Modelling of silver adhesion on MgO(100) surface with defects
Yu F Zhukovskii†‡, E A Kotomin†§¶, P W M Jacobs‡, A M Stonehamkand J H Hardingk
† Institute of Solid State Physics, University of Latvia, Kengaraga 8, LV-1063 Riga, Latvia ‡ Department of Chemistry, University of Western Ontario, London, Ontario N6A 5B7, Canada § Max Planck Institut f¨ur Festk¨orperphysik, Heisenberg strasse 1, D-70569 Stuttgart, Germany kCentre for Materials Research, Department of Physics and Astronomy,
University College London, Gower Street, London WC1E 6BT, UK E-mail:[email protected]
Received 11 August 1999, in final form 6 October 1999
Abstract. We show how surface defects (especially F0
s and V0scentres) can play a major role in
the adhesion of Ag (at 1:4 and 1:1 coverages) on the MgO(100) surface. Our calculations use a periodic (slab) model and an ab initio Hartree–Fock approach with a posteriori electron correlation corrections. We are able to analyse the interatomic bond populations, effective charges and multipole moments of ions, in combination with the interface binding energy and the equilibrium distances. Both surface defects cause strong redistributions of the electron density which increase the binding energy of metal atoms by more than an order of magnitude. This implies radiation-induced strengthening of metal adhesion on oxide substrates and clarifies defect mechanisms in nucleating film growth. We compare our atomistic predictions with those from simpler methods which might be used for complex technologically interesting systems. There is good general agreement with the image interaction model; differences arise partly from different treatments of dispersion and partly from subtle but significant charge redistribution in the Ag. Further, a simple Born–Haber analysis of charge transfer is consistent with the several cases predicted in the atomistic calculations.
1. Introduction
The control of metal/oxide interfaces in their many technological applications depends on the quantitative understanding of adhesion and of the kinetics and mechanisms of metal film growth on oxide surfaces (see [1–6] and references therein). This understanding contains significant gaps, despite many theoretical studies, mostly of the adhesion of noble and transition metals on MgO substrates [7–17]. Partly this is because of the very sensitive balance between the various energy contributions. Perhaps the successes are more surprising than the inconsistencies. This is especially true when the range of atomistic methods is noted. Cluster models [8, 9, 15] and slab models (periodic in two dimensions) [11–14] have been applied to defect-free interfaces; a few calculations have attempted proper embedding [18]. An ab initio Hartree–Fock formalism has been used in cluster models of Me/MgO interfaces [8, 9]. For metal adhesion on MgO surfaces, methods applied include the local density approximation (LDA) as a full-potential linearized muffin-tin orbital method (FP LMTO) in a slab model [11, 12], full-potential linearized augmented plane waves (FP LAPWs) [13] and self-consistent local orbitals (LOs) [14, 15]. We have recently made Hartree–Fock calculations for the Ag/MgO(100) interface
¶Corresponding author.
using a slab model [16]. The modelling of the complex, technological metal/oxide systems requires simpler methods. Examples are the image interaction model (IIM) (Stoneham and Tasker [2], [7]) and the shell model (SM; this model is also used in many IIM calculations) [17].
Tests of predictions come from several types of experiment. Results for atomic structure nowadays could be compared directly with high-resolution electron microscopy data, at least for a few metal/oxide (e.g. Ag/MgO) interfaces [25] at near-atomic resolution. For liquid metal/oxide interfaces, there are data for adhesion energies and wetting angles. Wetting (small wetting angle) often implies a chemical reaction between the liquid metal and the oxide substrate. For non-reactive liquid metals, there are systematic trends of wetting angle with substrate [7]. Yet there is significant adhesion even without reaction. For Ag/MgO, our calculations (both HF [16] and IIM [20]) showed negligible chemical reaction of Ag with perfect MgO surfaces. The major term in adhesion comes from polarization of the metal by the oxide ions (the major IIM mechanism), plus electronic density accumulation in gap positions between atoms in the metal film, as discovered in the HF calculations.
For thin solid metal films on oxides, what is seen depends strongly on growth conditions. Thus, Ag growth on MgO usually yields 3D islands [19, 21]. Yet a recent low-energy-electron-diffraction study showed a layer-by-layer growth mode could occur for silver deposited on vacuum-cleaved MgO(100) surfaces, even though the structure is metastable [22]. Partly, this is a matter of kinetics: the rate of Ag deposition, and competition between surface processes [23]. It is clear that defects can be critical in determining the growth mode, since metal clusters nucleate and grow mainly at defect sites [1]. Experiment shows defects on an MgO(100) surface serve as nuclei for the growth of metal clusters on oxide substrates [5, 24], a continuous metal film forming after clusters overlap. This agrees with calculations [10] showing a delicate energy balance between Ag island and monolayer-mode growth.
As first suggested by Stoneham and Tasker (1986) [25], charged (e.g. radiation-induced) defects increase the adhesion energy via the image interaction between charged oxide defects and the metal. Calculations [20] predict a maximum interaction between a vacancy in MgO and a metal of≈2 eV per defect. Similar values were found in atomistic SM calculations for ten layers of Ag on an MgO(100) surface with O vacancies [15]. Our main concern here is with effects which go beyond these simpler yet useful approaches, so as to understand electron transfer and redistribution for metal atoms at defect sites on the oxide surface. We find our quantum calculations both support the earlier approaches and point to new and simple ideas which will make analysis of more complex systems practical.
2. Theory
We use the ab initio Hartree–Fock code CRYSTAL95 for periodic systems [26], with electron correlation corrections (HF-CC method) calculated using density-functional theory [27]. Such terms are needed, as standard HF theory underestimates binding energies and overestimates molecular bond lengths. We use a posteriori corrections in the non-local Perdew–Wang generalized gradient approximation (PWGGA) [28]. The MgO basis set (BS), optimized previously [29], consists of all-electron 8-61G and 8-51G functions (s and sp shells) for Mg and O atoms, respectively. To reduce computation, we employed Hay–Wadt small-core (HWSC) pseudopotentials for Ag atoms [30], so reducing the total number of electrons per Ag to 19 (4s24p64d105s1). An initial guess for the valence BS of silver (311-31G for sp and d shells) was taken from AgCl calculations [31], and the outer exponents were re-optimized [16]. Calculations starting from the Ag 4d95s2configuration give essentially the same results. We have estimated a posteriori electron correlation corrections to the total energy in our HF calculations. They prove to be about 0.5% of the HF total energy, and are almost independent of interface distance.
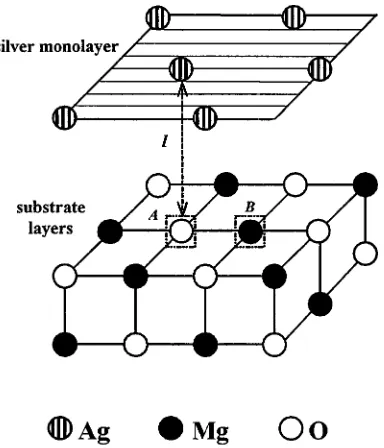
Figure 1. Sketch of Ag/MgO(100) interface, with Ag atoms placed at a distancelabove either the surface Fscentres (A) (or O2−ions on the perfect substrate), or above the Vscentres (B) (or the
regular Mg2+ions). The surface concentration of defects is 1:4.
vary along thezaxis, perpendicular to the interface. The distances between different silver planes within the metal slab are also free to change.
For all perfect and defective configurations, the binding energy was calculated as difference of the two total energies, that for the interface with optimized geometry and that for the two separate slabs. To find the binding energy correctly, we calculated carefully the interface potential energy curve versus the interface distance. As the distance increased to large values, we used as an initial guess for SCF procedure the density matrix of the interface configuration with slightly closer separation. In this way, we could avoid the so-called ‘polarization catastrophe’ for widely separated slabs. If one calculates the binding energy for a defective interface using some of the traditional formulae, the interface energy inevitably includes an error.
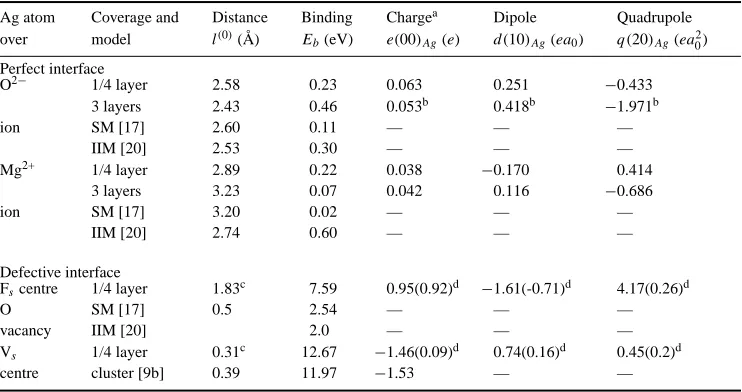
Results in table 1 are for the most likely sites for Ag adsorption: above surface O2−ions and above surface Mg2+ions. Surface defects on the MgO substrate are modelled by removing
one of four O or Mg atoms from the 2×2 extended surface unit cell (figure 1). By removing neutral atoms, we retain neutral supercells, with neutral surface F0
s centres, or V0s centres. Charged defects are not discussed in this paper. We leave in the vacancy the basis set of the missing atom (the so-called ghost [26]); without this part of the basis set, we should have a poor representation of the neutral defect. Atomic relaxation of the MgO structure around these defects has been found for the isolated MgO slabs and reoptimized for the Ag/MgO interface with defects.
3. Results and discussion
3.1. Perfect interface
First, we consider the favoured adsorption site for Ag on the perfect MgO surface. For full (1:1) coverage, HF-CC predicts adsorption over the surface O atoms is favoured energetically. This agrees with recent experiments [32] and with three previous LDA-type calculations [11–13]. It contradicts those IIM results [18, 33] for which the most accurate treatment of the dispersion forces was included, but agrees with an image model (IM) approach which used a less accurate treatment of dispersion. The main difference from the IIM appears arise from the strength of the long-range dispersion forces between the Ag and the O neighbours of the Mg site below the Ag. Shell model calculations [17], broadly equivalent to the IIM, predict preferential adsorption over O2−, with a low adhesion energy of only 0.11 eV. This value, smaller than
our predictions, may result from neglect of the electron density redistribution within the metal plane. Our present results show that a large part of the differences in detail between full electronic structure calculations and these approaches stems from relatively subtle shifts in Ag charge density.
When Ag is above O, all microscopic methods give similar spacings between the outer MgO plane and the Ag which faces it, with values in a narrow range 2.4 to 2.7 Å. The image model result agrees (2.53 Å), as does the SM (2.60 Å). Our HF-CC value of 2.43 Å for three metal layers agrees with recent experimental data [32]. For a three-layer Ag film with Ag over O, the HF-CC adhesion energy of 0.46 eV is smaller than the value of 0.88 eV from FP LAPW calculations [11]; an even lower value is found for Ag over Mg by HF-CC (table 1). The relevant experimental estimate is 0.26 eV [19], which is probably lower than the value for a defect-free surface because of misfit dislocations arising from the 3% difference in lattice parameters of Ag and MgO.
metal by the ions. Calculated Mulliken charges on Ag atoms (e(00)column in table 1) show negligible charge transfer between MgO and Ag. The bond populations across the interface (between Ag atoms and ions of the perfect MgO(100) substrate) are practically zero. We note that the existence of a good fit of the interfacial energy versus interface distance [14] to the so-called universal binding energy relation (similar to the potential energy curve for diatomic molecules) does not necessarily imply chemisorption between metal and substrate (see more in [16]).
Table 1. Energies, distances and charge distribution parameters of the perfect and defective
Ag/MgO(100) interfaces. Note that the shell model [17] has ten Ag layers atop 31 MgO planes; for the image interaction model [20] of NiO, the optimum vacancy site is in the second oxide layer. Ag atom Coverage and Distance Binding Chargea Dipole Quadrupole over model l(0)(Å) Eb(eV) e(00)Ag(e) d(10)Ag(ea
0) q(20)Ag(ea02)
Perfect interface
O2− 1/4 layer 2.58 0.23 0.063 0.251 −0.433
3 layers 2.43 0.46 0.053b 0.418b −1.971b
ion SM [17] 2.60 0.11 — — —
IIM [20] 2.53 0.30 — — —
Mg2+ 1/4 layer 2.89 0.22 0.038 −0.170 0.414
3 layers 3.23 0.07 0.042 0.116 −0.686
ion SM [17] 3.20 0.02 — — —
IIM [20] 2.74 0.60 — — —
Defective interface
Fscentre 1/4 layer 1.83c 7.59 0.95(0.92)d −1.61(-0.71)d 4.17(0.26)d
O SM [17] 0.5 2.54 — — —
vacancy IIM [20] 2.0 — — —
Vs 1/4 layer 0.31c 12.67 −1.46(0.09)d 0.74(0.16)d 0.45(0.2)d
centre cluster [9b] 0.39 11.97 −1.53 — —
aA positive sign means an excess of electron density as compared to a neutral Ag atom. bFor the interfacial silver layer.
cThe distance between the Ag atom and the centre of the vacancy.
dThe numbers in parentheses are the multipoles of ghost orbitals centred on the vacancy.
There is significant redistribution of charge within the metal; bond populations between nearest Ag atoms within metal planes parallel to the interface are typically 0.1eper atom, and are not sensitive to the adsorption site. The concentration of Ag electron density at the bridge position between nearest metal atoms has been confirmed in inelastic He scattering studies [34]: only by introducing negative pseudo-charges in these positions in metal films could a good fit be found for theoretical surface phonon-dispersion curves to experimental data. The effective atomic charges and their definitions are discussed further in [35]. Bond population analysis for the three-layer Ag on MgO(100) shows why Ag adsorption over O2− ions is favoured. This stems from electrostatic attraction with enhanced electron density near the hollow position in the interfacial Ag layer (0.07efrom table 1). The extra charge has an attractive interaction with the substrate Mg2+ion below it. For the Ag adsorption over the Mg2+
ions, there is instead a repulsion between the electron density localized between Ag atoms, and the substrate O2−ion below it.
mixing. The negativeq(20)Agfound for thicker layers means an axial Ag contraction in the zdirection or expansion in the xy plane. The Ag atoms adsorbed on MgO(100) surfaces are considerably deformed. This deformation is the origin of much of the adhesion, and its trends with numbers of Ag layers underlie trends in adhesion energies for defect-free surfaces [16]. Partial (1:4) Ag coverage of the MgO(100) surface shows differences from three-layer (1:1) coverages. For the low (1:4) coverage there is negligible interatomic electron density concentration between Ag atoms; the interatomic charge density is too small to play a role. For Ag adsorption over O2−or Mg2+ions, there is a single nearest substrate ion (either O2−or Mg2+) and four next-nearest substrate ions of the opposite type (either Mg2+ or O2−). Adsorption energies for these two cases are close, suggesting compensation of electrostatic attraction and repulsion between the slightly charged Ag adatoms and substrate ions.
For the 1:4 surface coverage, the small charge transfer (0.06e) from the substrate to each isolated Ag atom is almost the same for three Ag layers atop MgO, but the value of the dipole moment is larger for the thicker Ag layer. The isolated Ag atom charge density is deformed along thezaxis, as expected in the IM model. The difference in quadrupole deformation (table 1) may be come from mismatch of Ag and MgO lattice spacings, which has a bigger effect at full coverage. Difference electron density maps, analysed by Zhukovskii et al (1999) [16] for low Ag coverage, show that Ag atoms are more polarized above O2−
substrate ions. Charge transfer from the substrate is still very small, and it is not clear that chemical bonding is a major factor. The main effects seem to be intra-ionic polarization of Ag and oxide ions. Second neighbours are important, as seen on comparing the effects on MgO(100) and (110) (Zhukovskii et al 1999 [16]). The effect is even more pronounced for Ag on the O-terminatedα-Al2O3(0001) surface (Zhukovskii et al 1998 [16]). Our estimates
of the basis-set-superposition error (BSSE) [36] show it to be very small, e.g. for Ag above O2−it is only 0.03 eV.
3.2. Surface defects
The results of defect calculations for low (1:4) Ag coverage are summarized in table 1. We first optimized the geometry of a (bare) pure MgO(100) slab around the neutral F0
s and V0s centres, as well as around empty (charged) Mg2+or O2−vacancies. When a ghost orbital was
included in the O2−vacancy, its population was 1.72e, typical of F0centres in ionic oxides.
The remaining 0.28eis distributed over two spheres of nearest ions. The F0centre thus mimics
the O2−ion which it replaces, and there is negligible atomic relaxation of the surrounding ions.
Its energy level lies 4.2 eV above the top of the valence band. If we neglect ghost orbitals, the O2−vacancy becomes doubly charged, with the two electrons distributed on nearest-neighbour
ions. Four surface Mg2+ions are repelled from the vacancy and shift outwards by 0.16 Å; the axial subsurface Mg2+ion is displaced downwards by 0.23 Å. In the V0s centre calculations the effect of the ghost orbitals is small, since no electron density can be localized inside the Mg2+vacancy. The O2−ions surrounding the Mg2+vacancy shift outwards from the vacancy: four equivalent surface O2−ions move outwards by 0.12 Å, and the axial subsurface O2−ion moves downwards by 0.16 Å. The four surface O2−ions share 1.8 of two holes associated with
the V0
s centre. This agrees with HF cluster calculations (Ferrari and Pacchioni 1995 [9]). In the further interface calculations with Ag present, we re-optimized atomic relaxations continuously for every position of an adsorbed Ag atom over the vacancies at which ghost orbitals are centred. When an Ag atom approaches the V0
s centre, it donates two of its valence electrons to the four O2−ions surrounding the Mg vacancy. These fill the two holes localized
on the O− ions of the free V0
than +2eby 0.5edue to partial electron delocalization and weak bonding with these O2−
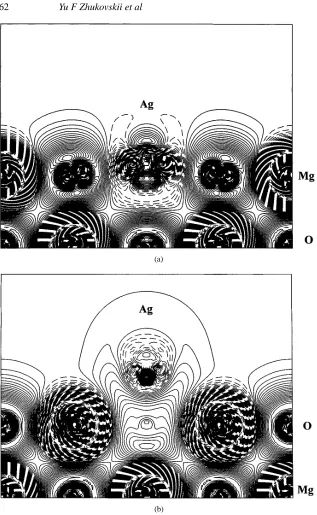
ions.) This Ag2+ion substitutes almost perfectly for the Mg2+host ion removed, being only 0.31 Å above the surface plane. This agrees with the conclusions of two previous HF cluster calculations for Cu and Ag on MgO(100) [8, 9]. Large dipole and quadrupole moments, and the difference electron density maps in figure 2(a), indicate that the Ag2+ion is quite strongly polarized and deformed (cf similar data for Ag atom adsorption on the perfect surface given in table 1). The binding energy is very high, 12.67 eV, again agreeing well with a cluster calculation [9]; this is due to a very large electrostatic stabilization effect. We use Born–Haber cycles in section 4 to understand the electron transfers between Ag and the neutral metal and neutral oxygen vacancies.
For the Ag atom adsorption over F0s centres, we observe an opposite effect: one electron is transferred from the surface defect to the Ag atom. This results in a pair of opposite charges, Ag−/F+
s, with the mutual separation of 1.83 Å, much smaller than for the Ag atom above O
2−
on a regular surface. The population 0.5eof the Ag−–F+
s bond is quite considerable, whereas all other bonds have negligible populations. The optimized lattice relaxation includes 0.06 Å outward on-plane displacements of Mg2+from the Fscentre, and 0.08 Å vertical displacement of the Mg2+in the plane below the Fs centre. The considerable binding energy can again be supported by electrostatic estimates for the interaction between Ag−with nearest surface ions. Large dipole and quadrupole moments indicate that Ag−is contracted and strongly deformed as is also seen from figure 2(b). IIM calculations [20] give the interaction energy between the doubly charged vacancy (preferentially in the subsurface plane) and a neutral Ag atom to be ≈2 eV which addresses a quite different situation. The latter estimate is close to an atomistic SM prediction of 2.54 eV [17] which also neglects charge transfer.
We also performed calculations for an Ag monolayer over an array of the Fscentres in the same concentration (1:4). The binding energy per 2×2 extended unit cell increases by 0.75 eV, i.e. there is an additive effect in the interaction of four Ag atoms with the defect and three regular O2−ions. The adhesion energy for each of the three Ag atoms is 0.75/3=0.25 eV, which is very close to that for a perfect surface with monolayer coverage [16]. The quadrupole moments of Ag atoms in the monolayer are large, which demonstrates their deformation. It is likely that such a monolayer is unstable against faceting.
In any periodic boundary calculation, there is inevitably some band width (energy dispersion) associated with even highly localized defect states. When such ‘band’ states are only partly occupied, there is some argument for saying the system is conducting. Any real system will have features which inhibit long-range transport, such as the stress and electric fields of random defects, or perhaps special correlation or polaron effects beyond those already included. According to our CRYSTAL calculations, both the perfect MgO(100) slab and the MgO(100) slab with F-centres (for 1/4 surface coverage) are insulating. As to the MgO(100) slab with V-centres (for the same 1/4 coverage), our calculations do suggest it may be conducting. Whether real MgO surfaces with these Mg defects would conduct is an open question, and the few experimental data give no guidance. Both the perfect and defective Ag/MgO(100) slabs are conducting, even for 1/4 Ag coverage; it is probably not surprising that Ag has this effect, even at 1/4 coverage.
4. Charge transfer processes
Our calculations have shown at least three cases in which electrons are transferred to or from the Ag. These are the transfer of an electron from the F0
s centre to Ag, the transfer of two electrons from Ag to the V0
s centre, and the loss of two electrons by Ag
0to form Ag2+when
(a)
(b)
Figure 2. The difference electron density maps (total density minus superposition of atomic
densities) for the cross-section perpendicular to the (100) interface plane for 1:4 Ag adsorption: (a) over a surface Vs centre, showing effectively an Ag2+substituting for Mg2+and (b) over a
surface Fscentre, showing noticeable Ag−–F+s bonding. Isodensity curves are drawn from−1 to
+1e a−03with an increment of 0.0025e a−03.
are more transparent. Such simpler approaches can be used to identify trends over a range of materials, or to discuss complex systems, or to generalize to other metals.
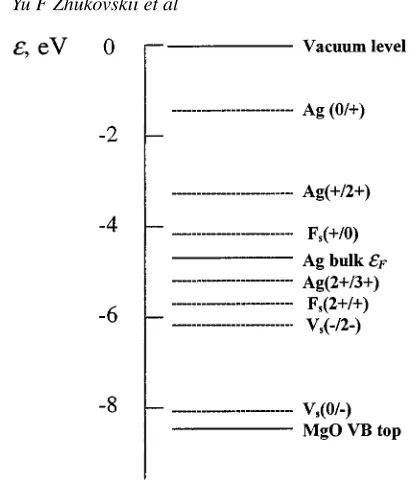
We shall assume that the Ag behaves like bulk Ag, with a work function of 4.4 eV, i.e. that the Fermi levelεF is 4.4 eV below vacuum. We now discuss energies which we write in forms such asε(0/+). An electron with this energy could be added to the ‘+’ state of the centre considered to form the neutral state without absorbing or releasing energy (for a fuller discussion, see [37]). The important question is whether a particular energy, likeε(0/+), lies above or below the Ag Fermi level. If it lies above, the ‘+’ state will be stable, i.e. the neutral centre will transfer an electron to the Ag. We now estimate some of the energies relevant for the charge transfers predicted in our full calculations. We emphasize that there may be significant uncertainties (perhaps 1 eV) in the rough estimates. Figure 3 shows the important energy levels schematically.
4.1. Electrons from the surfaceF0
s centre to Ag
Here we use the results of Ferrari and Pacchioni (1996) [9], corrected for some missing but substantial polarization energy terms [38]. These results show:
F0s goes to F+s + e (free) costs 4.2 eV, soεF c(+/0)= −4.2 eV F+s goes to F2+s + e (free) costs 5.5 eV, soεF s(2 +/+)= −5.5 eV
The neutral F0centre should transfer an electron to the Ag; the remaining electron in the F+
centre should not transfer. This agrees with our full-scale calculations.
4.2. Electrons from Ag to the surfaceVs0centre
Again we use the results of Ferrari and Pacchioni (1996) [9] as corrected in [38]
V0
scaptures free electron costs 7.8 eV, soεV c(0/−)= −7.8 eV V−s captures free electron costs 5.9 eV, soεV s(−/2−)= −5.9 eV.
In both cases, we expect electrons to be transferred from Ag to the cation vacancy, i.e. we expect theVs2−state to be obtained. This again agrees with our full-scale calculations.
4.3. Substitutional Ag in an Mg vacancy
Here we ask first of all which charge states of substitutional Ag will be stable at an Mg site in bulk MgO. This type of question was addressed in [37] and [39]. These authors showed in their ionic shell model description that the main terms in the energy are the Madelung energy, the polarization energies and the free ion ionization potentials. There is also a need to know the electron affinity of MgO. Experimentally, this is very uncertain. The results of [39] and [40] showed that the known stable states of transition metal ion dopants in MgO could all be predicted correctly, along with their optical ionization spectra, with a negative electron affinity. Values in the range−1 eV to−3 eV were acceptable. We shall choose−1.0 eV, since this ensures that the known stable charge states of the bulk cation and anion vacancies are also correctly predicted. This value is consistent with known V-centre ionization energies and with estimates from MIES spectra (A L Shluger and V Kempter, private communication). In [37] reasons are discussed for differences between this negative value and certain measurements. The ionization potentials for Ag are similar to the corresponding transition metal values (the first three Ag ionization potentials in eV are 7.57, 21.48 and 34.82) [41]. Following [37] and [39] we find for AgMgthe values:
Figure 3. Schematic positions of the basic energy levelsεifor defects, for Ag and for MgO crystal bands.
The accuracy of these values is not high. For a surface defect, there will be other terms in the energy as well; these terms are of both signs. Nevertheless, if we ask what we should expect if these (bulk) energies do apply for the Ag atom at a surface Mg2+site, we see that the ‘2+’
state is to be expected. This agrees with the conclusions of our full-scale calculations. There are three comments we should make. First, the simple energies are relatively close and not very accurate. In particular, relatively modest changes in metal work function could lead to changes in what is expected: the ‘3+’ charge state of Ag might be stabilized, or the F0
s centre might retain both its electrons. Secondly, there may be other defects which can change charge state. This was addressed in [42] for Ag/NiO. In such cases, the space charge must be treated properly. Thirdly, there is an opportunity to control adhesion by controlling charge states. This we shall address in a separate paper.
5. Conclusions
One important general conclusion to be drawn from our ab initio Hartree–Fock calculations is that chemical bond formation is not important for the perfect Ag/MgO(100) interface. This is true also for Ag/MgO(110) (Zhukovskii et al 1999 [16]). Physical adhesion associated with polarization and charge redistribution is dominant. The adhesion energy is enhanced by the interaction of the substrate ions with the extra electron density near the interatomic positions of the interfacial Ag layer. This favours silver atoms placed above either surface O2−ions. The difference in predicted optimal Ag adsorption sites observed for this microscopic work and the image model stems mainly from the different treatments of long-range dispersion (van der Waals) interactions. However, these two kinds of calculation complement each other, since they address different situations: the atomistic modelling focuses on several metal planes whereas the image model prediction is for a thick metal layer atop an oxide substrate.
supported by experimental studies [18, 43, 44]. We find, in full agreement with previous HF cluster calculations [8, 9], an Ag atom placed above a Vs centre donates two of its valence electrons to the four O atoms surrounding the Mg vacancy, transforming into an Ag2+ ion, which then is drawn towards the surface and, in effect, substitutes for the missing Mg2+ion. The relevant binding energy is very high, about 12 eV, since there is a very large electrostatic stabilization effect.
In contrast, we also find that Ag atoms atop F0scentres attract one of the defect electrons to form a complex Ag−/Fs+of oppositely charged defects. Here, unlike Ag on the perfect surfaces, there is considerable covalent bonding, which localizes an additional 0.5e. The binding energy is 7.6 eV. Similar HF cluster calculations (Ferrari and Pacchioni 1996 [9]) do not find this effect for a neutral Fs0centres but only for the charged F+s centres. The discrepancy could arise from interaction between Fscentres (in our work, the concentration is high), different Fscentre basis sets or the cluster model boundary effects (unlike us, Ferrari and Pacchioni (1996) [9] find Ag atoms bound neither to O2−nor Mg2+on a perfect MgO surface, although Ag+ions are bound
to O2−, as one would expect).
One important result of our work is the validation of the ideas ([20], Stoneham and Tasker 1985 [25]) that charged defects can enhance adhesion. This was one of the main ideas developed in the image interaction model (Stoneham and Tasker 1985 [25]), although the early papers were not able to exploit full electronic structure methods to handle defects which we discuss here.
Acknowledgments
This study was supported by a British–Latvian Royal Society Joint Project Grant for collaborations with the former Soviet Union, and also by EPSRC grants GR/L02678 and GR/L70615. YZ greatly appreciates the support of the Centre for Chemical Physics of the University of Western Ontario (Canada) for a Senior Visiting Fellowship. Many thanks are due to M Alfredsson for assistance in some calculations and preparation of figure 2 as well as A B Kunz for stimulating discussions.
References
[1] Heinrich V E and Cox P A 1994 The Surface Science of Metal Oxides (Cambridge: Cambridge University Press) [2] Stoneham A M and Tasker P W 1985 J. Phys.: Condens. Matter 18 543
Stoneham A M 1981 J. Am. Ceram. Soc. 64 64 [3] Finnis M W 1996 J. Phys.: Condens. Matter 8 5811 [4] Ernst F 1995 Mater. Sci. Eng. R 14 97
Campbell C T 1997 Surf. Sci. Rep. 27 1 Renaud G 1998 Surf. Sci. Rep. 32 1
[5] Ertl G and Freund H-J 1999 Phys. Today No 1 32
[6] Proc. 4th Int. Symp. on Atomically Controlled Surfaces and Interfaces 1998 Appl. Surf. Sci. 130–132 Stoneham A M, Harding J H and Harker A H 1996 MRS Bull. 21 29
[7] Stoneham A M 1983 Appl. Surf. Sci. 14 249 [8] Bacalis N C and Kunz A B 1985 Phys. Rev. B 32 4857 [9] Ferrari A M and Pacchioni G 1995 J. Phys. Chem. 99 17 010
Ferrari A M and Pacchioni G 1996 J. Phys. Chem. 100 9032 [10] Stoneham A M and Harding J H 1998 Acta Metall. Mater. 46 1155
[11] Sch¨onberger U, Andersen O K and Methfessel M 1992 Acta Metall. Mater. 40 S1 [12] Goniakowski J 1998 Phys. Rev. B 57 1935
Goniakowski J 1998 Phys. Rev. B 58 1189
[13] Li C, Wu R, Freeman A J and Fu C L 1993 Phys. Rev. B 48 8317 [14] Hong T, Smith J R and Srolovitz D J 1994 J. Adhesion Sci. Technol. 8 837
[15] Pacchioni G and R¨osch N 1996 J. Chem. Phys. 104 7329
Yudanov I V, Vent S, Neyman K, Pacchioni G and R¨osch N 1997 Chem. Phys. Lett. 275 245 Matveev A V, Neyman K, Pacchioni G and R¨osch N 1999 Chem. Phys. Lett. 299 603 [16] Heifets E, Zhukovskii Yu F, Kotomin E A and Caus´a M 1998 Chem. Phys. Lett. 283 395
Zhukovskii Yu F, Alfredsson M, Hermansson K, Heifets E and Kotomin E A 1998 Nucl. Instrum. Methods 141 73
Zhukovskii Yu F, Kotomin E A, Jacobs P W M, Stoneham A M and Harding J H 1999 Surf. Sci. 441 373 [17] Purton J, Parker S C and Bullett D W 1997 J. Phys.: Condens. Matter 9 5709
[18] Duffy D M, Harding J H and Stoneham A M 1995 Acta Metall. Mater. 43 1559
[19] Trampert A, Ernst E, Flynn C P, Fischmeister H F and R¨uhle M 1992 Acta Metall. Mater. 40 S 227 Guenard P, Renaud G, Vilette B, Yang M-H and Flynn C P 1994 Scr. Metall. Mater. 31 1221 [20] Duffy D M, Harding J H and Stoneham A M 1994 J. Appl. Phys. 76 2791
[21] Schaffner M-H, Patthey F and Schneider W-D 1998 Surf. Sci. 417 159 [22] Harada T, Asano M and Mizutani Y 1992 J. Cryst. Growth 116 243
Didier F and Jupille J 1994 Surf. Sci. 307–309 587
[23] Duffy D M, Harding J H and Stoneham A M 1996 Acta Metall. Mater. 44 3293 [24] Harding J H, Stoneham A M and Venables J A 1998 Phys. Rev. B 57 6715
[25] Stoneham A M and Tasker P W 1986 Ceramic Microstructures ’86: Role of Interfaces ed J A Pask (New York: Plenum) p 155
Stoneham A M and Tasker P W 1985 J. Phys. C: Solid State Phys. 18 L543
[26] Dovesi R, Saunders V R, Roetti C, Caus´a M, Harrison N M, Orlando R and Apr´a E 1996 CRYSTAL-95 User
Manual (Turin: University of Turin)
[27] Caus´a M and Zupan A 1994 Chem. Phys. Lett. 220 145 [28] Perdew J P and Wang Y 1992 Phys. Rev. B 45 13 244
[29] Caus´a M, Dovesi R, Pisani C and Roetti C 1986 Surf. Sci. 175 551 [30] Hay P J and Wadt W R 1985 J. Chem. Phys. 82 284
[31] Apr´a E, Stefanovich E V, Dovesi R and Roetti C 1991 Chem. Phys. Lett. 186 329 [32] Guenard P, Renaud G and Villette B 1996 Physica B 221 205
[33] Duffy D M, Harding J H, and Stoneham A M (appendix by J R Willis) 1993 Phil. Mag. A 67 865 [34] Kaden C, Ruggerone P, Toennies J P, Zhang G and Benedek G 1992 Phys. Rev. B 46 13 509 [35] Caus´a M, Kotomin E A, Pisani C and Roetti C 1987 J. Phys. C: Solid State Phys. 20 4391 [36] Grimes R W, Catlow C R A and Stoneham A M 1989 J. Phys.: Condens. Matter 1 7367 [37] Stoneham A M and Ramos M M D 1993 J. Solid State Chem. 106 2
[38] Stoneham A M and Harding J H 1999 Ceramics into the 2000s Part C: Proc. 1998 World Ceramic Congr.,
CIMTEC
[39] Stoneham A M and Sangster M J L 1980 Phil. Mag. B 43 609
[40] Sangster M J L, Stoneham A M and Tasker P W 1981 Phil. Mag. B 44 603
[41] Moore G E 1971 Atomic Energy Levels (Ser. Natl Bur. Stand. US 35) vol 1 (Washington, DC: US Government Printing Office)
[42] Duffy D M, Harding J H and Stoneham A M 1995 J. Appl. Phys. 76 2791 [43] He J-W and Møller P J 1986 Chem. Phys. Lett. 129 13