Antigen processing and T cell priming
by mouse dendritic cells
Alexander Hal Drakesmith
A thesis submitted for the degree of
Doctor of Philosophy
March 1998
ProQuest Number: U643410
All rights reserved
INFORMATION TO ALL USERS
The quality of this reproduction is dependent upon the quality of the copy submitted.
In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed,
a note will indicate the deletion.
uest.
ProQuest U643410
Published by ProQuest LLC(2016). Copyright of the Dissertation is held by the Author.
All rights reserved.
This work is protected against unauthorized copying under Title 17, United States Code. Microform Edition © ProQuest LLC.
ProQuest LLC
789 East Eisenhower Parkway P.O. Box 1346
X2&2.7I
. V |
Abstract
For a vertebrate to survive and reproduce, it must be able to fight infection efficiently
throughout its life-span. The vertebrate immune system performs this function by
recognising, responding to, killing and remembering pathogens. A crucial component of
immunity is the dendritic cell, which responds to inflammatory signals such as
cytokines which are produced because of infection. The invading pathogen is captured
and partially degraded by dendritic cells via a mechanism termed antigen processing.
Dendritic cells are specialised to prime specific T cells against small regions (called
determinants) derived from the pathogen by antigen processing. The activated T cells
then control the immune response which fights the infection.
This thesis is a study of how antigen processing by dendritic cells can be affected by
cytokines. The determinants processed and presented from a model antigen are shown
to vary considerably depending upon which cytokines the dendritic cells are exposed to.
In particular, determinants that are normally not presented (cryptic determinants) can
become very immunogenic: the cytokine exposed dendritic cells activate T cells against
these cryptic deteminants both in vitro and in vivo.
How one particular cytokine, interleukin-6, can cause these effects is further
investigated. Although the exact mechanism is not elucidated, interleukin-6 is shown to
differentiate dendritic cells in novel ways, including acidifying certain intracellular
compartments which are involved in antigen processing. Because pH influences many
aspects of antigen processing, the display of cryptic determinants by interleukin-6
Acknowledgements
I have had an extremely enjoyable time in the Tumour Immunology Unit. This is
entirely due to the people I have been lucky enough to work with.
Dr. Hans Stauss was my first supervisor, has been ever ready with support and advice,
and shows that comprehensive knowledge of cytotoxic T lymphocytes and European
football are not mutually exclusive. Professor Peter Beverley has been my main
supervisor and even in complicated times has always been inspiring, great fun, a
reasonable off-spinner, and an estimable supplier of fine Chardonnay. Professor Benny
Chain at UCL also supervised me, and his energy, enthusiasm and interesting ideas for
experiments contributed much to this thesis.
In the lab, Maria Dahl, Christine Hughes, Marcos Timon, Diana Wallace, Anne Marit-
Sponaas, Ray Hicks and Lindsey Goff helped me when I didn't have a clue. Peter Noble
always had a question. Pip O'Brien was a fantastic friend who made the lab a more
unpredictable place. The four who were with me at the end, like in 'Zulu' or (perhaps
more accurately) the Alamo, lovely but mad Mala Maini, Christian Zilch and my main
men Harry White and Sorena Kiani-Alikhan, were understanding in the extreme as I
hogged the computers and went slowly insane.
At UCL Immunology, Debbie O'Neil, Patrick Medd, Mike Binks, Ness Woodhead, Tim
Stonehouse, Torben Lund, Tom MacDougal, Peter Bunyard and the wondrous Lucienne
Lopes all helped me out technically, told me something important that I didn't know or
bought beer. Upstairs, Maureen Cohen, Simon McAdam, Steve Furrow, John Bingham,
Caroline O'Hare, Inusha De Silva, Victoria Spanswick, Lucia Christodoulides, John
Hartley and Peter McHugh also helped me and generally added to the camaraderie.
At the La Jolla Institute for Allergy and Immunology, California, Susanne Schneider
was extremely generous and hospitable, and Eli Sercarz was fun and gave good advice.
Contents
Title page 1
Abstract 2
Acknowledgements 4
Contents 5
List of Figures and Tables 11
Abbreviations 14
Chapter 1
General Introduction: dendritic cells, antigen processing
and hierarchy of T cell determinants 18
Introduction 19
Origins of dendritic cells 23
First sightings 23
A novel cell type in lymphoid organs 25 Dendritic cells prime naive T cells and T-dependent inunune reponses 25 Dendritic cell maturation and migration 26 Differentiation of dendritic cells from precursors 29 Molecular events of dendritic cell maturation and T cell priming 31 Generation of the T cell repertoire 33 MHC class II restricted processing of exogenously acquired antigen 36 Accessory proteins in MHC class II restricted processing 37 Endocytic compartments involved in antigen processing 38 Dominant and cryptic T cell determinants 40 Determinant capture and protection 42 Cryptic determinants, tolerance and autoimmunity 47 Antigen processing by different cell types 48
Chapter 2
Materials and Methods 50
Plastics 51
Mice 51
Reagents 51
Cytokines 51
Radioactive Isotopes 51
Peptides 52
Media 54
Sera 54
Buffers 54
Antibodies 55
Cell lines 56
Cell counting 58
Cry op reservation and retrieval of cells 58
Immunofluorescence staining of cell surface markers 58
Enriching dendritic cells from spleen 59
Grovying dendritic cells from bone marrow precursors 59
Allogeneic mixed lymphocyte proliferation assays 60
Induction of cytotoxic T lymphocytes 61
Using dendritic cells to generate allogeneic CTL in vitro 61
Using dendritic cells to generate syngeneic CTL in vitro 61
CTL assay 61
Target cells 61
Effector cells 62
MHC Class I peptide binding assay 62
Antigen processing assays 63
Immunisation of mice with dendritic cells 63
Antigen presenting cell separation 64
Endocytosis assay 65
Intracellular staining and confocal microscopy 65
Mounting cells onto slides 66
Confocal microscopy 66
Enzyme Linked Immunosorbent Assay (ELISA) 66 Pulsing dendritic cells with radioactive antigen 67
Messenger RNA differential display 68
RNA extraction 68
Cleaning extracted RNA 69
Reverse transcription of RNA 69
Polymerase chain reaction 69
6% denaturing polyacrylamide gel electrophoresis 70
Chapter 3
Characterisation of mouse dendritic cells 71
Introduction 72
Results 74
50-fold enrichment of dendritic cells from mouse spleen 74
Spleen dendritic cells are strong stimulators of allo-responses 76
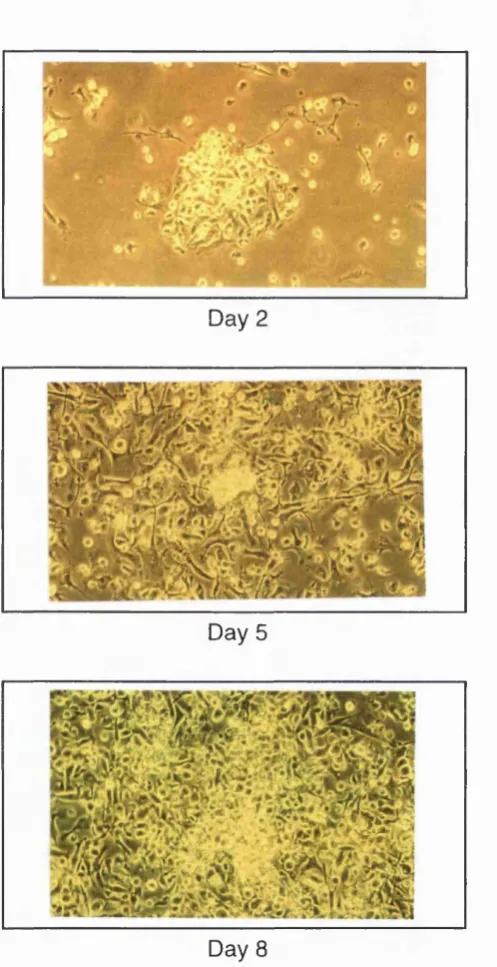
Culturing bone marrow to grow dendritic cells in vitro 78
Mature bone marrow derived dendritic cells induce strong responses 80
Phenotype of day 8 dendritic cells 82
Day 8 dendritic cells can stimulate allo-specific CTL 84
Relative stabilisation abilities of three H-2Kb binding peptides 85
Comparison of splenic and bone marrow derived dendritic cells
in inducing syngeneic peptide specific CTL 86
Discussion 89
Comparison of splenic and bone marrow derived dendritic cells 89
Using dendritic cells to prime anti-cancer responses 90
Summary 92
Chapter 4
Effects of cytokines on antigen processing and
T cell priming by dendritic cells 93
Introduction 94
Results 98
Surface markers of T-T cell hybridomas 98
Effects of cytokines on the processing of HEL in vitro 98
Effects of cytokines on the differentiation/maturation of dendritic cells 104
Interleukin-6 treated dendritic cells prime T cells in vivo 108
Injection of EL-6 with HEL in adjuvant alters the anti-HEL T cell response 110
Dendritic cells draining HEL plus IL-6 immunisations present the cryptic
determinant HEL 2-16 112
Discussion 114
IL-4 114
TNF-a 115
IFN-y 116
Effects of IL-6 on dendritic cells 118
Mechanisms of escape from self tolerance 120
Molecular mimicry 120
Superantigen effects 121
CD2 ligation 121
Direct priming 121
Chapter 5
Action of IL-6 on antigen processing pathways
of dendritic cells 124
Introduction 125
Results 128
Endocytic progress of dextran through a dendritic cell 128
Characteristics of fluorochromes at different pH 130
IL-6 treated dendritic cells quench the fluorescence of endocytosed pH
sensitive markers 132
Visualisation of acidic vesicles in dendritic cells and colocalisation with
endocytosed mated al 136
Lysosome associated membrane protein-1 is peripherally located
in IL-6 treated dendritic cells 139
Levels and distribution of other molecules associated with
antigen processing in IL-6 treated and control dendritic cells 141
Antigen proteolysis by IL-6 treated and control dendritic cells 142
Altered gene expression in dendritic cells after IL-6 treatment 144
Discussion 145
IL-6 regulated acidification of peripheral vesicles 145
Role of pH in antigen processing 148
Protein unfolding 148
Proteolytic degradation 149
Peptide loading 150
Additional factors which may affect determinant selection by
dendritic cells 151
Chapter 6
Conclusions and perspectives:
multiple pathways of dendritic cell differentiation 156
Introduction 157
Many types of receptor molecules on dendritic cells 157
Antigen receptors (pattern recognition receptors) 157
Chemokine receptors 158
The diverse roles of cytokine receptors on dendritic cells 159
Induction of IL-12 secretion and TH-1 -type responses 159
Th-2-type responses or tolerance 160
Direct effects of pathogen on dendritic cells 161
Abnormal dendritic cell function and disease 163
Summary 165
Conclusions 168
Appendix I
169Plastic ware 169
Appendix II
170Chemical and biological reagents 170
Recombinant cytokines 172
Appendix III
173Buffers and solutions 173
Elisa buffers 173
Molecular biology buffers 174
List of Figures
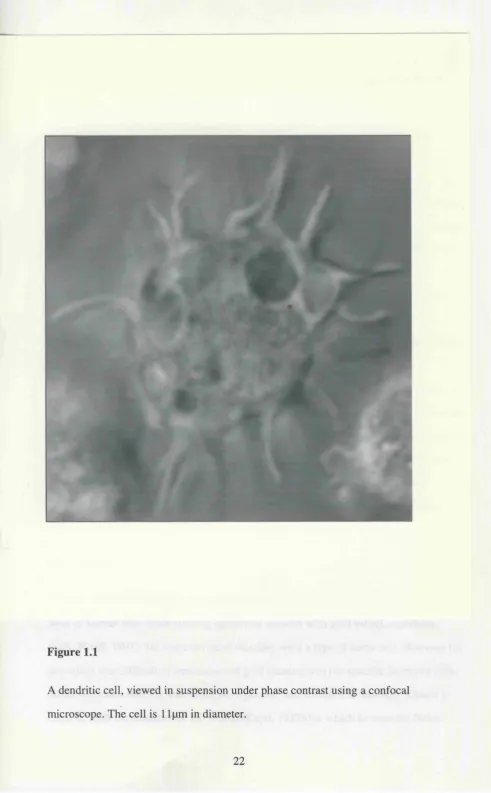
Figure 1.1 A dendritic cell 22
Figure 1.2 Migration patterns of dendritic cells in vivo 28
Figure 1.3 Scheme for determinant capture and protection 46
Figure 3.1 Constituents of the dendritic cell enriched cell fraction 74
Figure 3.2 FACS analysis of the dendritic cell enriched cell fraction 75
Figure 3.3 Splenic dendritic cells stimulate an allogeneic mixed
lymphocyte reaction 76
Figure 3.4 Splenic dendritic cells prime allo-specific CTL 77
Figure 3.5 Development of bone marrow cultures over time 78
Figure 3.6 Number of viable cells in developing bone marrow cultures 79
Figure 3.7 Day 7 bone marrow derived dendritic cells in culture 80
Figure 3.8 Time course of allogeneic mixed lymphocyte reaction 81
Figure 3.9 Time course of syngeneic mixed lymphocyte reaction 82
Figure 3.10 FACS analysis of day 8 bone marrow derived dendritic cells 83
Figure 3.11 Bone marrow derived dendritic cells prime allo-specific CTL 84
Figure 3.12 MHC class I stabilisation by various H-2 binding peptides 85
Figure 3.13 Comparison of the abilities of splenic and bone marrow derived
dendritic cells in inducing peptide specific syngeneic CTL 87
Figure 3.14 Blocking B7 costimulatory molecules on dendritic cells inhibits
CTL priming 88
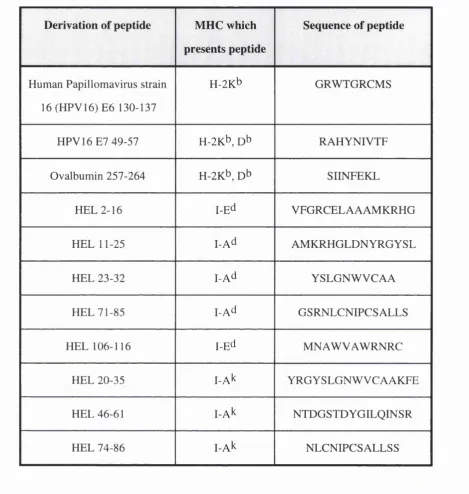
Figure 4.1 MHC class II binding determinants of Hen Eggwhite Lysozyme 95
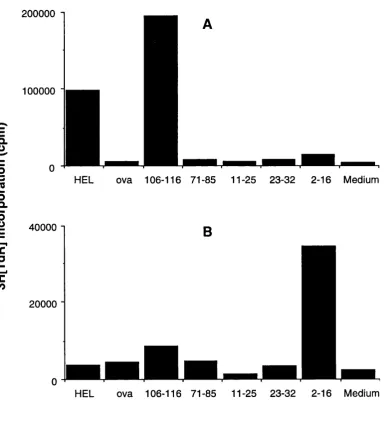
Figure 4.2 Comparison of responses to cryptic and dominant determinants 96
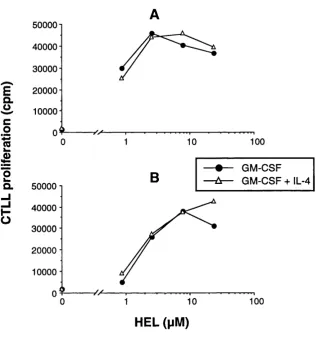
Figure 4.3 Effect of IL-4 on the processing of HEL by dendritic cells 99
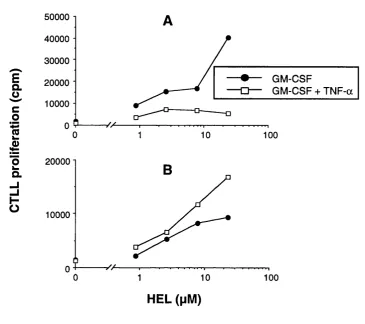
Figure 4.4 Effect of TNF-a on the processing of HEL by dendritic cells 100
Figure 4.5 Effect of IFN-y on the processing of HEL by dendritic cells 101
Figure 4.7 Effect of cytokines on fluid-phase endocytosis by dendritic cells 105
Figure 4.8 Surface levels of MHC class II on cytokine treated dendritic cells 106
Figure 4.9 Effect of cytokines on dendritic cell viability 107
Figure 4.10 Immune response to injected IL-6 and HEL treated dendritic cells 109
Figure 4.11 Immune response to injected IL-6 plus HEL in adjuvant 111
Figure 4.12 Dendritic cells from lymph nodes draining IL-6 plus HEL
immunisation sites present cryptic determinants 113
Figure 5.1 Confocal analysis of dendritic cells endocytosing dextran 129
Figure 5.2 Behaviour of FTTC at different pH 131
Figure 5.3 Fluid-phase endocytosis of FITC conjugated markers by
IL-6 treated dendritic ells 132
Figure 5.4 Effect of fixation on the fluorescence of IL-6 treated
dendritic cells 133
Figure 5.5 Receptor-mediated endocytosis of FITC conjugated markers 134
Figure 5.6 Pulse-chase endocytosis of IL-6 treated dendritic cells 135
Figure 5.7 Structure of DAMP 136
Figure 5.8 Location of acidic vesicles in dendritic cells 137
Figure 5.9 Colocalisation of DAMP and endocytosed dextran 138
Figure 5.10 Positions of lysosome associated membrane protein-1 and
the transferrin receptor in dendritic cells 140
Figure 5.11 Degradation of iodinated bovine serum albumin by dendritic cells 143
Figure 5.12 Differential display of mRNA from dendritic cells 145
Figure 5.13 Effects of chloroquine on dendritic cell viability 153
List of Tables
Table 2.1 Storage procedures for certain reagents
Table 2.2 Details of peptides used
Table 2.3 First layer antibodies
Table 2.4 Second layer antibodies and streptavidin cychrome C
Table 2.5 Cell lines used as targets in CTL assays
Table 2.6 Summary of T cell hybridomas
52
53
55
56
57
57
Table 5.1 Characteristics of markers used in endocytosis assays
Table 5.2 Intracellular staining of dendritic cells for levels of
proteolytic enzymes and MHC class II
130
Abbreviations
A aa Ab APC ATP ATTC bp Bq BSA C CD cDNA CFA Ci CIIV CLIP cpm CQ Cr CTL Da DAMP DCadenine or adenosine
amino acid
antibody
antigen presenting cell
adenosine triphosphate
The American Type Culture Collection
basepairs
becquerel (1 disintegration/sec)
bovine serum albumin
cytosine
cluster of differentiation
complementary DNA
complete Freund's adjuvant
curie (3.7 x lO^® Bq)
MHC class II containing vesicle
MHC class II associated invariant chain peptide
counts per minute
chloroquine
chromium
cytotoxic T lymphocyte
Dalton
N-(3-((2,4-dinitrophenyl)amino)propyl)-N-(3- aminopropyl)methylamine dihydrochloride
DC/IL-6 DEPC DMSO DNA DNAse DNP dNTP dpi DTT EBV ECACC EDTA ELISA E:T ratio EtBr EtOH FACS PCS FITC g G [3H]TdR HCl HEL HIV HLA
as for DC, except IL-6 added for the last 18 hours of culture diethyl pyrocarbonate dimethylsulfoxide deoxyribonucleic acid deoxyribonuclease dinitrophenol deoxyribonucleoside triphosphate
dots per inch
dithiothreitol
Epstein-Barr virus
The European Collection of Animal Cell Cultures
ethylenediamine tetraacetic acid (disodium salt)
enzyme linked immunosorbent assay
effector to target cell ratio
ethidiumbromide
ethanol
fluorescence activated cell sorter
foetal calf serum
fluorescein isothiocyanate
gram or gravitational force
guanidine
tritiated thymidine
hydrochloric acid
hen eggwhite lysozyme
human immunodeficiency virus
HPLC high-performance liquid chromatography
HPV human papillomavirus
ICRF Imperial Cancer Research Fund
IFA incomplete Fruend's adjuvant
IFN interferon
Ig immunoglobulin
li invariant chain
IL interleukin
IMDM Iscove's modified Dulbecco's medium
kb kilobase
KCl potassium chloride
kDa kilodalton
LAMP lysosome associated membrane protein
LCMV lymphocytic choriomeningitis virus
M molar
mAh monoclonal antibody
2-ME p-Mercaptoethanol
MEM minimal essential medium
MHC major histocompatibility complex
MIIC MHC class II containing compartment
MMLV Moloney murine leukemia virus
mRNA messenger RNA
M-tropic macrophage infecting HIV strain
Mw molecular weight
NaAc sodium acetate
NOD non-obese diabetic
OD260 optical absorbance at 260 nm
OD450 optical absorbance at 450 nm
ova ovalbumin
PAGE polyacrylamide gel electrophoresis
PBSA phosphate buffered saline
PCR polymerase chain reaction
PHA phyto haemagglutinin
RNA ribonucleic acid
RNAse ribonuclease
rpm revolutions per minutes
SDS sodium dodecyl sulphate
T thymidine
TCR T cell receptor
TE tris-EDTA buffer
TEMED N, N, N', N', tetramethylethylenediamine
Tf-R Transferrin receptor
TH-1 T helper type 1 cell
TH-2 T helper type 2 cell
TNF tumour necrosis factor
TR-Dx Texas Red conjugated dextran
TRITC tetramethylrhodmamine isothiocyanate
T-tropic T cell infecting HIV strain
V volt
vol volume
Chapter 1
General introduction:
dendritic cells,
antigen processing
Introduction
Vertebrate immunity, consisting of the innate and adaptive immune systems, has
evolved under selection from both infection and the needs of the host. Innate immunity
is based on the recognition of a limited number of common features of pathogens, is
fast acting, but does not improve over time in its ability to fight pathogen. Acquired
immunity is slower acting, more specific to particular antigens, and improves on
repeated exposure to the same infection(Austyn and Wood, 1993; Roitt et al, 1985).
Innate immunity consists of several basic biochemical and physical defences against
pathogens, including the skin, the acidic environment of the gut and the activity of
lysozyme, and some more advanced molecular and cellular mechanisms. This latter
category includes phagocytes, which are cells such as eosinophils, basophils,
neutrophils, Kupffer cells and macrophages which recognise, internalise and destroy
infectious material. Natural killer cells are able to lyse infected cells (and some tumour
cells), providing another mechanism of innate defense. Finally the levels of some serum
proteins increase in response to infection, for instance C-reactive protein, which binds
the C protein of pneumococci bacteria, and promotes subsequent attatchment of
complement(Austyn and Wood, 1993; Roitt et al, 1985).
The complement system is made up from a number of soluble protein factors which are
involved in several arms of both innate and adaptive immunity. Some complement
factors can directly bind polysaccharides found on a number of microbes and aid their
capture by phagocytes possessing complement receptors, in a process called
opsonisation. Another complement factor, C5a, attracts neutrophils to sites of infection.
Complement can also lyse bacteria via the formation of the membrane attack complex
which punches a hole in bacterial cell walls. This complex is formed during two
multistep enzymatic cascades, termed the classical and the alternative pathways. The
pivotal event in both these sequences is the proteolytic cleavage of factor C3 to yield
activated C3 and C3a. Activated C3 generates other complement components, building
Intrinsic to many of the mechanisms of innate immunity is the ability to recognise
pathogen as being foreign. This can occur because most invading microorganisms have
clear structural differences compared to higher organisms, such as the components of
their cell walls. Receptors encoded by higher organisms can recognise these differences
and alert both innate and adaptive immunity(Medzhitov and Janeway, 1997).
Adaptive immunity, unlike innate immunity, is not 'ready to go': it requires activation
signals which often originate from the innate immune system. The adaptive immune
system's main components are lymphocytes, particularly thymus derived lymphocytes
(T cells) and bone marrow derived lymphocytes (B cells). These two cell types encode
receptors which unlike innate receptors are clonal and can rearrange, giving a greater
diversity and specificity to pathogen recognition. T cells expressing T cell receptors
(TCR) consisting of one a and one p chain can be restricted by Major
Histocompatability (MHC) class I or class II molecules presenting pathogenic antigen
in the form of short peptides, termed determinants (Townsend et al, 1986; Zinkemagel
and Doherty, 1974). B cells secrete immunoglobulin (Ig) in many different forms,
which can bind specific three-dimensional conformations of pathogenic antigen. Ig also
binds and activates complement, which results in the destruction of the bound pathogen
by phagocytosis or lysis(Austyn and Wood, 1993; Roitt et al, 1985).
Clearly, it is important that innate immunity and adaptive immunity cooperate. This
occurs by many mechanisms, including complement and Ig, chemotactic signals
released after pathogen recognition, and importantly, the recruitment and maturation of
dendritic cells. The first cell type of the adaptive immune system to be activated is
usually the CD4“^ helper T cell. These cells are the most important components of
adaptive immunity as they control Ig release by B cells, cell killing by CD8+ cytotoxic
T cells (CTL), and immunological memory. Together, these features of the adaptive
immune system are crucial for the longer life-span of vertebrates(Medzhitov and
This introductory chapter will deal with the discovery, lineage and properties of
dendritic cells, in particular their special ability to prime naive T cells in lymph nodes
against antigen found in the periphery. How antigen is processed so that determinants
are presented by MHC class II molecules to helper T cells is outlined. Although an
antigen can contain many potential determinants, T cell immune responses tend to be
focussed on just one or two of these, termed the dominant deteminants. Additionally, T
cell self tolerance is also targeted to dominant self determinants. This phenomenon is
called immunodominance(Sercarz et al, 1993); how immunodominance arises and its
É ^ '
Figure 1.1
A dendritic cell, viewed in suspension under phase contrast using a confocal
Origins of dendritic cells
"Really, if the lower orders don't set us a good example, what on earth is the use of
them?"
-Oscar Wilde (The Importance of Being Earnest)
The invertebrate immune system, which is the forerunner of the innate immune system
of vertebrates, does not have T or B cells(Roitt et al, 1985). In invertebrates, pathogen
recognition results in host defence mechanisms such as the secretion of anti-microbial
peptides appropriate to the particular type of invading pathogen(Medzhitov and
Janeway, 1998). In vertebrates, pathogen recognition leads to an innate response, but
the adaptive immune system including pathogen-specific lymphocytes can also be
activated(Medzhitov and Janeway, 1997; Roitt et al, 1985); dendritic cells play a major
role in this process. Dendritic cells are able to recognise and capture infectious
pathogens, but are also specialised to prime T cells using MHC and other molecules of
the adaptive immune system(Steinman, 1991). Thus dendritic cells have characteristics
of both invertebrate and vertebrate immunity: they are a lower order cell made good. As
described later, dendritic cells are seeded in peripheral tissues of the body where they
act as 'sentinels' ready to be activated at the point of entry of infection. The first
peripheral dendritic cells to be identified were those of the skin, called Langerhans
cells.
First sightings
In 1868, Paul Langerhans saw cells with striking dendritic processes in the suprabasal
layer of human skin while staining epidermal sections with gold salts(Langerhans,
1868; Wolff, 1991). He was convinced that they were a type of nerve cell. However his
procedure was difficult to reproduce and gold staining was not specific for nerve cells.
Silver chromate staining was much more precise, and revealed to Santiago Ramon y
Prize for Medicine in 1906, along with Camillo Golgi). Silver chromate does not stain
Langerhans cells: for this reason and others Billingham and Medawar proposed in 1953
that Langerhans cells were not nerve cells, but were effete or dying melanocytes’ en
route to being shed at the skin surface(Wolff, 1991). This theory was discounted in
1961 by the findings of Birbeck et al, showing that Langerhans cells had a distinctive
marker lacked by melanocytes (the Birbeck granule) and were present in vitiligous
tissue where melanocytes were absent(Birbeck et al, 1961). However a (more distant)
relationship between melanocytes and Langerhans cells was still believed to exist until
limb bud transplantation experiments showed different lineages for these two cell types:
melanocytes did not reappear after transplantation of limb buds (as they are derived
from the neural crest), but Langerhans cells did, indicating a mesenchymal
origin (Wolff, 1991).
The first circumstantial evidence for a role in immune responses came from an
observation that Langerhans cells were found in the dermis and dermal lymphatics after
epidermal challenge of animals passively sensitised to dinitrophenol(Wolff, 1991). If
Langerhans cells were involved in immunity, they might possess known immunological
molecules. Three papers published contemporaneously in 1977 found this to be the
case, showing MHC class II, complement receptors and Fc receptors to be
expressed(Klareskog e ta l, 1977; Rowden e ta l, 1977; Stingl e ta l, 1977). The
capacity of Langerhans cells to stimulate proliferative and cytotoxic T cell responses
was demonstrated shortly afterwards(Stingl et al, 1978; Wolff, 1991). Because of their
surface Fc and complement receptors, and their ATPase activity, Langerhans cells were
thought to be of the macrophage system, even though they lacked other macrophage
markers such as CD 14, and expressed some non-macrophage markers such as NLDC-
145. The Langerhans cell lineage problem was finally resolved in 1985, when Schuler
and Steinman found that after an in vitro culture of 2-3 days, Langerhans cells
differentiated to become almost indistinguishable from lymphoid dendritic cells, which
A novel cell type in lymphoid organs
In January 1973 the first of five papers describing a novel cell type in peripheral
lymphoid organs was published(Steinman and Cohn, 1973). These papers established
the term, dendritic cell, as applying to a rare adherent cell type in mouse spleen, of low
buoyant density, possessing a distinctive morphology including very mobile processes,
few lysosomes, and lacking both lymphocytic markers and the endocytic activity of
macrophages(Steinman and Cohn, 1973; Steinman and Cohn, 1974). Dendritic cells
were shown to have a bone marrow origin and had a low rate of proliferation but turned
over rapidly in the spleen, at the rate of about 10% of the dendritic cell population per
day(Steinman et al, 1974). They were found in situ in the white pulp of spleen and
were cytologically very similar to previously identified interdigitating cells (Steinman et
al, 1975). The adherence of dendritic cells was found to be transient, allowing greater
purification which enabled further investigations into both phenotype (revealing surface
topography and expression of MHC class II) and immune function(Steinman et al,
1979).
Dendritic cells prime naive T cells and T-dependent immune reponses
The first direct evidence of an immunostimulatory property came when lymphoid
dendritic cells were found to be extremely potent inducers of the mixed allogeneic
lymphocyte reaction(Steinman and Witmer, 1978). Later, dendritic cells were shown to
be critical accessory cells in forming primary antibody responses in vitro{lnaba. et al,
1983); dendritic cells, T cells and B cells all cluster together during this process(Inaba
et al, 1984). In the case of antibody responses against hapten-conjugated peptides,
dendritic cells primed naive helper T cells from resting lymphocytes, which then
stimulated B cell growth and differentiation(Inaba and Steinman, 1985). As well as
activating helper CD4"^ T cells, dendritic cells were also directly primed allogeneic
CD8+ CTL in vitro, in both mouse and human systems(Inaba et al, 1987; Young and
Steinman, 1990). Additionally, dendritic cells primed syngeneic influenza virus-specific
Dendritic cells were also found to prime T cells in vivo. Splenic dendritic cells were
pulsed in vitro with protein antigens such as conalbumin or ovalbumin and injected into
the hind footpads of mice. T cells isolated from the draining popliteal lymph nodes
proliferated ex vivo to the protein in an MHC restricted manner(Inaba et ai, 1990).
Reciprocally, after protein antigen was administered intravenously or intraperitoneally,
dendritic cells in the mouse spleen were the major source of immunogenic
protein(Crowley et al., 1990). These findings have been extended by Guery et at, who
found that after injection of protein emulsified in adjuvant, dendritic cells but not B
cells in draining lymph nodes were able to present processed protein to T cells ex
v/vo(Guery et at., 1996). In parallel, other investigations aimed to elucidate the
properties of dendritic cells that could account for this ability to prime T cells in
lymphoid tissues against peripherally administered antigens.
Dendritic cell maturation and migration
Although splenic dendritic cells had been shown to turn over rapidly, the first clue that
dendritic cells could have a multi-stage life-cycle came from Schuler and Steinman who
showed that Langerhans cells differentiate during three days of in vitro culture to
become as immunostimulatory as lymphoid dendritic cells (Schuler and Steinman,
1985). This immediately suggested a relationship between the two cell types which was
further investigated. Freshly isolated Langerhans cells were found to synthesise and
express MHC class U(Pure et ai, 1990), phagocytose bacterial and yeast products (Reis
e Sousa et al, 1993) and to process antigen without efficiently priming naive T
cells(Schuler and Steinman, 1985). Upon ex vivo culture, these features altered so that
little new MHC class U was synthesised, phagocytosis and antigen processing were
reduced, Birbeck granules were lost and the capacity to stimulate naive T cells was now
very high: thirty of these cells stimulated three hundred thousand T cells (Schuler and
Steinman, 1985). As dendritic cells isolated from spleen had previously been shown to
be non-phagocytic, it was proposed that Langerhans cells might differentiate into
lymphoid dendritic cells in vivo. This process was termed the maturation of dendritic
Migration of Langerhans cells to lymph nodes via afferent lymph was directly shown by
skin explant experiments(Larsen et al, 1990a), and found to be induced by
inflammatory stimuli such as LPS, TNF-a and IL-lp(Cumberbatch and Kimber, 1992;
Roake et al, 1995). Similar observations were reported for the migration of rat gut
epithelial dendritic cells into afferent lymph(Liu and MacPherson, 1993; MacPherson et
al, 1995). These migrating cells had sheet-like membrane processes, and were named
veiled cells. They were non-phagocytic, but retained previously phagocytosed material.
Other routes for dendritic cell migration have been defined. In solid organs such as
heart, kidney and spleen, dendritic cells are found in interstitial spaces in connective
tissue. Austyn et al showed that when dendritic cells isolated from spleen were labelled
and injected intravenously into mice they homed back to specific regions of the
spleen (Austyn et al, 1988; Kupiec et al, 1988). In a murine cardiac allograft model,
dendritic cells were shown to migrate from the heart via the blood to the spleen (Larsen
et al, 1990b). This effect was increased by administration of LPS (Roake et al, 1995).
A third route for dendritic cell migration was suggested after intravenous administration
of particles into mice resulted in particle laden dendritic cells being recruited to liver
lymph nodes(Matsuno et al, 1996). The pathway was further investigated using
dendritic cells isolated from hepatic lymph: they were injected intravenously and
subsequently found in the paracortical regions of recipient mouse liver lymph nodes.
The translocation from blood to lymph did not occur at high endothelial venules, but at
liver sinusoids, with the probable involvement of Kupffer cells (Kudo et al, 1997). This
migratory route enables immunological sampling of antigen in hepatic blood. Dendritic
cells in the liver may also migrate to the spleen via the blood(Austyn, 1996).
Dendritic cells are found in different regions of the spleen: in the white pulp (both T cell
and B cell areas), in marginal zones and red pulp. These may represent stages of
migration through the spleen. Additionally, dendritic cells may also be able to go
directly into lymphoid tissue from blood without first going to peripheral sites(Austyn,
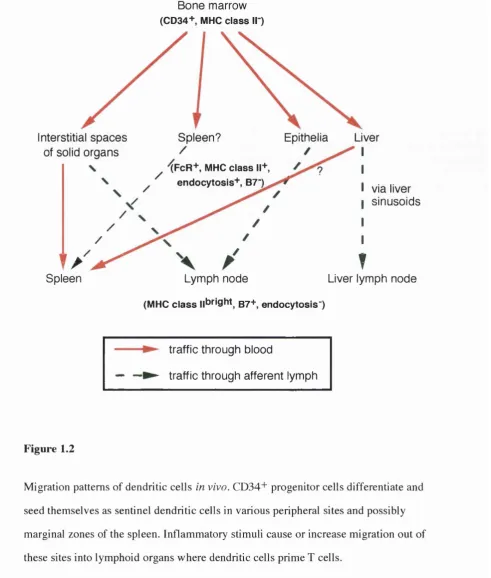
Bone marrow (CD34+ MHC c la s s II )
Interstitial spaces Spleen? Epithélia Liver
of solid organs
XFcR+, MHC c la s s 11+ endocytosis+ , B7"K
via liver sinusoids
Spleen Lymph node
(MHC c la s s ||bright g7+ endocytosis")
T
Liver lymph node
traffic through blood
— —► traffic through afferent lymph
Figure 1.2
Migration patterns of dendritic cells in vivo. CD34+ progenitor cells differentiate and
seed themselves as sentinel dendritic cells in various peripheral sites and possibly
marginal zones of the spleen. Inflammatory stimuli cause or increase migration out of
these sites into lymphoid organs where dendritic cells prime T cells.
A recurring theme in studying dendritic cells was that cells isolated from peripheral
sites were unable to prime naive T cells, but acquired this ability on migration to lymph
nodes or during in vitro culture. Elucidating the differentiation events that must occur
during this maturation process was tricky until methods became available to generate
large numbers of dendritic cells in vitro from their in vivo precursors.
Differentiation of dendritic ceils from precursors
The maturation of Langerhans cells into potent immunostimulatory cells in vitro could
be enhanced using the cytokine granulocyte-macrophage colony-stimulating-factor
(GM-CSF)(Witmer et al, 1987). As kératinocytes in epithelial tissue are a major source
of GM-CSF, this made sense. In the search for factors that could differentiate dendritic
cells in vitro from pre-Langerhans cell precursors, GM-CSF was the obvious choice.
Inaba et al showed that GM-CSF could differentiate mature dendritic cells from
precursors found in mouse blood and bone marrow, that the bone marrow precursor was
MHC class II" and that it could give rise to dendritic cells, granulocytes and
macrophages(Inaba et at., 1993; Inaba et ai, 1993; Inaba et al, 1992). In general, GM-
CSF is sufficient to generate dendritic cells from murine precursor cells, although
additional cytokines can improve the yield.
In the human, differentiation of mature T cell stimulatory dendritic cells from
precursors is evidently more complicated. Although GM-CSF and TNF-a can cooperate
to form dendritic cells from CD34+ MHC class II" cord blood precursors(Caux et ai,
1992), factors such as c-kit ligand(Szabolcs e ta l, 1995), IL-4(Sallusto and
Lanzavecchia, 1994), flt-3 ligand(Maraskovsky et aL, 1996) and other as yet
unidentified moieties in monocyte-conditioned medium can play other roles in dendritic
cell development. These roles include general enhancement of proliferation, inhibition
of macrophage differentiation, and stabilisation of the mature dendritic cell phenotype.
The requirements for these factors may also vary depending upon the starting precursor
cells: bone marrow cells, cord blood cells, peripheral blood CD34+ and CD 14"*" cells
have all been used.
Other differentiation pathways of dendritic cells have been shown to occur in vivo,
cells is important because of the potential use of dendritic cells in treating diseases: it is
vital to use dendritic cells which will have the appropriate migratory, antigen retaining
and T cell stimulating properties. Subsets of dendritic cells have been studied most
extensivley in the mouse, and have been defined phenotypically and functionally.
Differences between dendritic cells types were immediately obvious when it was found
that thymic dendritic cells, which delete thymocytes during negative selection, can
actually arise from the same precursor as thymocytes(Ardavin et al, 1993), and that a
subpopulation of both splenic and thymic dendritic cells expressed CD8a(Vremec et
al, 1992). It later became clear that CD8a-chain expression was a marker for thymic
and splenic dendritic cells arising from this early T cell/dendritic cell progenitor, which
seems expresses low levels of CD4(Wu et al, 1996). This lineage has been termed the
lymphoid lineage of dendritic cells and is separate from the myeloid lineage: lymphoid
dendritic cells can be differentiated in vitro from the precursor cells in the abscence of
GM-CSF(Saunders et al, 1996). CD8a expressing splenic dendritic cells do not
activate naive T cells; instead they kill CD4+ T cells by Fas/Fas ligand induced
apoptosis(Suss and Shortman, 1996), and limit production of IL-2 by CD8+ T cells,
reducing their proliferation(Kronin et al, 1996). In attempting to prime anti-cancer
immune responses, it would probably be a good idea to avoid using this lineage of
dendritic cells, as they could actually remove from the available repertoire T cells
specific for tumour antigen determinants. However, if one wanted to limit immune
responses for example in autoimmunity by inducing T cell tolerance to an autoantigen,
these cells could be useful.
The intracellular signalling and gene transcription events induced during the
differentiation of dendritic cells from their precursors are incompletely characterised.
The relB subunit of the NF-kB complex is likely candidate to be involved in controlling
maturation of dendritic cells from the myeloid lineage. RelB is expressed in splenic
dendritic cells, and transgenic mice with a disrupted relB gene had normal numbers of
Langerhans cells, but had impaired splenic antigen presenting function, indicating
The in vitro generation of dendritic cells (although not identical to dendritic cells
differentiating in vivo) enabled the phenotypical changes that accompany maturation
and T cell priming to be studied in greater detail.
Molecular events of dendritic cell maturation and T cell priming
The activation requirements of naive T cells have been studied independently of
dendritic cells. In particular, work in Jonathan Sprent's laboratory has used Drosophila
melanogaster cells transfected with various molecules to determine the minimum
equipment needed to prime a MHC class I restricted naive T cell of known specificity.
Three molecules were sufficient: the correct MHC class I-peptide complex, the
adhesion molecule ICAM-1, and the costimulatory molecule B7.1(Cai et al, 1996). As
noted below, dendritic cells acquire these molecules (and others) during their
maturation.
Sallusto et al used an in vitro model to investigate the maturation of dendritic
cells(Sallusto et al, 1995; Sallusto and Lanzavecchia, 1994). Their method involved
culturing human peripheral blood adherent cells in the presence of GM-CSF and IL-4.
After 8 days in these conditions, most cells were loosely adherent or non-adherent and
possessed motile dendritic processes. Phenotypically, these cells expressed high levels
of MHC class I, MHC class II, CD la, CD lb, CDlc, CDl lb, C D llc, CD40, CD44, B7,
ICAM-1, F c '^ n , lower levels of LFA-1, LFA-3 and invariant chain, but lacked CD 14
and T- or B cell markers. Functionally, they could stimulate a mixed allogeneic
leukocyte reaction (MLR), present tetanus toxoid to T cell clones, and could capture
antigen by means of macropinocytosis or mannose and Fc receptors, Intracellularly,
they possessed a large compartment accessible by endocytosis which contained MHC
class II, the proteolytic enzyme cathepsin D and the lysosome associated membrane
protein-1.
Because of their ability to internalise and process antigen and their surface phenotype,
these cells were likened to immature dendritic cells. When they were cultured with
some analagous to the maturation of Langerhans cells. Surface levels of MHC class I,
MHC class n, CD la, CD lb, CDlc, B7, CD40, ICAM-1, CD44 all increased as did the
capacity to stimulate an allogeneic MLR, while surface invariant chain, FcyRII,
macropinocytosis and the ability to process tetanus toxoid for presentation to T cells all
decreased. The intracellular MHC class II containing compartment disappeared. More
recent studies have delved further into the intracellular events that occur during
maturation, and are discussed in chapter 5. However the findings of the two Sallusto
papers summarised above show that mature dendritic cells possess characteristics
required for the activation of T cells, and that maturation is driven by inflammatory
stimuli present at sites of infection. The sensitivity of immature 'sentinel' dendritic cells
to environmental signals is further discussed in chapter 6.
T cell priming by mature dendritic cells is a multi-stage process. Initially, chemokines
secreted by dendritic cells induce migration of T cells, for example the C-C chemokine
DC-CKl preferentially attracts naive Tcells(Adema etal., 1997). Adhesion molecules
then allow antigen independent dendritic cell-T cell clustering(Inaba et al, 1989),
before specific MHC/TCR interactions with the help of B7/CD28 ligation act to trigger
the T cell. CD40 ligand on T cells binds CD40 on dendritic cells resulting in IL-12
release by the dendritic cell, which can influence the primed CD4'*' T cell to become a
Th-1 helper cell(Kelsall et al, 1996; Koch et al, 1996). CD40 ligation also protects
dendritic cells from apoptosis(Bjorck et al, 1997; Ludewig et al, 1995). The lifespan
of dendritic cells in lymphoid tissue and the mechanisms of dendritic cell death are not
yet fully understood.
The nature of the responding CD4+ T cells can sometimes determine whether or not an
infection is controlled. The pattern of cytokines produced by the responding cells in
addition to IL-2 is an important factor in mobilising other components of the immune
response. TH-1 cells secrete IFN-yand TNF-P while TH-2 cells make IL-4, IL-5, IL-6,
IL-10 and IL-13. Generally, but by no means always, TH-1 type immune responses are
associated with cell mediated immunity for example CTL killing virally infected cells,
antibodies and neutrophils. Although this dichotomy of T cell responses is more
obvious in the mouse than in the human, and the relationship between TH-1/TH-2 and a
particular type of infection is often blurred, the efficacy of the T cell response is largely
determined by the cytokines that T cells make(Abbas et al, 1996; Seder and Paul,
1994). The molecular events in which dendritic cells may be involved that could control
the TH-I/Th-2 decision are further discussed in chapter 6.
As well as priming T cells in the periphery, dendritic cells can also delete developing T
cells during negative selection in the thymus(Matzinger and Guerder, 1989). This and
other related issues are outlined below.
Generation of the T cell repertoire
Despite the fact that T cell immune reponses can be primed by dendritic cells presenting
determinants derived from foreign antigen, the majority of determinants displayed by
MHC molecules are in fact self in origin(Rudensky et al, 1991). As clinically
recognisable autoimmunity is not an everyday occurrence, there must be mechanisms
which avoid T cell self reactivity and invoke a state of T cell self tolerance: developing
self reactive T cells in the thymus are in fact deleted by negative selection. In order to
combat infection, the T cells that remain available in the repertoire must have TCR that
adequately recognise foreign determinants in the context of self MHC molecules: this
can be accounted for by thymic positive selection.
The earliest T cell progenitors are the multipotent and self renewing CD34+ (in
humans) haematopoeitic stem cells of adult bone marrow. In an incompletely
understood process, these stem cells can differentiate, change phenotype and lose the
ability to generate cells of other lineages (for example the myeloid lineage) while
becoming more committed to the lymphoid lineage. The identification of a self
renewing but lymphoid committed stem cell has not yet been forthcoming(Shortman
and Wu, 1996). Additionally, the T progenitors which traffic to and seed the thymus are
unknown, as are the factors which regulate their movement. In the mouse thymus, the
Shortman's group. This cell can also form thymic dendritic cells, natural killer cells and
B cells(Shortman and Wu, 1996). Committment to the T lineage probably occurs before
the functional rearrangement of the P or y TCR chains. The pre TCR alpha chain, in
association with a successfully rearanged p chain, rescues thymocytes from apoptosis.
This pre TCR dimer then induces differentiation, so that a functionally rearranged alpha
chain may be expressed(von Boehmer and Fehling, 1997). This cell expresses low
levels of mature TCR and is CD4 and CDS double positive. The vast majority (95-97%)
of double positive cells die in the thymus: much of this is due to neglect, in other words,
a lack of interaction with MHC molecules(von Boehmer and Fehling, 1997).
Early indications that positive selection of thymocytes recognising self MHC molecules
must take place came from bone marrow chimaera experiments (Jameson et al, 1995).
Inbred homozygous mice (A) were lethally irradiated and then rescued with bone
marrow from mice of mixed parentage, of which one parent was genetically identical to
the irradiated mouse (AxB Fi mice). The T cells that developed in the irradiated mice
after bone marrow transfer were almost entirely restricted to the MHC molecules of the
recipient and of the shared parent (A) and not to the MHC haplotype of mouse B. This
showed that non-bone marrow derived cells regulated host MHC positive selection of
bone marrow derived thymocytes(Jameson et al, 1995). More information about the
process of positive selection has been elucidated, such as the cell types responsible
(thymic epithelial cells but almost certainly other cell types too) and its location (the
thymic cortex)(Anderson et al, 1996). Experiments using TCR transgenic mice showed
that the same peptide/MHC complex could induce positive selection or negative
selection depending on the concentration of the peptide present: low concentrations of
peptide caused positive selection(Sebzda et al, 1994). Additionally, peptides which
were unable to activate mature T cells could mediate positive selection of developing
thymocytes(Hogquist et al, 1994a), while agonist peptides for T cell activation could
not induce positive selection(Hogquist et al, 1994b). It appears that thymocytes are
positively selected on the basis of having low avidity for self peptide/self MHC
peptide/self MHC complexes or allogeneic MHC. During positive selection, the TCR
on double positive thymocytes will come into contact with MHC class I or MHC class
II molecules: this will determine whether a CDS or a CD4 single positive thymocyte
will result(Jameson et al, 1995).
Thymocytes need to avoid negative selection in the thymus; this process has also been
investigated using TCR transgenic mice(Kisielow and von Boehmer, 1995). Negative
selection is the induction of apoptosis in thymocytes with high avidity for self peptides.
Avidity is the sum of the affinity of the T cell receptors, the density of peptide/MHC
complexes and the density of other costimulatory molecules. Perhaps because of this,
the cells that mediate negative selection, for instance thymic dendritic cells, tend to
have high levels of costimulatory molecules(Matzinger and Guerder, 1989). Generally,
negative selection deletes thymocytes recognising abundant self peptides with
intermediate or high affinity and thymocytes with TCR of high affinity for other self
peptides(Kisielow and von Boehmer, 1995). In turn, this implies that some thymocytes
may escape negative selection. If the TCR of these thymocytes have a low affinity for
self this should not be a problem, as the activation requirements for mature naive T cells
are more stringent than the threshold to induce negative selection in
thymocytes(Pirchner et al, 1991, Sebzda et al, 1994). However some T cells
recognising self peptide with high affinity may escape negative selection because the
self peptide is not expressed in the thymus. There could be two reasons for this: either
the peptide is not adequately processed from the protein it derives from by thymic
antigen presenting cells, or the protein is not expressed in the thymus. In the first case T
cells could be activated against the self peptide if it is processed efficiently elsewhere
by a different antigen presenting cell. In the second case, other mechanisms probably
exist to tolerise T cells against the large numbers of proteins whose expression is either
tissue or developmentally regulated to preclude thymic expression. This is called
peripheral tolerance, and although there is evidence to suggest that peripheral lymphoid
The development of a self-MHC recognising, but non-self reactive T cell repertoire is
dependent upon generating self peptides which are presented by MHC molecules.
Likewise, the ability of dendritic cells to prime T cells against MHC class II restricted
pathogenic antigen not only requires migration and costimulatory molecules, but also
involves the processing of protein antigen into a form capable of binding MHC class II,
and regulating the binding event itself. Antigen processing for MHC class II
presentation is discussed below, although it must be noted that only recently has antigen
processing by dendritic cells been directly studied.
MHC class II restricted processing of exogenously acquired antigen
In 1981 Ziegler and Unanue found that in order for Listeria monocytogenes antigens to
be recognised in the context of MHC class II molecules (la) on macrophages by CD4+
T cells. Listeria had to be phagocytosed and partially catabolised(Ziegler and Unanue,
1981). This catabolism was termed antigen processing and was temperature and energy
dependent. Further experiments showed antigen processing could be inhibited by prior
fixing of the macrophages, by lysosomo- and acido-tropic compounds such as
chloroquine and ammonia, or by the protease inhibitor leupeptin(Streicher et al, 1984).
Importantly, the degree of catabolism inhibition was reflected in similar blocking of
antigen presentation to T cells. Other studies showed that in vitro enzymatic or
chemical fragmentation of antigen could substitute for cellular
processing(Shimonkevitz et al, 1983), and that MHC class II molecules could form
stable complexes with synthetic peptides(Babbitt et al, 1985). This latter finding was
also shown to be the case for MHC class I molecules(Townsend et al, 1986), although
the normal origin of peptides for the two types of MHC molecules appeared to be
different: MHC class I presented endogenously derived peptide(Townsend and Bodmer,
1989) while peptides presented on MHC class II tended to be from exogenous
antigen(Braciale et al, 1987). Although there are important exceptions to both these
MHC class n was shown to be accessible to internalised antigen (Cress well, 1985)and
intracellular transport of MHC class II intersected the endocytic route(Neefjes et al,
1990). Other work showed that interection of MHC class II dimers with peptide led to
enhanced MHC class II stability^ vitro and in vivo , and that as a result(Germain and
Hendrix, 1991; Sadegh Nasseri and Germain, 1991), peptides can affect the lifespan of
MHC class II molecules within cells(Nelson et al, 1994), A major question raised by
these findings was how peptide binding to MHC class II was prevented in early
compartments such as the endoplasmic reticulum (ER) or the golgi body (where many
peptides would be present), but allowed in later compartments which contained peptides
derived from captured exogenous antigen. In fact MHC class II molecules do present
many self peptides(Rudensky et al, 1991), but that some MHC class II does remain
accessible to endocytosed and processed foreign antigen is due to the functions of
invariant chain, HLA-DM, HLA-DO and associated proteases.
Accessory proteins in MHC class II restricted processing
The stabilisation of MHC class II by peptides leads to the conclusion that newly
synthesised MHC class II was probably incompletely folded(Germain et al, 1996);
usually mechanisms in the ER would be expected to degrade such molecules. In 1979
Invariant chain (li) was found to coprecipitate with MHC class II from cell
lysates(Jones et al, 1979): li is a non-MHC encoded protein whose expression is
nevertheless co-regulated with MHC class II(Ceman and Sant, 1995). li exists in two
forms in the mouse, p31 and p41, and four forms in humans, p22, p35, p41 and
p43(Ceman and Sant, 1995). After translation in the ER, li trimerises and then binds
three MHC class II dimers: a nonanmer is formed(Roche et al, 1991). In the absence of
li, MHC class II was found in the ER in large misfolded aggregates or with ER proteins
complexed in the MHC class II binding site: this implied that li facilitated MHC class II
folding, and prevented premature access to the binding site(Bonnerot et al, 1994;
Marks et al, 1995). It also appears that trimérisation of li controls the intracellular
Mutations in the MHC class Il-like heterodimer HLA-DM were found to abolish
presentation of certain determinants from exogenously acquired antigen even though
MHC class II was successfully synthesised(Riberdy et al, 1992; Sette et al, 1992).
Peptides eluted from MHC class II molecules in these cells were shown to correspond
to amino acids 82-107 of li; this region was termed CLIP (Class II associated invariant
chain peptide). Further work showed that the function of normal HLA-DM was to
facilitate the exchange of CLIP from MHC class II for other more tightly binding
peptides(Denzin and Cress well, 1995; Sherman e ta l, 1995; Sloan e ta l, 1995). It
should be noted however that CLIP and HLA-DM have different binding affinities for
and regulatory activities on different alleles of MHC class II. HLA-DO has recently
been shown to counteract the function of HLA-DM(Denzin et al, 1997); the ratio of
DM/DO in a cell may therfore be crucial in determining how available MHC class II
molecules are for peptide loading. Additionally, proteases such as cathepsin S are
responsible for degrading non CLIP regions of li prior to CLIP removal(Riese et al,
1996), enabling DM/DO to gain access the MHC class II/CLIP complex.
The exact location of peptide loading onto MHC class II is still not clear, for one main
reason, that different determinants have different processing requirements, and so could
be loaded under different conditions. Moreover, present knowledge of the intracellular
location of antigen processing and peptide loading comes largely from studies with B
cell lines, not with dendritic cells, which are arguably the most important antigen
processing cell type. A summary of compartments thought to play a role in antigen
processing is given below.
Endocytic compartments involved in antigen processing
Generating MHC class II determinants from an antigen is a degradative process, while
transport of MHC class II involves membrane transport. Normally these two processes
are kept separate to protect surface receptors from being degraded, and to ensure that
degradative enzymes remain within the cell. The integration of these processes which
The first intracellular structure to be identified as containing both MHC class II and
molecules needed for antigen degradation was the MIIC, found by Peters et a l MÜCs
were multilamellar, close to the trans-Golgi network and although not directly accessed
by transferrin receptors (found in early endosomes), could be reached by
endocytosis (Peters eta l, 1991). They contained lysosome associated glycoproteins and
acid hydrolases. Whether MHC class II in MIIC was associated with li was unclear, but
generally only small amounts of li were present in these compartments. Attempts to
purify MIICs using density gradient centrifugation and free flow electrophoresis of
murine A20 B cell lysates identified an anodally deflected set of vesicles which were
termed CIIV(Amigorena et al, 1994). CHV and MIIC share many characteristics,
including accessibilty by endocytosis and having the ability to form stable MHC class
Il-peptide complexes. However CITV contain transferrin receptors, and are of lower
density than MIIC, suggesting that CIIV may be more endosomal than lysosomal.
The route of MHC class Il/Ii into the endocytic pathway has been studied in A20 cells.
This pathway is normally rapid, so in order to slow it down and reveal transport
intermediates, leupeptin was used, which inhibits li degradation. These experiments
showed that MHC class Il/Ii first travelled to early endosomes, and from there to
CIIV(Amigorena et al, 1995). During this process li was degraded so that MHC class II
was available for loading at the CIIV stage. This finding and the morphological
relationship with MIIC suggest that MHC class Il/Ii leave the golgi body, go to early
endosomes (and possibly even the surface) and then 'bounce back' via CIIV, then MIIC,
and then ultimately to perinuclear lysosomes for degradation if peptide loading has not
occurred(Castellino and Germain, 1995). If MHC class II has been stabilised by
peptide, the complex is transported to the surface, via a route which is at present very
uncertain.
The experiments outlined above, which show that both low and high density vesicles
contain stable MHC class Il/peptide complexes are biochemical in nature. If specific T
cells or antibodies against particular MHC class Il-peptide complexes are used to
appear, with either high or low density vesicles being identified (but not both),
depending on the determinant being investigated (Watts, 1997). This reflects the fact
that different determinants have different processing requirements and so may be
revealed in one compartment type but not in others, even though many compartments
can in principle load peptide. As discussed in chapter 5 some determinants can be
processed and presented independently of invariant chain, by loading onto recycling
MHC class II in early endosomes(Pinet et al, 1995). Overall, the intracellular sorting of
MHC class II (in B cells at least) seems to be regulated to allow maximum possible
exposure to the determinants generated in various different compartments as an antigen
is processed.
Given the ability of MHC class II to scan intracellularly most of the endocytic route for
determinants to bind to, the phenomenon of immunological dominance in T cell
reponses is perhaps surprising. This is the empirical observation that an in vivo T cell
response to an injected foreign antigen is usually narrowly focussed on one or two
determinants derived from that antigen, even though many more determinants may have
the potential to stimulate T cells(Sercarz et al, 1993). How this phenomenon may arise,
and its consequences for immunity are discussed below.
Dominant and cryptic T cell determinants
Immunodominance was first used in the context of B cells, referring to determinants to
which most of the immunoglobulins in a response were directed(Sercarz et al, 1993).
The term was later employed to describe a murine proliferative response to injected Hen
Eggwhite Lysozyme (HEL): T cells from mice immunised with whole HEL were found
to respond ex vivo to one cyanobromide fragment of HEL, despite the the fact that other
fragments were immunogenic when injected into mice(Maizels et al, 1980). Similar
observations were made for responses to other antigens including cytochrome c, lambda
repressor protein, insulin, myoglobin, ovalbumin and staphylococcal nuclease(Sercarz
Determinants are strictly defined as being dominant, subdominant or cryptic in the
following manner: a native foreign protein antigen is emulsified in adjuvant and
injected into mice. Ten days later, T cells isolated from draining lymph nodes are
cultured with synthetic peptides derived from the primary sequence of the antigen. The
peptide determinant which elicits the largest proliferative response is the dominant
determinant. Subdominant peptide determinants induce weak responses from
lymphocytes primed against whole antigen, while cryptic determinants are not
responded to. All three types of determinant can induce determinant specific responses
when injected into mice in the form of synthetic peptide, but, as a corollary to the
above, only T cells primed against dominant peptides in vivo will proliferate strongly to
whole antigen in in vitro recall assays(Maizels et al, 1980). An important point is that
the positions of the determinants (dominant, subdominant and cryptic) in an antigen are
usually different for different MHC class II alleles, for instance HEL 106-116 is
dominant in BALB/c mice (H-2^), but HEL 46-61 is dominant in CBA mice (H-2^).
These differences are mainly due to the properties of the peptide binding grooves of the
two MHC class II dimers, but other factors such as CLIP/MHC class II affinity and
levels of proteases may also have an influence. Once the pattern of determinant
hierarchy is established after the injection of a foreign native antigen, it is stable and
does not usually change over time(Sercarz et al, 1993).
A major reservation for these definitions is that proliferative assays detect primarily TH-
1-type responses. Therefore determinants which are dominant in TH-2 responses will
not be identified. Another problem is that MHC class Il/peptide complexes formed by
loading of exogenously added peptide onto recycling MHC class II molecules (as in in
vitro in recall assays) will not necessarily have the same conformation as complexes
formed in later compartments following processing of the whole antigen. This seems to
be the explanation behind findings of Viner et al who showed that some T cell
hybridomas isolated following immunisation of mice with a dominant peptide could not
recognise naturally processed whole antigen in vitro(VinQr et al, 1996). Another study
on the basis of poor proliferative responses was in fact well displayed by B cells after
processing the whole antigen in vitro : the reason behind the crypticity could have been
at the level of the T cell response, not in the processing of the determinant(Viner et al,
1995). There is the possibility however that while this determinant was well processed
and presented by B cells, it may not have been efficiently displayed by dendritic cells in
vivo , the crucial antigen presenting cell type in forming T cell responses. Differences
between antigen presenting cell types are discussed later.
Usually, dominant determinants are well presented and less immunogenic determinants
are poorly presented. The mechanisms by which this occurs are thought to be as a
consequence of how MHC class II binds partially degraded protein during antigen
processing.
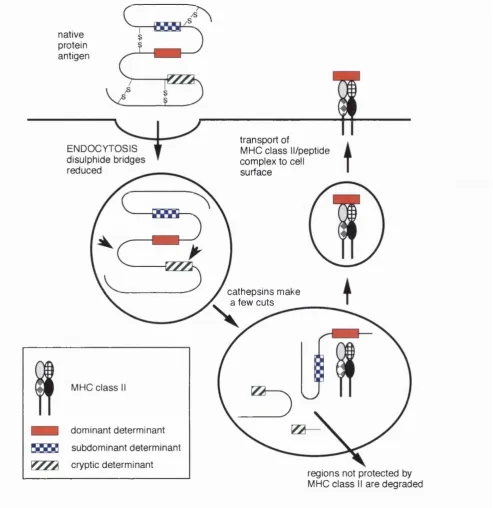
Determinant capture and protection
Buus et al found that peptides with high binding affinity for MHC class II molecules
were also well presented(Buus et al, 1987). Schaeffer et al also observed this
phenomenon using peptides covering the entire sequence of staphylococcal nuclease,
but went further by analysing the immunigenicity of the peptides(Schaeffer et al,
1989). They found that strongly binding peptides were very stimulatory for T cells,
whereas peptides of lower affinity showed decreased immunogenicity. Adorini et al
examined whether peptide determinants could compete with each other for MHC class
II binding: they showed that a strongly binding peptide from mouse lysozyme could
prevent in vivo priming against HEL peptides if the self peptide was injected into mice
simultaneously with, but in excess to the HEL peptide(Adorini et al, 1988). Further
experiments demonstrated that this in vivo prevention effect was limited to situations
where one of the peptides involved was clearly a weaker binder(Sercarz et al, 1993).
These findings and observations showing that changes in regions of an antigen distant
from a determinant could affect that determinants' immunogenicity led to the hypothesis
of determinant capture and MHC protection to explain immunodominance. This has