Rapamycin-treated human endothelial cells
preferentially activate allogeneic regulatory T
cells
Chen Wang, … , George Tellides, Jordan S. Pober
J Clin Invest.
2013;123(4):1677-1693. https://doi.org/10.1172/JCI66204.
Human graft endothelial cells (ECs) can act as antigen-presenting cells to initiate allograft
rejection by host memory T cells. Rapamycin, an mTOR inhibitor used clinically to suppress
T cell responses, also acts on DCs, rendering them tolerogenic. Here, we report the effects
of rapamycin on EC alloimmunogenicity. Compared with mock-treated cells,
rapamycin-pretreated human ECs (rapa-ECs) stimulated less proliferation and cytokine secretion from
allogeneic CD4
+memory cells, an effect mimicked by shRNA knockdown of mTOR or
raptor in ECs. The effects of rapamycin persisted for several days and were linked to
upregulation of the inhibitory molecules PD-L1 and PD-L2 on ECs. Additionally,
rapa-ECs produced lower levels of the inflammatory cytokine IL-6. CD4
+memory cells activated
by allogeneic rapa-ECs became hyporesponsive to restimulation in an alloantigen-specific
manner and contained higher percentages of suppressive CD4
+CD25
hiCD127
loFoxP3
+cells that did not produce effector cytokines. In a human-mouse chimeric model of allograft
rejection, rapamycin pretreatment of human arterial allografts increased graft EC expression
of PD-L1 and PD-L2 and reduced subsequent infiltration of allogeneic effector T cells into
the artery intima and intimal expansion. Preoperative conditioning of allograft ECs with
rapamycin could potentially reduce immune-mediated rejection.
Research Article
Immunology
Find the latest version:
Rapamycin-treated human endothelial cells
preferentially activate allogeneic
regulatory T cells
Chen Wang,1 Tai Yi,1 Lingfeng Qin,2 Roberto A. Maldonado,3 Ulrich H. von Andrian,3
Sanjay Kulkarni,2 George Tellides,2 and Jordan S. Pober1
1Department of Immunobiology and 2Department of Surgery, Yale University School of Medicine, New Haven, Connecticut, USA. 3Immune Disease Institute, Harvard Medical School, Boston, Massachusetts, USA.
Human graft endothelial cells (ECs) can act as antigen-presenting cells to initiate allograft rejection by host
memory T cells. Rapamycin, an mTOR inhibitor used clinically to suppress T cell responses, also acts on DCs,
rendering them tolerogenic. Here, we report the effects of rapamycin on EC alloimmunogenicity. Compared
with mock-treated cells, rapamycin-pretreated human ECs (rapa-ECs) stimulated less proliferation and cytokine
secretion from allogeneic CD4
+memory cells, an effect mimicked by shRNA knockdown of mTOR or raptor
in ECs. The effects of rapamycin persisted for several days and were linked to upregulation of the inhibitory
molecules PD-L1 and PD-L2 on rapa-ECs. Additionally, rapa-ECs produced lower levels of the inflammatory
cytokine IL-6. CD4
+memory cells activated by allogeneic rapa-ECs became hyporesponsive to restimulation in
an alloantigen-specific manner and contained higher percentages of suppressive CD4
+CD25
hiCD127
loFoxP3
+cells that did not produce effector cytokines. In a human-mouse chimeric model of allograft rejection,
rapamy-cin pretreatment of human arterial allografts increased graft EC expression of PD-L1 and PD-L2 and reduced
subsequent infiltration of allogeneic effector T cells into the artery intima and intimal expansion. Preoperative
conditioning of allograft ECs with rapamycin could potentially reduce immune-mediated rejection.
Introduction
Immune-mediated rejection represents a significant barrier to the success of solid organ transplantation. Host alloreactive CD4+
T cells that cross-react to recognize non-self (allogeneic) MHC molecules expressed on donor-derived APCs carried along in the graft are critical mediators of rejection (1). APC function requires expression of MHC-peptide complexes (the antigen or signal 1), expression of antigen-independent costimulators (signal 2), and, often, production of activating cytokines (signal 3). Human vascu-lar ECs in situ basally express MHC class I and class II molecules (2, 3); costimulatory molecules such as LFA-3 (CD58), ICOS ligand (CD275), OX40 ligand (CD252), 41BB ligand (CD137L), CD40, and GITR ligand (4); and cytokines such as IL-1α, IL-6, IL-15, and IL-18 (5–8) that can contribute to T cell activation and differen-tiation. This panoply of molecular signals enables human ECs to function as APCs and activate allogeneic memory, but not naive, CD4+ T cells to proliferate, secrete effector cytokines, and reject
human allografts in immunodeficient mouse hosts (4, 9–11). Human ECs can also deliver inhibitory signals via expression of PD-L1 (also known as B7-H1 or CD274) and PD-L2 (also known as B7-DC or CD273), which bind to PD-1 (CD279) on T cells (12, 13). The balance of positive and negative signals on ECs affects the net outcome of memory T cell responses.
Human peripheral blood CD4+ T cells in adults are
approxi-mately equally divided between naive and memory cells. Memory T cells are important mediators of rejection in clinical transplan-tation (14, 15), and a significant percentage of circulating memory cells in human transplant recipients, likely generated during prior microbial infections, cross-react to recognize donor graft
allo-antigens (16). Furthermore, the frequency of donor-specific mem-ory T cells correlates with the likelihood of rejection (17). Because alloreactive memory T cells can be activated by ECs, it is believed that allograft ECs are sufficient to trigger rejection by directly pre-senting alloantigen to and activating host alloreactive memory T cells (18) in the absence of graft-derived professional APCs. This is in marked contrast to the phenotype in rats, in which the pas-senger leukocytes, defined as professional APCs present within a solid organ allograft, have been shown to be necessary to initiate rejection (19). Despite this pathogenic role, there are no clinical therapies aimed at reducing EC alloimmunogenicity.
CD4+ effector T cells are a heterogeneous population that can
be divided into multiple subsets defined by their cytokine pro-files. Among these subsets are Th1 cells, which express the mas-ter transcription factor T-bet and secrete IFN-γ; Th2 cells, which express the master transcription factor GATA3 and secrete IL-4, IL-5, and IL-13; and Th17 cells, which express the master tran-scription factor RORγT and secrete IL-17A and IL-17F (20, 21). All three subsets are capable of causing allograft destruction (22–25). A fourth subset of CD4+ T cells, called Tregs, can suppress immune
responses and comprises at least two major populations: natural and inducible Tregs (26). Natural Tregs develop in the thymus, recognize self antigens, control autoimmunity, and express high levels of CD25 and the transcription factor FoxP3. Although activated human T effector cells also express CD25 and FoxP3, expression in natural Tregs is generally higher and more sustained, a difference attributable to reduced methylation of DNA in a crit-ical transcriptional control region of the FOXP3 gene known as the Treg-specific demethylated region (TSDR) (27). Inducible Tregs (iTregs) form in the periphery and develop or convert from conventional CD25-negative CD4+ T cells that may also have the
potential to become effector T cells (26). Such iTregs, which may
Conflict of interest: The authors have declared that no conflict of interest exists.
research article
or may not stably express FoxP3, are specific for non-self antigens (including alloantigens) and likely restrain immune responses to environmental antigens (28–30). Both natural and inducible Tregs prevent rejection and induce allograft tolerance in several models of transplantation (31–33).
The milieu in which a CD4+ T cell becomes activated largely
determines the acquisition of specific effector or regulatory func-tions (reviewed in refs. 20, 21). In the case of Tregs, development of some types of iTregs requires TGF-β and the absence of IL-6 (26), whereas other types are induced by IL-10 (34). In addition to cytokines, formation of iTregs can also be driven by cell surface signals provided by APCs (35), such as PD-L1, which promotes iTreg differentiation even in the absence of TGF-β (36). Although these observations are based on studies of naive T cell commit-ment, human memory CD4+ T cells display plasticity and are
capa-ble of acquiring regulatory activity following antigenic restimula-tion (5, 37–39). The relarestimula-tionship between human ECs and Tregs is incompletely understood, and it is unknown whether human ECs can be induced to express molecules that favor activation or dif-ferentiation of tolerance-promoting Tregs instead of pathogenic inflammatory effectors T cells.
Since its approval in 1999, rapamycin has been used as an immu-nosuppressant in human transplant recipients (40). Rapamycin acts by forming a complex with a cytosolic prolyl isomerase known as FK-BP12, and the drug-protein complex acts as an inhibitor of mTOR, a 289-kDa serine/threonine protein kinase that inte-grates environmental cues to regulate cell growth and division (41). More specifically, rapamycin targets mTOR as part of an mTOR signaling complex (called mTORC1) that also contains a protein called raptor that is critical for its function. Prolonged rapamycin treatment has been shown to inhibit another mTOR-containing signaling complex, known as mTORC2 (42), that lacks raptor but instead contains a protein called rictor that is critical for its function. In CD4+ T cells, mTOR is activated in response to
IL-2 or other growth-promoting cytokines, and it is believed that rapamycin achieves immune suppression by inhibiting growth fac-tor–driven lymphocyte proliferation. In addition to its effects on T cells, recent data suggest that rapamycin may also target mTOR in APCs (43), altering their function. Human and rodent bone marrow–derived DCs treated with rapamycin display decreased expression of MHC and costimulatory molecules, an effect resis-tant to maturation signals. These rapamycin-treated DCs poorly stimulate allogeneic T cells, induce alloantigen-specific anergy, and selectively expand FoxP3+ regulatory T cells (44–46). In rodent
models of allograft rejection, infusion of rapamycin-treated, allo-antigen-pulsed DCs into the host prior to transplant results in prolonged graft survival, associated with increased frequencies of FoxP3+ cells in the spleen and infiltrating into the graft (45, 47).
Based on these data, we hypothesized that rapamycin may also have tolerance-promoting effects on human ECs.
Here we show that pretreatment of cultured human umbilical vein ECs with rapamycin reduces proliferation and cytokine secre-tion of cocultured allogeneic memory CD4+ T cells, a phenotypic
change that can be replicated by shRNA knockdown of mTOR or raptor in the ECs. Rapamycin treatment or knockdown of mTOR or raptor affected both costimulator molecule expression and cytokine production, but Ab-blocking experiments linked reduced allostimulatory capacity to the upregulation of the inhibitory mol-ecules PD-L1 and PD-L2 on rapa-ECs. T cells activated by rapa-ECs became hyporesponsive to restimulation, in an alloantigen-specific
manner, and contained an increased ratio of CD4+CD25hiCD127lo
FoxP3+ to CD4+CD25+FoxP3– T cells. These FoxP3+ cells were
func-tionally suppressive, displayed demethylation of the TSDR within the FOXP3 gene, and did not secrete effector cytokines. Finally, using a humanized mouse model of artery allograft rejection, we demonstrate that treatment of human vessels with rapamycin prior to transplant reduced both the infiltration of allogeneic T cells into the vessel intima and intimal expansion, an effect that coincided with increased expression of PD-L1 and PD-L2 on treated graft ECs and was diminished by PD-1 blockade. These data show that rapamycin reduces alloimmunogenicity and confers tolerance-pro-moting effects on ECs in vitro and in vivo.
Results
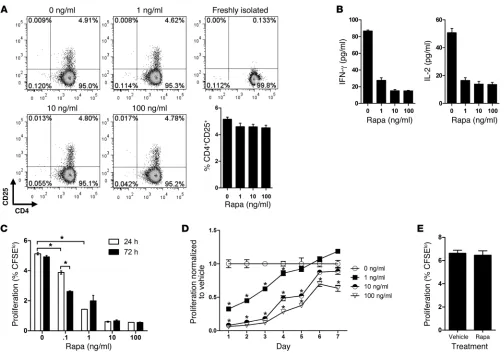
Rapamycin-pretreated ECs poorly stimulate allogeneic memory CD4+ T cells. To study the effects of rapamycin on EC alloimmuno-genicity, we treated MHC class II+ ECs with varying doses of
rapamycin (up to 100 ng/ml) for up to 72 hours, then washed away drug and cocultured pretreated ECs with purified alloge-neic resting memory CD4+ T cells. There was no difference in
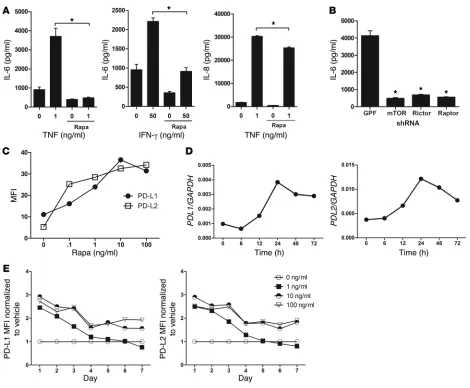
the ability of T cells to recognize allogeneic MHC class II mol-ecules on control ECs and rapa-ECs, as evidenced by a similar degree of induction of CD25, an activation marker expressed in response to TCR signaling, at 72 hours (Figure 1A). However, T cells stimulated by rapa-ECs secreted less IL-2 and IFN-γ at 24 hours (Figure 1B) and were poorly proliferative compared with T cells cocultured with control EC, an effect dependent on both drug dose and treatment duration (Figure 1C). To determine the longevity of this effect, ECs were treated with rapamycin for 72 hours, washed, cultured in the absence of drug for up to 7 days, and then cocultured with allogeneic memory CD4+ T cells.
We found that at low doses, rapa-ECs were poorly stimulatory compared with untreated ECs even after 4 days following drug washout, while at high doses, the effects of rapamycin treat-ment persisted for as long as 7 days (Figure 1D). To determine whether changes in cell surface or soluble factors were respon-sible for the decreased allostimulatory capacity of rapa-ECs, we cultured control ECs or rapa-ECs across a Transwell from cocultures of untreated autologous ECs and allogeneic memory CD4+ T cells. T cell proliferation, in the lower chamber, was
unaffected when either control ECs or rapa-ECs (Figure 1E) were plated in the upper chamber, suggesting that rapamycin likely reduces EC alloimmunogenicity by altering expression of cell surface molecules.
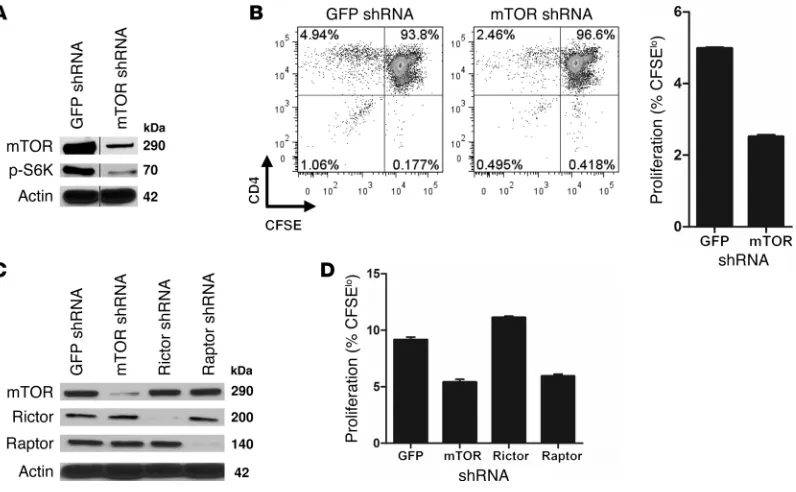
Because rapamycin is known to directly inhibit effector CD4+
T cells, it is possible that the observed inhibition of T cell allore-sponses resulted from carryover of rapamycin from the pretreat-ment period into the EC–T cell cocultures, where it directly acts on T cells. To address this possibility and to confirm that the effect of rapamycin was mediated by inhibition of mTOR in the ECs, we used shRNA to knock down mTOR in the ECs (Figure 2A). Since carry-over of shRNA is extremely unlikely, this approach allowed us to dis-tinguish the effects of specifically inhibiting mTOR in ECs. Similar to rapa-ECs, mTOR-knockdown ECs poorly stimulated prolifera-tion of allogeneic memory CD4+ T cells (Figure 2B), suggesting that
T cells proliferated less when stimulated by either mTOR- or raptor-knockdown ECs, but not when rictor was knocked down (Figure 2D). Interestingly, knockdown of rictor in ECs slightly increased proliferation of cocultured T cells. We conclude that disruption of mTORC1 signaling reduces EC alloimmunogenicity.
Allogeneic memory CD4+ T cells stimulated by rapa-ECs are hyporespon-sive to restimulation. To distinguish between immunologic ignorance and induction of tolerance, we investigated whether T cells were functionally altered by coculture with rapa-ECs by testing their ability to respond to restimulation. CD4+ memory T cells first
cocultured with allogeneic rapa-ECs, for 72 hours, proliferated less when restimulated by untreated ECs from either the same donor as used in the primary stimulation or third-party donors (Figure 3A); however, this hyporesponsiveness was more profound when matched ECs were used as restimulators. This suggests that T cells
cocultured with rapa-ECs become hyporesponsive in at least a partly alloantigen-specific manner. It is important to note that after 72 hours of primary stimulation, the percentages of T cells activated by control and rapa-EC (as judged by CD25+) were equal (Figure 1A).
[image:4.585.42.546.80.436.2]Thus, the decreased proliferation of T cells cocultured with rapa-ECs upon restimulation does not reflect differences in the degree of expansion during primary stimulation. The lesser reduced response to third-party ECs could imply some contribution of MHC-inde-pendent hyporesponsiveness, but since the ECs were not HLA typed, we cannot rule out the possibility of shared MHC alleles. In either case, the functional alteration was highly dependent on TCR recog-nition of MHC class II molecules on rapa-ECs, as addition of block-ing antibodies to HLA-DR in primary EC–T cell cocultures largely, but not completely, prevented the development of hyporesponsive-ness by rapa-EC–primed T cells during restimulation (Figure 3B).
Figure 1
Rapa-ECs poorly stimulate allogeneic memory CD4+ T cells. (A) CD4+ memory cells were cocultured with allogeneic control ECs or rapa-ECs for
72 hours and analyzed by FACS. Freshly isolated T cells were stained and analyzed immediately following isolation. Representative FACS plots are gated on total CD4+ cells. Similar results were seen in 3 independent experiments. (B) Production of IL-2 and IFN-γ by CD4+ memory cells
cocultured with allogeneic control ECs or rapa-ECs at 24 hours was assessed by ELISA. Similar results were seen in 3 independent experiments. (C) ECs were treated with the indicated doses of rapamycin (rapa) for 24 or 72 hours and cocultured with CFSE-labeled allogeneic CD4+ memory
T cells. Proliferation of T cells was assessed after 7 days via CFSE dilution. (D) ECs were treated with the indicated doses of rapamycin for 72
hours, washed, and maintained up to 7 days (in the absence of drug), then cocultured with CFSE-labeled allogeneic CD4+ memory T cells. T cell
proliferation was assessed after 7 days and normalized to vehicle control (0 ng/ml rapa). (E) Control ECs or rapa-ECs were cultured in a Transwell
across from cocultures of untreated ECs and CFSE-labeled allogeneic CD4+ memory T cells. Proliferation of CFSE-labeled T cells was assessed
research article
This incomplete blockade also may be interpreted as an MHC-inde-pendent contribution, but even though HLA-DR is the dominant determinant of direct CD4+ T cell allorecognition, we cannot rule
out small contributions from recognition of HLA-DP and HLA-DQ that at not blocked by our antibody.
Rapa-ECs induce an increased percentage of CD4+CD25hiCD127lo FoxP3+ cells. The observation that allogeneic CD4+ T cells
stimu-lated by rapa-ECs become hyporesponsive to restimulation could
be due to clonal anergy, to clonal deletion, or to activation or induction of Tregs. We did not detect any differences in cell death (assessed by Annexin V or propidium iodide) in T cells stimulated by control ECs or rapa-ECs (data not shown), arguing against clonal deletion. To address the possibilities of anergy or Treg induction, we cocultured memory CD4 T+ cells with allogeneic
[image:5.585.91.489.83.329.2]control ECs or rapa-ECs for 72 hours and FACS sorted activated T cells on the basis of CD4+CD25+ expression. Using qRT-PCR, we
Figure 2
mTOR and raptor knockdown decreases EC alloimmunogenicity. (A) ECs were transduced with shRNA targeting GFP (control) or mTOR as
described in Methods. Knockdown was confirmed by Western blot analysis (lanes were run on the same gel, but were not contiguous; breaks are indicated by black vertical line). (B) GFP or mTOR knockdown ECs were cocultured with CFSE-labeled allogeneic CD4+ memory T cells.
Proliferation was assessed after 7 days via CFSE dilution. Representative FACS plots are gated on total CD4+ cells. Similar results were seen in
3 independent experiments. (C) ECs were transduced with shRNA targeting GFP (control), mTOR, raptor, or rictor. Knockdown was confirmed by
Western blot analysis. (D) Knockdown ECs from C were cocultured with CFSE-labeled allogeneic CD4+ memory T cells. T cell proliferation was
assessed after 7 days. Similar results were seen in 2 independent experiments. Mean ± SEM is shown for representative experiments in B and D.
Figure 3
Allogeneic CD4+ memory cells stimulated by rapa-ECs become hyporesponsive to restimulation. (A) Allogeneic CD4+ memory T cells stimulated
by control ECs or rapa-ECs for 72 hours (at which time the percentage of activated CD25+ T cells in each group is equal) were rested, CFSE
labeled, and restimulated by fresh ECs from the same donor as used in primary stimulation or from third-party donors. Proliferation of restimulated T cells was assessed by CFSE dilution; percent proliferation (%CFSElo) is shown on left, and proliferation normalized to vehicle control is shown
on right. (B) Restimulation experiment, as described above, except 30 μg/ml of neutralizing HLA-DR or isotype control antibodies was added to primary cocultures, and T cells were only restimulated by matched ECs. Mean ± SEM of triplicate samples is shown from 3 (A, normalized data)
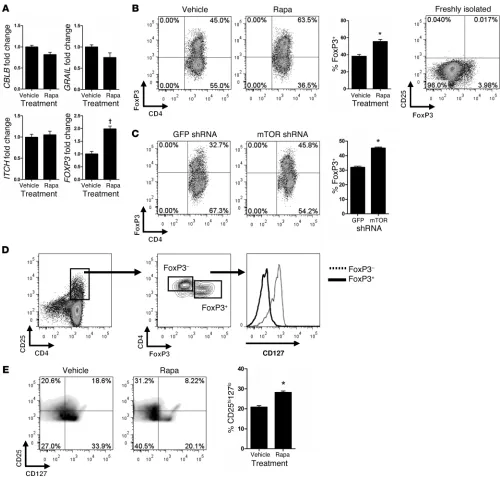
[image:5.585.95.481.547.658.2]found that T cells activated by rapa-ECs did not express increased transcript levels of the anergy-associated genes GRAIL, ITCH, and
CBLB but did contain significantly higher mRNA levels of the Treg master transcription factor FOXP3 (Figure 4A). We were intrigued by this result, given the observation that rapamycin-treated DCs
favor activation and expansion of Tregs. To determine whether rapa-ECs have a similar capability, we performed intracellular FACS analysis on allogeneic memory CD4+CD25– T cells
cocul-tured with rapa-ECs and found that activated (now CD25+)
T cells, which constituted about 5%–10% of the total CD4+
popu-Figure 4
Rapa-ECs increase the percentage of CD4+CD25hiCD127loFoxP3+ cells among CD25+ activated T cells. (A) Activated T cells cocultured with
control ECs or rapa-ECs were FACS sorted on the basis of CD25+ expression, and used for qRT-PCR analysis. All transcripts were normalized
to CD3, and expression was then normalized to vehicle control. (B) Allogeneic CD4+ memory T cells were cocultured with control ECs or
rapa-ECs for 7 days and analyzed by intracellular FACS. Freshly isolated T cells were stained and analyzed immediately following isolation. Repre-sentative FACS plots are gated on CD4+CD25+ T cells, except for freshly isolated sample, which is gated on total CD4+ cells. (C) CD4+ memory
T cells cocultured with allogeneic GFP or mTOR knockdown ECs were analyzed by intracellular FACS. Representative FACS plots are gated on CD4+CD25+ T cells. (D) CD127 expression was assessed on FoxP3+ and FoxP3– populations in CD25+ T cells activated by rapa-ECs. Similar
results were seen in 2 additional independent experiments. (E) Cocultures were performed as described in B. Representative plots are gated on
[image:6.585.41.541.81.563.2]research article
lation in various experiments, contained a greater proportion of FoxP3+ T cells compared with cocultures with control ECs (Figure
4B). These CD25+FoxP3+ T cells were CD127 low/negative
(Fig-ure 4D). Overall, CD25+ T cells activated by rapa-ECs contained
a greater percentage of CD25hiCD127lo T cells (Figure 4E)
com-pared with T cells activated by control ECs; in both groups these CD25hi CD127lo cells were greater than 95% FoxP3+ (data not
[image:7.585.35.541.83.539.2]shown). It is important to note that in our system, the memory
Figure 5
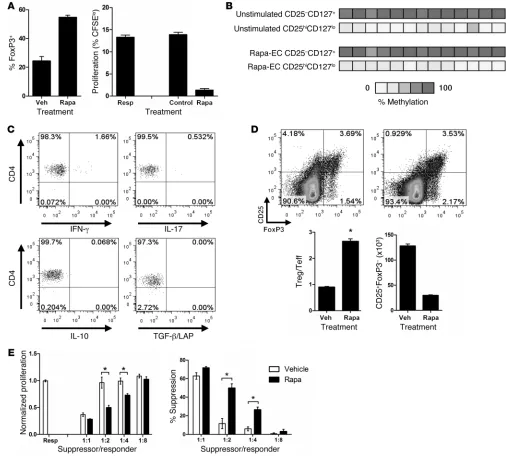
CD4+CD25hiCD127loFoxP3+ cells activated by rapa-ECs are functional Tregs. (A) Left: Percentage of FoxP3+ cells in CD25+ pool activated by
anti-CD3–loaded CD32+ control ECs and rapa-ECs. Right: FACS-sorted CD25hi T cells activated by anti-CD3–loaded CD32+ rapa-ECs were
added to fresh cocultures of untreated CD32-transduced ECs and CFSE- labeled responder CD4+ memory T cells at 1:1 ratios. In control wells,
equal numbers of freshly isolated T cells were added in place of rapa-EC–primed T cells. Similar results were seen in 3 independent experiments. (B) CD4+CD25hiCD127lo T cells activated by rapa-ECs were FACS sorted and used for DNA methylation analysis. Data are representative of 2
independent experiments. (C) CD4+ memory T cells cocultured with allogeneic rapa-ECs for 6 days were stimulated with PMA/ionomycin in the
presence of brefeldin A and analyzed by intracellular cytokine staining. Representative FACS plots are gated on CD4+CD25hiCD127lo T cells.
Sim-ilar results were seen in 2 independent experiments. (D) Representative FACS plots (gated on CD4+ T cells) of CD4+ memory T cells cocultured
with control ECs or rapa-ECs. Treg/effector T (Teff) ratio calculated as (%CD25+FoxP3+)/(%CD25+FoxP3–). Absolute numbers of CD25+FoxP3–
cells calculated by multiplying the percentage of CD25+FoxP3– cells in the CD4+ gate by the number of CD4+ T cells used in each coculture. (E)
FACS-sorted total CD25+ T cells, activated as in A, were titered at varying ratios into fresh cocultures of untreated CD32-transduced ECs and
T cells used to initiate the cocultures were depleted of preexisting CD25+ cells (reduced from about 2% of the memory CD4+
popu-lation to less than 0.02%; data not shown), a pool that contains the natural Tregs, prior to coculture. In other words, the CD25+
FoxP3+ cells induced by coculture with rapa-ECs arise from
previ-ously CD25– cells and thus could qualify as being iTregs. However,
about 4%–6% of human CD25– memory CD4+ T cells in
periph-eral blood expressed FoxP3 (Figure 4B), and we cannot determine whether CD25hiFoxP3+ T cells arise from this population, a
previ-ously FoxP3– population, or both. Last, allogeneic memory T cells
activated by mTOR-knockdown ECs similarly contained a greater percentage of FoxP3+ cells (Figure 4C), suggesting that our
obser-vations with rapa-ECs were not due to carryover of rapamycin act-ing directly on T cells.
In humans, FoxP3 is not an exclusive marker of Tregs, as it is also transiently expressed on activated conventional CD4+
T cells (48, 49). To determine whether CD4+CD25hiCD127lo
FoxP3+ cells induced by rapa-ECs were Tregs, we tested their
functional characteristics using in vitro suppression assays. Because the frequency of alloreactive T cells capable of respond-ing to an EC donor is low (1%), we were limited in the ability to sort sufficient numbers of cells for use in suppression assays. To bypass this obstacle, we used a system previously developed in our laboratory (50) wherein ECs are retrovirally transduced to express the extracellular domain of CD32, an FcγR that binds and displays IgG antibodies, and then preloaded with the anti-CD3 antibody OKT3. These OKT3-loaded ECs are capable of polyclonally activating 100% of T cells. Using this system, we confirmed that T cells activated by anti-CD3–loaded CD32+
rapa-ECs contained higher percentages of CD4+CD25hi
CD-127loFoxP3+ cells (Figure 5A). These cells were then sorted on
the basis of high CD25 expression and added to cocultures of freshly isolated CFSE-labeled T cells and OKT3-loaded ECs. Addition of these cells potently suppressed proliferation of the CFSE-labeled responder T cells (Figure 5A). Further-more, analysis of bisulfite-converted DNA revealed that CD4+
CD25hiCD127lo T cells activated by rapa-ECs displayed strong
demethylation of the TSDR within the FOXP3 gene (Figure 5B). Together these data suggest that FoxP3+ cells activated
by rapa-ECs are indeed Tregs. Analysis of the cytokines pro-duced by these cells by intracellular cytokine staining revealed
the absence of effector cytokines such as IFN-γ and IL-17, as well as the absence of inhibitory cytokines TGF-β and IL-10 (Figure 5C). It is important to note, how-ever, that such Tregs were also found in cocultures with control ECs; they sim-ply represent a significantly smaller per-centage of the total activated (CD25+)
T cells compared with their frequency after coculture with rapa-ECs.
These experiments establish that the population of CD25+ T cells activated by
rapa-ECs contains a greater percentage of Tregs. This shift in the balance could arise from an absolute expansion of Tregs, a reduction in the expansion of T effector cells, or both. FACS analysis suggest that the change in ratio largely resulted from a decrease in the number of effector T cells (i.e., FoxP3–) present within the CD25+ population (Figure 5D).
To determine whether this change in the ratio of Tregs to T effec-tor cells among activated T cells was functionally significant, we repeated our suppression assays using the total CD25+ population
rather than focusing only on the CD25hi subset. The net effect of
this altered balance of effector versus Tregs is that the total CD25+
population activated by rapa-ECs was more suppressive than the equivalent population activated by control ECs (Figure 5E).
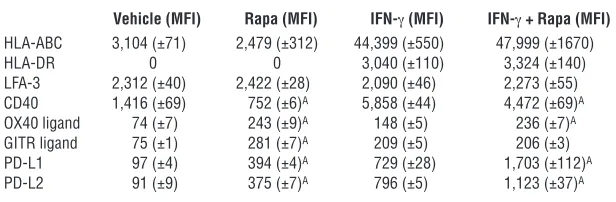
Rapamycin alters EC expression of costimulatory molecules and secre-tion of IL-6. To identify specific mechanisms by which rapamycin alters EC alloimmunogenicity, we investigated how the drug affects endothelial expression of immunologically relevant mole-cules under basal and IFN-γ–stimulated conditions (Table 1). As expected, MHC class I was expressed under basal conditions and upregulated by IFN-γ, while MHC class II was only detected after IFN-γ treatment. Rapamycin did not alter expression of either molecule, consistent with our observation that equal frequen-cies of T cells were initially activated by recognition of allogeneic MHC class II molecules on control ECs and rapa-ECs (Figure 1A). CD40 was downregulated on rapa-ECs, while expression of the costimulatory molecules OX40 ligand and GITR ligand was increased under both basal and IFN-γ–stimulated conditions (OX40 ligand only). Furthermore, compared with control ECs, rapa-ECs expressed significantly higher levels of the inhibitory molecules PD-L1 and PD-L2 under basal conditions and also fol-lowing optimal IFN-γ treatment. In addition to cell surface mole-cules, we also investigated soluble mediators released by ECs that may influence the response of allogeneic T cells. Interestingly, we found that rapamycin profoundly inhibited EC secretion of the T cell–activating cytokine IL-6, in response to IFN-γ or to TNF (Figure 6A). In contrast, rapamycin only mildly reduced secretion of IL-8 in response to TNF (Figure 6A), suggesting that the reduc-tion in IL-6 was not due to decreased viability or a generalized block of protein secretion in rapa-ECs.
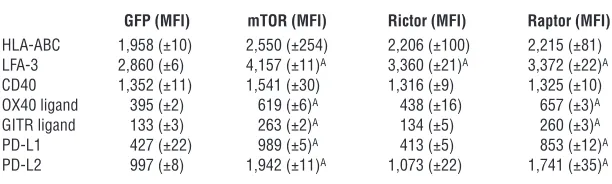
[image:8.585.52.360.113.214.2]To determine which component of mTOR signaling mediated these changes, we knocked down mTOR, raptor, and rictor in ECs. Similar to rapamycin treatment, mTOR knockdown increased expression of OX40 ligand, GITR ligand, PD-L1, and PD-L2, an effect that was phenocopied by raptor, but not rictor, knockdown (Table 2). mTOR knockdown also decreased EC secretion of IL-6 in response to TNF, and interestingly, both raptor and rictor
Table 1
Flow cytometric analysis of EC surface costimulatory molecules on control and Rapa-ECs
Vehicle (MFI) Rapa (MFI) IFN-γ (MFI) IFN-γ + Rapa (MFI)
HLA-ABC 3,104 (±71) 2,479 (±312) 44,399 (±550) 47,999 (±1670)
HLA-DR 0 0 3,040 (±110) 3,324 (±140)
LFA-3 2,312 (±40) 2,422 (±28) 2,090 (±46) 2,273 (±55) CD40 1,416 (±69) 752 (±6)A 5,858 (±44) 4,472 (±69)A
OX40 ligand 74 (±7) 243 (±9)A 148 (±5) 236 (±7)A
GITR ligand 75 (±1) 281 (±7)A 209 (±5) 206 (±3)
PD-L1 97 (±4) 394 (±4)A 729 (±28) 1,703 (±112)A
PD-L2 91 (±9) 375 (±7)A 796 (±5) 1,123 (±37)A
research article
knockdown reproduced this effect (Figure 6B). These data suggest that disruption of both mTOR signaling complexes is involved in mediating the effects of rapamycin.
PD-L1 and PD-L2 are induced by rapamycin and contribute to the decreased alloimmunogenicity of rapa-ECs. Of the cell surface changes induced by rapamycin (Table 1), we chose to focus on changes in PD-L1 and PD-L2, as these molecules are known to send inhibi-tory signals to T cells. Using flow cytometric analysis, we found that rapamycin increased EC cell surface expression of PD-L1 and PD-L2 in a dose-dependent manner with maximal induc-tion occurring at 72 hours with 10 ng/ml drug (Figure 6C). Using qRT-PCR, we found that PDL1 and PDL2 mRNA transcripts were upregulated by rapamycin treatment, peaking at 24 hours but still remaining elevated at 72 hours (Figure 6D). To study the longevity
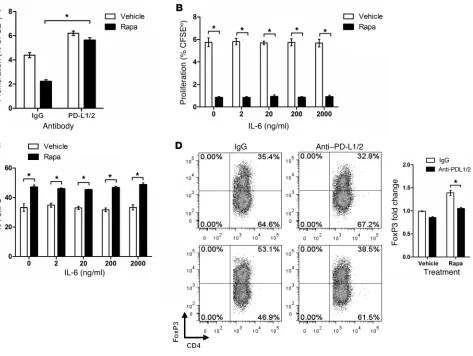
of this effect, we cultured rapamycin-treated ECs in the absence of drug. With lower treatment doses, PD-L1 and PD-L2 remained ele-vated 4 days after withdrawal of drug, while at higher doses these molecules were still elevated at 7 days (Figure 6E). Furthermore, shRNA knockdown of mTOR increased EC surface expression of PD-L1 and PD-L2 (Table 2), implying that the effect of rapamy-cin was due to specific inhibition of mTOR signaling. Last, addi-tion of PD-L1– and PD-L2–blocking antibodies into cocultures restored the ability of allogeneic memory CD4+ T cells to
prolifer-ate in response to rapa-ECs (Figure 7A). Because rapa-ECs secreted reduced amounts of IL-6 (Figure 6A), we investigated whether this deficit may also contribute to their weak allostimulatory abil-ity. However, in contrast to PD-L1/PD-L2 blockade, addition of recombinant IL-6 to replace the missing cytokine did not
com-Figure 6
Rapamycin decreases EC secretion of IL-6 and increases expression of PD-L1 and PD-L2. (A) ECs were treated with vehicle or rapamycin for
2 hours, at which time the indicated doses of TNF-α or IFN-γ were added to cells. After 24 hours, supernatants were assessed for IL-6 or IL-8 by ELISA. (B) GFP-, mTOR-, rictor-, and raptor-knockdown ECs were treated with 1 ng/ml TNF. After 24 hours, supernatants were assessed for IL-6.
(C) ECs treated with the indicated dose of rapamycin for 72 hours were analyzed by FACS. Similar results were seen in 3 independent
experi-ments. (D) mRNA expression in ECs treated with rapamycin for the indicated times. Transcript levels were normalized to GAPDH. Similar results were seen in 3 independent experiments. (E) ECs were treated with the indicated dose of rapamycin for 72 hours, washed, and maintained for up
[image:9.585.53.523.79.467.2]pensate for the rapamycin effect, as allogeneic T cells still poorly proliferated in response to rapa-ECs (Figure 7B). Collectively, these data suggest that rapamycin reduces EC alloimmunogenicity, at least in part, by increasing cell surface expression of PD-L1 and PD-L2. The reduction in IL-6 does not appear to have been func-tionally significant in these in vitro assays.
It has been previously shown that PD-L1 and PD-L2 on APCs mediate peripheral tolerance and can enhance generation of Tregs (51). Additionally, the presence of IL-6 is known to antag-onize the function and formation of Tregs (52). Since rapa-ECs have elevated expression of PD-L1 and PD-L2 and poorly secrete IL-6, we explored whether either of these effects contributed to the increased percentages of Tregs activated by rapa-ECs. We found that addition of recombinant IL-6 into cocultures had little effect on the percentage of FoxP3+ cells (Figure 7C). In contrast,
serolog-ical blockade of PD-L1 and PD-L2 partially reversed the increased percentage of FoxP3+ cells among activated T cells in rapa-EC
ver-sus control EC cocultures (Figure 7D). Thus, we conclude that the elevated expression of PD-L1 and PD-L2 on rapa-ECs significantly contributed to the preferential activation of CD25+FoxP3+ cells.
Rapamycin pretreatment reduces T cell–mediated injury in human artery allografts. To investigate whether pretreatment of allograft ECs with rapamycin can provide protective effects in vivo, we used a human-mouse chimeric model of artery allograft rejection recently developed by our laboratory (53). In this model, short seg-ments of human artery are implanted into the infrarenal aortae of immunodeficient SCID/beige mice and left to heal for 30 days, at which point the graft is harvested and re-transplanted into a sec-ond SCID/beige mouse previously receiving adoptive transferred of human PBMCs allogeneic to the artery graft. In these mice, adoptive transfer of human PBMCs yields stable engraftment of human CD3+ T cells capable of mediating graft rejection but does
not result in engraftment of human myeloid (monocytes, macro-phages, DCs, neutrophils) or NK cells. This re-transplant design was selected for three reasons. First, it assures that only the artery is exposed to drug, avoiding confounding effects of rapamycin on T cells. Second, by allowing the arteries to quiesce in the primary mouse host, the model eliminates variability (injury status, time from harvest/spent on ice) in the state of human arteries at the time the arteries first encounter allogeneic T cells, which could influence the subsequent T cell alloresponse. And third, because
we can perform transplant into the sec-ond host at a time of our choosing, we can ensure that recipients will have optimal levels of circulating human T cells at trans-plant. This is a feature we cannot otherwise control, since circulating T cell numbers require 1–2 weeks to reach adequate levels but begin to cause pathological changes in the host after 4–5 weeks, and we do not know in advance when human artery seg-ments will become available. This design also provides a closer recapitulation of clin-ical transplantation, where allografts are transplanted into donors with circulating T cells in a “physiological” range.
To test whether rapamycin pretreatment has a protective role in vivo, we treated mice with healed artery grafts with vehi-cle or rapamycin for 72 hours, at which time grafts were harvested, perfused, and re-transplanted into PBMC-inoculated mice. Because these secondary recipients are not exposed to rapamycin, the system allows us to determine the effects of rapamycin on graft alloimmunogenicity, independent of any direct effects on the circulating human lymphocytes. Fur-thermore, at the time of re-transplant, the human artery graft consists of human luminal ECs, medial (and rare intimal) human smooth muscle cells, human adventitial fibroblasts and microves-sels, but no detectable human (passenger) leukocytes (54). In other words, human ECs are the only functional allogeneic APCs in the graft. Using this model, we found that rapamycin pretreatment of grafts reduced the total number of T cells infiltrating into the vessel intima (Figure 8A) and concomitantly reduced both the total vessel area and intima expansion (Figure 8B) at 10 days following re-transplant. At this early stage, there was no loss of lumen in either group, as the outward expansion of the control vessels compensated for intimal expansion (Figure 8B and ref. 55). To determine the durability of this protective effect, we extended our experiments to 3 weeks following re-transplantation. At this later time, we found that rapamycin-pretreated grafts still had reduced total vessel areas and intima sizes compared with control grafts (Figure 8C); however, the magnitude of this effect appeared to be diminished relative to earlier time points. Together, these data suggest that pretreatment of allografts with rapamycin can reduce subsequent T cell–mediated rejection, although the effect may wane with time.
Based on our observations that rapamycin reduces the alloimmu-nogenicity of cultured ECs via induction of PD-1 ligands, we exam-ined graft expression of these molecules via immunofluorescence. We found that rapamycin-pretreated grafts, harvested 10 days after re-transplant into PBMC-inoculated mice, had pronounced EC expression of both PD-L1 and PD-L2 (Figure 9A). These molecules were barely detectable on the ECs lining replicate control-treated grafts re-transplanted into paired mice that had been adoptively transferred with the same human PBMCs. To determine whether this induced expression of PD-L1 and PD-L2 could be contributing to the protective effects observed in rapamycin-pretreated grafts, we transplanted paired rapamycin-pretreated grafts into pairs of PBMC-inoculated mice and treated these animals with either a neu-tralizing F(ab′)2 fragment to PD-1 or an isotype-matched F(ab′)2 for
[image:10.585.53.359.124.216.2]14 days. (This anti–PD-1 F[ab′]2 fragment effectively reduced the
Table 2
Flow cytometric analysis of EC cell surface costimulatory molecules on control (GFP)–, mTOR-, rictor-, and raptor-knockdown ECs
GFP (MFI) mTOR (MFI) Rictor (MFI) Raptor (MFI)
HLA-ABC 1,958 (±10) 2,550 (±254) 2,206 (±100) 2,215 (±81) LFA-3 2,860 (±6) 4,157 (±11)A 3,360 (±21)A 3,372 (±22)A
CD40 1,352 (±11) 1,541 (±30) 1,316 (±9) 1,325 (±10) OX40 ligand 395 (±2) 619 (±6)A 438 (±16) 657 (±3)A
GITR ligand 133 (±3) 263 (±2)A 134 (±5) 260 (±3)A
PD-L1 427 (±22) 989 (±5)A 413 (±5) 853 (±12)A
PD-L2 997 (±8) 1,942 (±11)A 1,073 (±22) 1,741 (±35)A
research article
protective effect of rapamycin pretreatment of cultured ECs [data not shown].) We found that PD-1 blockade increased the vessel area and intima size of rapamycin-pretreated grafts (Figure 9B), as well as the number of infiltrating intimal T cells (Figure 9C), compared with animals treated with the isotype control. We conclude that PD-1 ligands contribute to the protection of rapamycin-pretreated grafts against T cell–mediated injury in vivo.
Discussion
We found that the mTOR inhibitor rapamycin modulates the alloimmunogenicity of human ECs. Specifically, rapa-ECs poorly stimulate alloreactive memory CD4+ T cells to proliferate and
secrete effector cytokines without affecting recognition of MHC class II molecules on the ECs (assessed by induction of CD25). Similar effects were observed with ECs in which mTOR had been knocked down by shRNA. The recognition of MHC class II
is significant, because T cells that had been cultured with rapa-ECs became hyporesponsive to restimulation by rapa-ECs from the same donor, an effect largely abrogated by blocking MHC class II recognition in the primary culture. Additionally, T cells activated by rapa-ECs contained an increased ratio of suppressive CD4+
CD25hiCD127loFoxP3+ Tregs to CD4+CD25+FoxP3– effector
T cells. Mechanistically, rapamycin treatment (or mTOR knock-down) altered endothelial expression of several costimulatory molecules, but not MHC molecules, and reduced EC secretion of the proinflammatory cytokine IL-6 in response to IFN-γ and TNF. The primary effect on the CD4+ T cell responses appears to
[image:11.585.54.527.82.438.2]be mediated via induction of PD-L1 and PD-L2 on rapa-ECs, an effect that appears to be much more substantial than the previ-ously described upregulation of these molecules in response to IFN-γ. In vivo, in a human-mouse chimeric model, pretreatment of human artery allografts with rapamycin led to upregulation of
Figure 7
Elevated expression of PD-L1 and PD-L2, but not hyposecretion of IL-6, contributes to altered alloimmunogenicity of rapa-ECs. (A) Control ECs
and rapa-ECs were cocultured with CFSE-labeled allogeneic CD4+ memory T cells in the presence of blocking antibodies to PD-L1 and PD-L2,
or isotype control. Proliferation of T cells was determined after 7 days via CFSE dilution. (B) Control ECs and rapa-ECs were cocultured with
CFSE-labeled allogeneic CD4+ memory T cells in the presence of indicated dose of recombinant IL-6. Proliferation of T cells was determined after
7 days via CFSE dilution. (C) Intracellular FACS analysis of allogeneic CD4+ memory T cells cocultured with control or rapa-ECs in the presence
of the indicated dose of recombinant IL-6. Numbers represent percent CD4+FoxP3+ cells in the CD4+CD25+ gate. (D) Intracellular FACS analysis
of allogeneic CD4+ memory T cells cocultured with control or rapa-ECs in the presence of blocking antibodies to PD-L1 and PD-L2, or isotype
control. Representative FACS plots are gated on CD4+CD25+ T cells. Data are normalized to FoxP3 expression in the control group (vehicle with
PD-L1 and PD-L2 on graft ECs and protected allografts, for up to 3 weeks, against subsequent T cell–mediated infiltration and rejec-tion, while blockade of PD-1 worsened immune injury in rapamy-cin-pretreated grafts.
The exclusive use of human materials in our studies provides two major advantages over murine systems. First, human ECs can activate resting allogeneic effector memory CD4+ T cells
(10), whereas mouse ECs only activate CD4+ Tregs (56). Given
their expression of MHC molecules and their direct contact with the host circulation, vascularized graft ECs are the first donor-derived cells to encounter and interact with the recipient immune system. Thus, it is likely that the EC–CD4+ T cell
inter-action is important in human allogeneic responses, and strong evidence suggests that such an interaction plays a pathologic role in the development of cardiac allograft vasculopathy (18, 57, 58). Second, human transplant recipients have high num-bers of alloreactive memory T cells, which are not present in the circulation of rodents housed under pathogen-free conditions (14). Alloreactive memory T cells act as important mediators of rejection and are believed to be more resistant to immune sup-pression and tolerance induction (15). Because our model repro-duces both of these phenomena, which are specific to human
transplantation and commonly overlooked in rodent studies, we propose that our data can more accurately predict clinical outcomes than can results from conventional mouse models.
[image:12.585.76.494.83.397.2]The mTOR inhibitor rapamycin was initially believed to act as an immunosuppressant by directly blocking proliferation of T lym-phocytes. Given that mTOR is ubiquitously expressed, recent data demonstrating that rapamycin has effects on other cell types is not surprising (43, 59). Both human and murine DCs differentiated in the presence of rapamycin poorly stimulate allogeneic T cells and express low levels of MHC class I, MHC class II, CD80, and CD86 (45, 46). We provide the first evidence to our knowledge that rapamy-cin reduces the alloimmunogenicity of human ECs. In contrast to DCs, human ECs do not express CD80 or CD86, and rapamycin did not alter EC expression of MHC molecules, but did lead to increased expression of PD-L1 and PD-L2, associated with reduced allostim-ulatory capacity. PD-L1 and PD-L2 are inhibitory molecules that bind the PD-1 receptor, expressed on activated T cells (60). Upon ligation by either PD-L1 or PD-L2, PD-1 antagonizes TCR signal-ing via recruitment of SHP-1 and SHP-2 phosphatases that inhibit phosphorylation of CD3ζ, ZAP70, and PKCθ (51, 61). PD-1 ligation also inhibits PI3K activity and downstream activation of Akt (36) and mTOR. Together, these signaling events reduce proliferation
Figure 8
Pretreatment of human arterial allografts with rapamycin reduces T cell–mediated graft injury in vivo. (A) Human artery interposition grafts were
pretreated with vehicle or rapamycin and then re-transplanted into PBMC-reconstituted hosts and harvested after 10 days. Representative immunofluorescence detection of infiltrating CD3+ T cells in graft vessel intima. ECs are stained with Ulex and visualized in green; CD3+ cells are
visualized in red. Intimal T cells were counted. Original magnification, ×200. (B) Arterial graft sections (treated as above) were stained with EVG
and the total vessel area (area bounded by external elastic lamina and lumen), intimal area (area bounded by internal elastic lamina and lumen), and lumen area quantified using ImageJ. Original magnification, ×200. (C) As in B, except grafts were harvested 3 weeks after re-transplant into
research article
and cytokine secretion in activated T cells. In allogeneic heart (62), corneal (63), and skin (64) transplant models, blockade or absence of PD-1 signaling accelerated rejection, while administration of PD-L1.Ig (a PD-1 agonist) reduced cardiac allograft vasculopathy (65). Collectively, these data suggest a role for PD-1 ligands in main-taining allograft tolerance. Thus, the induction of PD-1 ligands on allograft ECs, by rapamycin, would be expected to provide protective effects by downregulating host alloreactive effector T cells.
Rapamycin inhibits EC secretion of IL-6 in response to IFN-γ
and TNF. IL-6 is a proinflammatory cytokine that has numer-ous effects on T cell activation, differentiation, and recruitment (66–68). In the context of transplantation, increased levels of
IL-6 correlated with adverse outcomes in human allograft recip-ients (69, 70). IL-6 is known to inhibit the function of natural Tregs (71), as well as the induction of iTregs (52). Given that neutralizing IL-6 in such cocultures increases Treg formation (5) and that rapa-ECs poorly secrete IL-6, we were surprised to find that addition of recombinant IL-6 into cocultures did not alter the proportions of FoxP3+ cells. We suspect that the
[image:13.585.51.525.81.483.2]explana-tion resides in the observaexplana-tion that rapamycin treatment did not completely eliminate IL-6 production by the ECs in our culture system. Nevertheless, the downregulation of IL-6 release could be important in the in vivo setting, where, due to perfusion, local cytokine levels may be more limited.
Figure 9
PD-1 ligands protect rapamycin-pretreated grafts from T cell–mediated graft injury in vivo. (A) Human artery interposition grafts were
pre-treated with vehicle or rapamycin and then re-transplanted into PBMC-reconstituted hosts and harvested after 10 days. Representative immunofluorescence detection of PD-L1 and PD-L2 on graft ECs is shown. ECs are stained with Ulex and visualized in green; PD-L1 and PD-L2 are visualized in red. Quantification of PD-L1 and PD-L2 immunofluorescence is normalized to staining in vehicle-pretreated grafts. Original magnification, ×200. (B) Rapamycin-pretreated grafts were transplanted into PBMC-reconstituted mice and subsequently treated
with neutralizing PD-1 F(ab′)2 or control F(ab′)2 fragments. Grafts were harvested after 2 weeks and stained with EVG. Original magnification,
×100. (C) Representative immunofluorescence detection of infiltrating CD3+ T cells in grafts treated as in B. ECs are stained with Ulex and
visualized in green; CD3+ cells are visualized in red. Intimal T cells were counted. Original magnification, ×200. Mean ± SEM are shown for
non may further contribute to the protective effects of rapamycin in vivo. Overall we conclude that pretreatment of allografts with rapamycin downmodulates human T cells responses to allogeneic artery grafts in vivo. We envision that our data may be translated clinically by including rapamycin in the preservation fluid that is flushed or perfused through an explanted organ, or alternatively, by treating brain-dead cadaver donors with rapamycin prior to organ harvest. However, we caution that such an intervention requires additional preclinical study, as rapamycin has also been reported to have proinflammatory effects on monocytes, DCs, and CD8+ T cells (75, 76). If such effects on leukocytes prove to
be significant, then rapamycin pretreatment could be coupled to pre-transplant depletion of donor leukocytes to eliminate donor myeloid and lymphoid cells from allografts.
The data presented in this report raise several interesting ques-tions. Presently, we do not fully understand the specific signaling events downstream of mTOR that control EC alloimmunoge-nicity. In the cell, mTOR exists in two distinct signaling com-plexes, known as mTORC1 and mTORC2, which have differing upstream inputs and downstream outputs (77). Our data show that disruption of mTORC1 signaling (by raptor knockdown) caused changes in EC surface molecules similar to those induced by rapamycin, and such ECs poorly stimulated allogeneic T cells. In addition, we have found that inhibition of both mTORC1 and mTORC2 signaling reduces the ability of ECs to secrete IL-6 in response to TNF. We are currently investigating whether any of the known mTORC1 and mTORC2 targets are involved in mediating these effects. Additionally, because blockade of PD-1 ligands only partially reduced the ability of rapa-ECs to preferen-tially activate Tregs, it is likely that there are other mechanisms behind this observation. Although ligation of GITR on Tregs by GITR ligand was initially thought to abrogate their suppressive activity (78), recent studies suggest that GITR ligand may act to maintain and expand Tregs (79–81). Additionally, the interaction between OX40 and OX40 ligand has been shown to be important in Treg cell development in the gut (82), and an OX40 agonist enhanced Treg proliferation (83). Given that both GITR ligand and OX40 ligand are highly expressed on rapa-ECs, we are explor-ing whether either of these molecules contributes to the ability of rapa-ECs to activate Tregs.
Methods
Isolation and culture of human cells. PBMCs were isolated by density centrifuga-tion of leukapheresis products obtained from anonymized healthy adult vol-unteers using Lymphocyte Separation Medium (MP Biomedicals). PBMCs were stored in liquid nitrogen in 10% DMSO/90% FBS until ready for use.
CD4+ T cells were isolated from PBMCs by positive selection using
Dynabeads (Invitrogen) magnetic beads and released using DETACHa-BEAD, according to the manufacturer’s instructions. Activated T cells, preexisting Tregs, and monocytes were removed by incubating the iso-lated CD4+ T cells with 5 μg/ml mouse CD14 (BioLegend),
anti-CD25 (eBioscience), and anti–HLA-DR (LB3.1; gift of J. Strominger, Harvard University, Cambridge, Massachusetts, USA) antibodies for 20 minutes, followed by depletion using magnetic pan-mouse IgG beads (Invitrogen). Memory CD4+ T cells were isolated following further
neg-ative selection using mouse anti-CD45RA (eBioscience) and pan-mouse IgG beads. Isolates were routinely >99% CD4+CD25–HLA-DR–CD45RA–
by flow cytometry. It is important to note that these populations are not only devoid of activated memory T cells, but are also depleted of CD25-expressing natural Tregs.
In rodent models, infusion of either natural or inducible Tregs into recipients prior to transplantation prevented rejection of allografts (72). In humans, increased numbers of Tregs are found in the circulation and within allografts of recipients that have achieved immunosuppression-free graft survival (operational tolerance) (1, 73, 74). We found that allogeneic T cells activated by rapa-ECs contained a higher percentage of suppressive CD4+
CD25hiCD127loFoxP3+ T cells, and such cells displayed
signifi-cant demethylation of the TSDR within the FOXP3 locus. While the increase in FoxP3+ cells activated by rapa-ECs compared with
control ECs (Figure 4B) may appear modest, it is not known how many of these FoxP3+ cells actually represent suppressive Tregs,
since conventional human T cells transiently express FoxP3 upon activation. However, we demonstrate that this increase is function-ally significant, as the CD25+ T cell pool primed by rapa-EC was far
more suppressive than control, especially at lower Treg/responder ratios (Figure 5E). Since our highly purified CD4+ memory T cells
are depleted of preexisting CD25+ T cells, a subpopulation that
in humans contains natural Tregs, one may consider the FoxP3+
cells induced by rapa-ECs as iTregs. The fundamental effect of rapamycin treatment appears to be a loss of the capacity of rapa-ECs to activate effector cells, resulting in an increase in the ratio of regulatory to effector T cell populations. We demonstrate that PD-1 ligands partially contribute to this effect, as their blockade reduces the preferential activation of CD25+FoxP3+ cells by
rapa-ECs. Importantly, we show that shRNA knockdown of mTOR in ECs phenocopies the increased ability of rapa-ECs to favor Treg activation, thereby ruling out the possibility that carryover of rapamycin may be directly inducing Tregs. The enhanced ability of rapamycin-treated allograft ECs to activate Tregs would be ben-eficial in the context of transplantation by reducing rejection and promoting tolerance.
phenome-research article
surface-stained cells were then permeabilized and restained using a FoxP3 Staining Kit (eBioscience), according to the manufacturer’s instruc-tions, with anti–human FoxP3-APC (clone PCH101; eBioscience). For intracellular cytokine staining, T cells cocultured with allogeneic ECs for 6 days were stimulated for 6 hours with 40 ng/ml PMA and 2 μM ionomy-cin in the presence of Brefeldin A (eBioscience). Stimulated T cells were sur-face stained with anti–human CD4, CD25, CD127 fluorescent antibodies, then permeabilized using FoxP3 Staining Kit and stained with FoxP3-APC and IL-17–FITC (eBioscience), IL-10–FITC (eBioscience), IFN-γ–FITC (eBioscience), or TGF-β/LAP–FITC (BioLegend). To ensure antibodies were functional, positive staining was analyzed in the following controls: CD14+
monocytes stimulated with LPS (IL-10), CD4+ T cells stimulated by MHC
II+ ECs (IFN-γ and IL-17), and CD4+ T cells stimulated with anti-CD3 and
anti-CD28 in the presence of IL-2 (LAP). All stained cells were analyzed on an LSR II (BD Biosciences) using FlowJo software (Tree Star).
In vitro suppression assays. The frequency of T cells that recognize alloge-neic cells is too limited to generate significant numbers of responder cells in a primary culture. Therefore, to determine whether ECs could gener-ate iTregs, we first modified EC stimulators to serve as polyclonal acti-vators of T cell responses by retroviral transduction of the extracellular domain of Fcγ receptor IIA (CD32), as previously described (50). CD32+
ECs were treated with vehicle (DMSO) or rapamycin and plated into 24-well plates, as described above. Treated ECs were then loaded with 2.5 μg/ml anti-CD3 (OKT3; BioLegend) for 4 hours and washed 3 times with RPMI complete medium. 2 × 106 allogeneic memory CD4+ T cells
were added to each well. After 72 hours of stimulation, T cells were col-lected, washed, stained with CD4-FITC (Beckman Coulter) and CD25-APC (BD Biosciences), and sorted on a FACSAria (BD Biosciences) to isolate total CD4+CD25+ or CD4+CD25hi populations. Sorted populations were
then added into cocultures of freshly isolated CFSE-labeled CD4+ memory
T cells and untreated OKT3-loaded ECs at indicated suppressor/responder ratios. Proliferation of CFSE-labeled responder T cells was assessed after 4 days via CFSE dilution, a time point at which responder cells that had divided could be readily distinguished from both labeled cells that had not divided and unlabeled Tregs. Percent suppression was calculated as follows: ([proliferation of responders alone – proliferation of responders with Tregs]/proliferation of responders alone) × 100).
qRT-PCR analysis. ECs treated with rapamycin for up to 72 hours were collected at various time points by trypsinization, lysed, and used to make cDNA using a Cells-to-CT kit (Ambion), according to the manufacturer’s instructions. For T cell experiments, memory CD4+ T cells stimulated by
allogeneic ECs for 72 hours were collected, washed, stained with fluorescent antibodies, and FACS sorted for CD4+CD25+ cells. cDNA was made from
these cells using the Cells-to-CT kit. All qRT-PCR reactions were assem-bled with TaqMan 2× PCR Master Mix and predeveloped gene expression probes from Applied Biosystems. Samples were analyzed on a CFX96 Real-Time system using CFX Manager Software (Bio-Rad). Gene expression lev-els were normalized to GAPDH or CD3, as indicated. All Taqman probes were purchased from Applied Biosystems: GAPDH (Hs99999905_m1), CD3 (Hs00167894_m1), FoxP3 (Hs01085834_m1), PD-L1 (Hs01125301_m1), PD-L2 (Hs00228839_m1), GRAIL (Hs00226053_m1), CBL-B (Hs00180288_ m1), and ITCH (Hs00395201_m1).
shRNA knockdown of mTOR, raptor, and rictor. mTOR, raptor, or rictor was silenced in ECs using lentivirus-mediated transduction of shRNA. To gener-ate lentivirus, plasmids encoding mTOR shRNA (mTOR_2 shRNA), raptor shRNA (raptor_2 shRNA), rictor shRNA (rictor_1 shRNA), or control GFP shRNA (pLKO.1 GFP shRNA) were transfected into 293T cells together with the CMV-dR8.2 packaging and CMV-VSVG envelope plasmids, using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instruc-tions. Virus containing supernatants was collected at 48 and 72 hours Human ECs were isolated from umbilical cords following collagenase
digestion and serially cultured on 0.1% gelatin-coated tissue culture plates in M199 (Invitrogen) supplemented with 20% FBS, l-glutamine (2 mM),
penicillin (100 U/ml), streptomycin (100 μg/ml), 0.1% EC growth supple-ment (Collaborative Biomedical Research), and porcine heparin (100 μg/ml; Sigma-Aldrich). EC cultures were used at passage levels 2–5, at which time the cultured cells are uniformly positive for the EC marker CD31 and are devoid of CD45+ contaminating leukocytes.
In vitro rapamycin treatment. ECs retrovirally transduced to express CIITA were grown on 10-cm gelatin-coated plates and allowed to reach conflu-ence. Post-confluent ECs were treated with varying doses (up to 100 ng/ml) of rapamycin (Sigma-Aldrich) or vehicle (DMSO) for up to 72 hours. Fresh medium with rapamycin was added every 24 hours. Vehicle- and rapamy-cin-treated ECs were then washed 3 times with HBSS, collected by trypsi-nization, washed an additional 3 times with HBSS, and plated into 24-well plates. Viable (assessed by Trypan blue staining) control ECs and rapa-ECs were counted to ensure that equal numbers of cells were plated. In other experiments, post-confluent ECs grown in 10-cm plates were treated with rapamycin and 50 ng/ml recombinant human IFN-γ simultaneously for 72 hours, then plated as above.
T cell activation in vitro. CD4+ memory T cell activation by allogeneic ECs
requires recognition of non-self MHC class II molecules, principally HLA-DR, on the ECs. To reinduce EC expression of MHC class II, which is lost on serially cultured cells, ECs were retrovirally transduced with the CIITA transactivator, as previously described (50). In other experiments, expres-sion was restored by treating ECs with 50 ng/ml recombinant human IFN-γ
(Invitrogen) for 72 hours. For in vitro stimulation, MHC class II+ ECs were
washed 3 times with HBSS, plated into gelatin-coated 24-well plates, and allowed to reach confluence. 2 × 106 allogeneic memory CD4+ T cells were
added to each well in RPMI 1640 (Invitrogen), supplemented with 10% FBS,
l-glutamine (2 mM), penicillin (100 U/ml), and streptomycin (100 μg/ml). To assess proliferation, T cells were labeled with 250 nM CFSE (Molecular Probes, Invitrogen) prior to coculture. Proliferation was assessed on day 7 via CFSE dilution, as previously described (84). In some experiments, recombi-nant human IL-6 (BioLegend) or 10 μg/ml of blocking antibodies to PD-L1 and PD-L2 (BioLegend) was added into allogeneic cocultures.
For restimulation experiments, unlabeled memory CD4+ T cells were
cocultured with MHC class II+ allogeneic control ECs or rapa-ECs for
72 hours, collected, washed 3 times in HBSS, and rested for 3 days in the presence of 10 U/ml recombinant human IL-2 (Invitrogen), which serves as a survival factor. Recovered T cells were washed in HBSS, labeled with 250 nM CFSE, and restimulated with either fresh MHC class II+ ECs from
the same donor as used in primary stimulation or from third-party donors. Proliferation was assessed (via CFSE dilution) at day 3 for T cells restim-ulated by matched EC donors and day 5 for T cells restimrestim-ulated by third-party EC donors. In some experiments, blocking antibodies (30 μg/ml) to HLA-DR or isotype control IgG (30 μg/ml) were added to the primary stimulation cocultures.
FACS analysis. To analyze ECs, cells were suspended with trypsin-ver-sene, washed in PBS, and stained (on ice) in PBS supplemented with 1% BSA with antibodies against human HLA-A,B,C–FITC, HLA-DR–FITC, or CD40-PE (all from Beckman Coulter), LFA-3–PE, PD-L1–PE, or PD-L2–PE (all from BD Biosciences), OX40 ligand–PE or GITR ligand–PE (from R&D Systems), or ICOS ligand–PE (from BioLegend).