberetal.,2002℄.BeideKomponentenwerdenimFolgendenbeshrie-ben.
Das VSN-Modellbasiert auf dem Fehlermodell von Roke und Durbin [Roke and
Durbin, 2001℄. Bei diesem Fehlermodell wird angenommen, dass die gemessene
Si-gnalintensität y eine Realisierung der ZufallsvariablenY ist,die wie folgt deniert
ist:
Y
=
α
+
βe
η
+
ν,
(2.17)α
istdabeiderOset undβ
dietatsählihgemessene Expression.e
η
und
ν
sindder multiplikativeundder additiveFehlerterm,wobeiη
mitN
(0
,
1)
undν
mitN
(0
, s
ν
)
verteilt ist.AlsErwartungswert von Y ergibt sihdamit:
E
(
Y
) =
E
(
α
+
βe
η
+
ν
)
=
E
(
α
) +
E
(
βe
η
) +
E
(
ν
)
=
α
+
βm
η
(2.18)
wobei
m
η
der Erwartungswert vone
η
V ar
(
Y
) =
E
(
Y
−
E
(
Y
))
2
=
E
(
α
+
βe
η
+
ν
−
(
α
+
βm
η
))
2
=
E
(
ν
+
β
(
e
η
−
m
η
))
2
=
E
(
ν
2
+ 2
νβ
(
e
η
−
m
η
) +
β
2
(
e
η
−
m
η
)
2
)
=
E
(
ν
2
) +
β
2
E
(
e
η
−
m
η
)
2
=
s
2
ν
+
β
2
s
2
η
(2.19) Dabei sinds
2
ν
unds
2
η
die Varianzen vonν
unde
η
.
Wenn man die Gleihung 2.18 nah
β
umformt,β
=
E
(
Y
)
−
a
m
η
(2.20)
und
β
in die Gleihung 2.19 einsetzt, so erhält man die Varianz als Funktion und damitdie quadratishe Abhängigkeit der VarianzvomMittelwert:v
(
u
) = (
c
1
u
+
c
2
)
2
+
c
3
,
mitc
3
>
0
(2.21)
v(u)ist dieVarianzals FunktionvomErwartungswert
u
(
→
E
(
Y
))
, wobeic
1
=
s
η
m
η
, c
2
=
−
αs
η
m
η
, c
3
=
s
2
ν
(2.22)gegeben ist. Gesuht wird eine Transformation h(y), für die Var(h(y)) = konstant
gilt.Die Methode, dieman indiesem Fallanwendet ist dieDelta-Methode (2.4).
Bei dieser Methode wird h(y) um den Erwartungswert u mit den ersten beiden
Gliedernder Taylor-Reiheapproximiert:
h
(
y
)
≈
h
(
u
) + (
y
−
u
)
h
′
(
u
)
≈
h
(
u
)
−
uh
′
(
u
) +
yh
′
(
u
)
(2.23)