Copyright2001 by the Genetics Society of America
Mutations in the
YRB1
Gene Encoding Yeast Ran-Binding-Protein-1 That Impair
Nucleocytoplasmic Transport and Suppress Yeast Mating Defects
Markus Ku
¨nzler,*
,†Joshua Trueheart,*
,1Claudio Sette,* Eduard Hurt
†and Jeremy Thorner*
*Department of Molecular and Cell Biology, Division of Biochemistry and Molecular Biology, University of California, Berkeley, California 94720-3202 and†Ruprecht-Karls-Universita¨t Heidelberg, Biochemie-Zentrum Heidelberg, D-69120 Heidelberg, Germany
Manuscript received July 13, 2000 Accepted for publication November 21, 2000
ABSTRACT
We identified two temperature-sensitive (ts) mutations in the essential gene,YRB1, which encodes the yeast homolog of Ran-binding-protein-1 (RanBP1), a known coregulator of the Ran GTPase cycle. Both mutations result in single amino acid substitutions of evolutionarily conserved residues (A91D and R127K, respectively) in the Ran-binding domain of Yrb1. The altered proteins have reduced affinity for Ran (Gsp1)in vivo.After shift to restrictive temperature, both mutants display impaired nuclear protein import and one also reduces poly(A)⫹RNA export, suggesting a primary defect in nucleocytoplasmic trafficking. Consistent with this conclusion, bothyrb1tsmutations display deleterious genetic interactions with mutations in many other genes involved in nucleocytoplasmic transport, includingSRP1(␣-importin) and several -importin family members. Theseyrb1tsalleles were isolated by their ability to suppress two different types of mating-defective mutants (respectively,fus1⌬andste5ts), indicating that reduction in nucleocytoplasmic transport enhances mating proficiency. Indeed, in both yrb1ts mutants, Ste5 (scaffold protein for the pheromone response MAPK cascade) is mislocalized to the cytosol, even in the absence of pheromone. Also, bothyrb1tsmutations suppress the mating defect of a null mutation inMSN5, which encodes the receptor for pheromone-stimulated nuclear export of Ste5. Our results suggest that reimport of Ste5 into the nucleus is important in downregulating mating response.
M
ATING in the yeastSaccharomyces cerevisiaeis the nucleus in response to pheromone, as observed for theregulated import and export of other nuclear proteins culmination of a complex series of events
re-quired for cellular and nuclear fusion of two haploid (KaffmanandO’Shea1999).
Proteins and protein-RNA complexes cross the
nu-cells of opposite mating type (Spragueand Thorner
1992). Mating pheromones (secreted peptides) bind clear envelope through nuclear pores comprised ofⵑ50
different proteins, termed nucleoporins (Ryan and
to G-protein-coupled receptors, stimulating a
mitogen-activated protein kinase (MAPK) cascade (Bardwellet Wente 2000). Nucleocytoplasmic transport also
re-quires soluble factors. Transport receptors for both
im-al.1994) that evokes dramatic changes in gene
transcrip-tion, cell cycle arrest, and pronounced alterations of port and export (-importin and its relatives) bind their
cargo and shuttle between the cytosol and the
nucleo-cell morphology and nuclear reorganization (Leberer
et al.1997;Stoneet al.2000). Pheromone-activated and plasm (Go¨ rlichandKutay1999). The small Ras-like GTPase Ran and its associated factors confer directional-pheromone-induced gene products required for cell
fusion are deposited at a localized site on the plasma ity to transport (Macara et al. 2000). In S. cerevisiae,
GSP1 and GSP2 encode Ran isoforms (Belhumeur et
membrane at the leading edge of a mating projection
(“shmoo tip”;MaddenandSnyder1998). Proteins re- al. 1993; Kadowaki et al. 1993). Ran exists
predomi-nantly in its GTP-bound form in the nucleus; in the quired for nuclear fusion are recruited to the nucleus
(Rose 1996). How the signal initiated at the plasma cytosol, Ran is mainly GDP-bound. This asymmetry is
imposed by the subcellular distribution of Ran regula-membrane is transmitted into the nucleus to activate
gene expression is still unclear. Two components of the tors: the Ran-specific guanine-nucleotide exchange
fac-tor (RanGEF1), thePRP20/SRM1/MTR1gene product
pathway, Ste5 (PryciakandHuntress1998;Mahanty
et al. 1999) and Far1 (Blondel et al. 1999), shuttle in S. cerevisiae, is confined to the nucleus, whereas the Ran-specific GTPase-activating protein (RanGAP1), the between the nucleus and the cytosol, are predominantly
nuclear in naı¨ve cells, but are rapidly ejected from the RNA1 gene product in S. cerevisiae, is located in the
cytoplasm. GSP1, PRP20, and RNA1 are all essential
genes, and recessive mutations in all three block nuclear
Corresponding author:Markus Ku¨ nzler, Ruprecht-Karls-Universita¨t protein import and poly(A)⫹ RNA export (Corbett Heidelberg, Biochemie-Zentrum Heidelberg (BZH), Im
Neuen-andSilver1997;Okiet al.1998).
heimer Feld 328, 4. OG, D-69120 Heidelberg, Germany.
Transport receptors bind specifically to the
GTP-E-mail: [email protected]
1Present address:Microbia, Inc., Cambridge, MA 02139. bound form of Ran via a conserved domain at their N
termini (Go¨ rlichandKutay1999). RanGTP-binding isolated (Katzet al.1987) as an extragenic suppressor
of missense mutations inSTE5, which encodes a scaffold
to an export receptor enhances its affinity for an export
substrate; conversely, binding of RanGTP to an import protein for the pheromone-activated MAPK cascade
(Elion1998). As described here, both yrb1mutations receptor prevents the binding of an import substrate.
Hence, high RanGTP in the nuclear compartment po- cause clear defects in nucleocytoplasmic trafficking of
various proteins, including Ste5, and are able to sup-tentiates association of export cargo with export
recep-tors and triggers release of import cargo from import press the mating defect of amsn5⌬mutant. These data
suggest that preventing efficient reimport of Ste5 after receptors. Ran-binding-protein-1 (RanBP1) is another
protein that binds specifically to RanGTP (Ku¨ nzleret its pheromone-induced release from the nucleus
sus-tains the mating-competent state.
al.2000;PlafkerandMacara2000). RanBP1 contains
a conserved Ran-binding domain (RBD) ofⵑ140
resi-dues (Beddowet al. 1995;Vetter et al. 1999), which
MATERIALS AND METHODS
is necessary and sufficient for high-affinity binding of RanGTP and for nuclear export of RanBP1, at least in
Strains and growth conditions: Yeast strains used in this
yeast (Ku¨ nzler et al. 2000). Homologous RBDs are study are listed in Table 1. Strains JTY2483 and JTY2484 were
found in other nuclear proteins, like vertebrate obtained by backcrossing strain 381G-42E-P1 three times
against either YPH499 or YPH500.msn5⌬::TRP1strain HMK30
RanBP2/NUP358 (Yokoyamaet al.1995) and RanBP3
was derived from strain LH90 (Blondelet al.1999) by three
(Muelleret al.1998), andS. cerevisiaenuclear proteins,
consecutive backcrosses against the W303-1A derivatives, CRY1
S. cerevisiaeNup2 (Boothet al.1999) and Yrb2 (Taura
or CRY2. DNA-mediated transformation of yeast cells was
per-et al. 1998). Transport receptors block stimulation of formed using a modified version of the lithium acetate method
Ran-mediated GTP hydrolysis by RanGAP1; in contrast, (Gietzet al. 1992). Thefus1⌬mutation deletes 90% of the
coding sequence (from theFUS1promoter to codon 460) and
RanBP1 acts as a coactivator of RanGAP1-stimulated
was constructed by a two-step gene disruption method (Boeke GTP hydrolysis by Ran and, moreover, is required for
et al.1987). Heterozygous diploid strain JTY2501 was derived
nucleotide hydrolysis when RanGTP is bound to a trans- from CRY3 by transplacing theYRB1locus on one homolog
port receptor (Go¨ rlichandKutay1999). These bio- of chromosome IV via transformation with anEcoRI-XbaI
frag-chemical activities, and the fact that RanBP1 is abun- ment containing theyrb1⌬::HIS3construct excised from
plas-mid pMK112n (Table 2). To construct strain HMK21, JTY2501
dant, shuttles between the nucleus and the cytoplasm,
was transformed with plasmid pMK103, sporulated, and a
but is found almost exclusively in the cytosol at steady
MATaHis⫹Ura⫹5-fluoro-orotic acid (5-FOA)-sensitive spore
state (Ku¨ nzleret al.2000;PlafkerandMacara2000),
was chosen. Strain JTY2486 was obtained by transformation of
suggest that RanBP1 has a major role in the cytoplasm CRY1 with anEcoRI-SpeI fragment containing thenup2::HIS3
both in recycling of transport receptors and in release construct excised from plasmid pJON115 (Loebet al.1993).
Strain HMK29 was constructed analogously using a Bam
HI-of export cargo (Petersonet al.2000). Consistent with
HindIII fragment containing agsp2⌬::LEU2construct (
Kado-this view,S. cerevisiae YRB1, encoding yeast RanBP1, is
wakiet al. 1993). Correct transplacements were verified by
essential for cell viability and is required for both
nu-Southern hybridization analysis.
clear protein import and poly(A)⫹ RNA export Unless indicated otherwise, yeast cells were propagated at
(Schlenstedtet al.1995). 30⬚. Rich medium (YP), synthetic complete medium (SC), and synthetic minimal medium (SM) were prepared as described
A link between yeast mating and the Ran GTPase
(Kaiseret al. 1994). Glucose (Glc) or raffinose (Raf) were
cycle was the identification of thesrm1-1mutation, now
added as carbon source at a final concentration of 20 g/liter
known to reside in RanGEF1, which suppressed the after autoclaving; induction with galactose (Gal) was
per-mating defect of cells lacking pheromone receptors and formed by adding Gal (final concentration 2%) to Raf-grown
increased the basal expression of a pheromone-respon- cells. Drop-out media (SC lacking the appropriate nutrients)
were used to maintain selection for plasmids. Agar plates
con-sive reporter gene (ClarkandSprague1989). Another
taining 5-FOA were prepared as described by Boeke et al.
connection between mating and nucleocytoplasmic
(1987).Escherichia colistrain DH5␣(Hanahan1983) was used
transport was the finding that theste21mutation,
identi-for propagation of plasmid DNAs. Bacteria were cultivated
fied in a screen for enhancers of the mating defect of using standard methods (Sambrooket al.1989).
a temperature-sensitive (ts) mutation inSTE4(encoding Quantitative mating assays:Quantitative mating assays were
performed as previously described (Sprague1991). Briefly,
the-subunit of the pheromone receptor-coupled
het-MATastrains to be tested andMAT␣tester strains were
pre-erotrimeric G-protein; Akada et al. 1996), resides in
grown at 26⬚ to midlogarithmic phase in selective and rich
MSN5, encoding the nuclear receptor for
pheromone-medium, respectively. Cells were washed with water and 106
stimulated export of Ste5 (Mahanty et al.1999) and cells of the MATa strains to be tested were mixed with 107
Far1 (Blondelet al.1999). As described here, we iso- cells of theMAT␣tester strain. In the case of the experiment
shown in Table 3, the mixture was spread directly onto
pre-lated a yrb1ts mutation as a suppressor of a mutant
cooled (14⬚) SMGlc plates, the plates were incubated for 3
(fus1⌬) defective in the cell fusion step of mating. Fus1
days at 14⬚, and then for 3 days at room temperature. The
is a pheromone-induced,O-glycosylated, integral
mem-resultant diploid colonies were counted and normalized to
brane protein that acts at a late stage in mating the titer of inputMATacells (determined by plating the same
(TrueheartandFink1989). While mapping this yrb1 dilutions on plates selective for theMATastrain to be tested
and incubating for 3 days at room temperature). In the case
TABLE 1
Yeast strains
Strain Characteristics Source
YPH499 MATaura3-52 trp1-⌬63 his3-⌬200 leu2-⌬1 lys2-801amade2-101oc SikorskiandHieter(1989) YPH500 MAT␣ura3-52 trp1-⌬63 his3-⌬200 leu2-⌬1 lys2-801amade2-101oc SikorskiandHieter(1989)
YPH501 MATa/MAT␣(YPH499⫻YPH500) SikorskiandHieter(1989)
CRY1 (W303-1A) MATaura3-1 trp1-1 his3-11,15 leu2-3,112 ade2-1 can1-100 GAL R. S. Fuller CRY2 (W303-1B) MAT␣ura3-1 trp1-1 his3-11,15 leu2-3,112 ade2-1 can1-100 GAL R. S. Fuller
CRY3 (W303D) MATa/MAT␣(CRY1⫻CRY2) R. S. Fuller
9399-7B MATaura3-52 his4-⌬29 GAL TrueheartandFink(1989)
JY416 MAT␣ura3-52 leu2-3,112 fus1⌬ This study
JTY2023 MATaura3-52 trp1-⌬63 his4⌬29 ade2-101ocGAL This study
JTY2024 JTY2023fus1⌬ This study
JTY2025 JTY2024yrb1-51 (sfo1-1) This study
JTY2488 MATaura3-52 trp1-⌬63 his4-⌬29 fus1⌬yrb1-51 This study
JTY2501 CRY3yrb1⌬::HIS3/YRB1 This study
HMK21 CRY1yrb1⌬::HIS3(pMK103) This study
JTY2026 MATaura3-52 trp1-⌬63 his3-⌬200 leu2-⌬1 ade2-101ocyrb1-51 This study JTY2027 MAT␣ura3-52 trp1-⌬63 his3-⌬200 leu2-⌬1 lys2-801amyrb1-51 This study
JTY2482 MATa/MAT␣( JTY2026⫻JTY2027) This study
381G-42E-P1 MATaade2-1 lys2octyr1ochis4-580amtrpamste5-3 yrb1-52 (stp52) CRY1 SUP4-3ts Katzet al.(1987) JTY2483 MATaura3-52 trp1-⌬63 his3-⌬200 leu2-⌬1 lys2-801amade2-101ocyrb1-52 This study JTY2484 MAT␣ura3-52 trp1-⌬63 his3-⌬200 leu2-⌬1 lys2-801amade2-101ocyrb1-52 This study
JTY2485 MATa/MAT␣( JTY2483⫻JTY2484) This study
JTY2500 MATaura3-52 his3-⌬200 leu2-3,112 trp1-901 canRgal4-542 gal80-538 Inouyeet al.(1997a) ADE2::PGAL-URA3 LYS2::lexop-lacZ
JTY2486 MATaura3-1 trp1-1 his3-11,15 leu2-3,112 ade2-1 can1-100 GAL nup2-5::HIS3 This study
HMK29 CRY1gsp2⌬::LEU2 This study
HMK30 CRY1msn5⌬::TRP1 This study
DC17 MAT␣his1 J. B. Hicks
of the experiment shown in Table 5, the mating mixture was DNA by complementation of the ts phenotype ofsfo1-1cells (see below) revealed that it is identical to theYRB1gene, and collected on a 0.45-m pore filter and incubated for 6 hr at
30⬚on YPGlc. After the incubation, cells were resuspended because sequence analysis (see below) demonstrated that both thesfo1-1andstp52mutations reside in theYRB1locus, these in SMGlc medium and plated in appropriate dilutions onto
SMGlc plates with appropriate nutrients to select for diploids. alleles were renamedyrb1-51andyrb1-52, respectively. Recovery and analysis ofyrb1tsalleles:The base sequence
As a control for the number of viableMATacells used in the
mating mixture, 106cells of theMATacells were collected on alterations corresponding to theyrb1-51andyrb1-52mutations were determined by cloning and sequencing of DNA isolated a separate filter, incubated as above, resuspended in YPGlc,
and plated on YPGlc plates at appropriate dilutions. Mating from the mutants. The polymerase chain reaction (PCR) was used to amplify 636-bp products comprising the entireYRB1 efficiency was expressed as percentage of the inputMATa
haploids that formed diploid colonies. open reading frame (ORF) using genomic DNA from JTY2026 (yrb1-51) and 381G-42E-P1 (yrb1-52) as the template and oligo-Isolation ofyrb1-51:JTY2024 (MATafus1⌬) was
mutagen-ized with ethyl methanesulfonate (108cells/ml; 3% EMS; 1 hr) nucleotide primers, 5⬘-GGG GAT CCG AAT GTC TAG CGA AGA TAA G-3⬘(OSFO1) and 5⬘-GGT CTA GAC GCA AGT to 25% survival, and spread on YPGlc plates. After 3 days at
28⬚, ⵑ72,000 colonies were replica plated onto precooled AAC AAG C-3⬘(OSFO5), which corresponded, respectively, to positions⫺2 to⫹18 and⫹635 to⫹616 of the 201-codon (14⬚) SMGlc plates containing uracil (20 mg/liter), on which
2A600 nmunits of JY416 (MAT␣fus1⌬) cells had been spread. YRB1 sequence (where⫹1 is the first base of the initiator codon of the ORF) and included restriction sites at their These plates were incubated for 4 days at 14⬚. Candidate clones
that gave a positive mating response (35 colonies total) were 5⬘-ends to facilitate cloning of the PCR products. Reaction products were isolated, digested withBamHI andXbaI, and restreaked from the master plate, retested for suppression,
and examined for their ability to grow at various temperatures. inserted intoE. colivector pUC19 for sequencing. Nucleotide sequence of multiple inserts was determined on both strands A single isolate ( JTY2025) displayed a ts phenotype that
coseg-regated with the ability to suppress the mating defect of the using the M13/pUC universal and M13/pUC reverse sequenc-ing primers (New England Biolabs, Beverly, MA) and, when fus1⌬cells at 14⬚(data not shown). The mutation conferring
these phenotypes was initially namedsfo1-1(suppressor offus necessary, sequence-specific primers. The single-base-pair mu-tations recovered were tested for their ability to confer a ts one). In the course of these crosses, it was shown, first, that
sfo1was tightly linked totrp1(no recombinants in 31 tetrads; phenotype by first substituting the mutantYRB1ORFs (excised asSalI fragments from the pUC19 derivatives) for the corre-distance ⱕ1.6 cM) and, second, by complementation tests,
that thesfo1-1mutation was allelic tostp52, another suppressor sponding segment in pMK103 and then introducing the entire yrb1-51 and yrb1-52 genes as EcoRI-XbaI fragments (excised of mating defects that was mapped to the same region (Katz
-TABLE 2
Plasmids
Plasmid Characteristics Source
YEp24 2-mURA3 Botsteinet al.(1979)
pRS314 CEN ARS TRP1 SikorskiandHieter(1989)
pRS424 2-mTRP1 SikorskiandHieter(1989)
pRS425 2-mLEU2 SikorskiandHieter(1989)
pUN100 CEN ARS LEU2 ElledgeandDavis(1988)
YEp352 2-mURA3 Hillet al.(1986)
pGAD424 2-mLEU2 ADH1p-GAL4TAD-MCS-ADH1t BartelandFields(1995) pBTM116 2-mTRP1 ADH1p-LexADBD-MCS-ADH1t BartelandFields(1995)
pRSETA AmpRT7-His6-MCS Invitrogen (Carlsbad, CA)
pNOPGFP2L pRS425-NOP1p-GFP K. HellmuthandE. Hurt
(unpublished results)
pJON115 nup2-5::HIS3 Loebet al.(1993)
pPS815 2-mURA3 ADH1p-SV40NLS-GFP-lacZ Leeet al.(1996) pPS817 2-mURA3 GAL1p-SV40NLS-GFP-lacZ Leeet al.(1996) pGADGFP 2-mLEU2 ADH1p-SV40NLS-GAL4TAD-GFP Shulgaet al.(1996) pNOPGFPAU-NPL3 2-mADE2 URA3 NOP1p-GFP-NPL3 Sengeret al.(1998) YEplac195AU-L25NLS-GFP 2-mADE2 URA3 RPL25NLS-GFP-MEX67t O. GadalandE. Hurt
(unpublished results) pKW430 2-mURA3 ADH1p-SV40NLS-PKINES-(GFP)2 Stadeet al.(1997)
pLDB419 2-mLEU2 YAP1-GFP Yanet al.(1998)
pNOPGFP2L-STE5 This study
pSB415 YEp24-NTH1-YRB1 This study
pMK102 pUC19-YRB1 This study
pMK103 YEp352-YRB1 This study
PMK104 pRSETA-YRB1 This study
pMK112n pRS316-yrb1⌬::HIS3 This study
pNOPPATA-GSP1G21V pUN100-NOP1p-ProtA-TEV-GSP1(G21V)-ADH1t Hellmuthet al.(1998)
pMK275 pRS314-YRB1 This study
pMK277 pRS314-yrb1-51 This study
pMK278 pRS314-yrb1-52 This study
pMK284n pRS314-YRB1-GFP(S65T) Hellmuthet al.(1998)
pMK294-51 pRS314-yrb1-51-GFP(S65T) This study
pMK294-52 pRS314-yrb1-52-GFP(S65T) This study
pMK291-wt pRS424-YRB1-GFP(S65T) This study
pMK291-51 pRS424-yrb1-51-GFP(S65T) This study
pMK291-52 pRS424-yrb1-52-GFP(S65T) This study
pMK199-wt pGAD424-YRB1 This study
pMK199-51 pGAD424-yrb1-51 This study
pMK199-52 pGAD424-yrb1-52 This study
pMK195-GV pBTM116-GSP1(G21V) This study
marked plasmids pMK277 and pMK278, respectively. Finally, complement the ts phenotype of the yrb1-51 mutant. The smallest original isolate (pSB415) contained theYRB1 gene HMK21 (yrb1⌬ [pMK103]) was transformed with either
pMK277, pMK278, or a control plasmid (pMK275) carrying as well as the neighboring geneNTH1encoding neutral treha-lase. Subsequent subcloning localized the complementing ac-the normalYRB1gene, plated on 5-FOA plates at 23⬚to select
against the residentURA3-marked YRB1-containing plasmid tivity to a 1.3-kb chromosomalEcoRI-XbaI fragment containing only YRB1, which was used to construct pMK102, pMK103, (pMK103), and the resulting isolates were analyzed for their
ability to grow at elevated temperature. and pMK275.
To construct a plasmid (pMK112n) carrying theyrb1⌬::HIS3 Construction of plasmids:Standard techniques were used
for the manipulation of recombinant DNA (Sambrooket al. deletion construct, an internal BglII fragment was excised from theYRB1-containing insert in pMK101 and replaced by 1989). Plasmid DNA from E. coli was isolated according to
Del Salet al.(1988). Unless specified otherwise, PCR amplifi- a BamHI fragment containing theHIS3gene, which was in-serted in the same transcriptional orientation asYRB1. Con-cations were performed using Vent DNA polymerase (New
England Biolabs). Correct sequence of PCR-generated con- struction of plasmid pNOPPATA-GSP1G21V, which expresses a GTPase-defective mutant form of Gsp1, Gsp1(G21V), fused structs was verified by nucleotide sequence analysis. Plasmids
used in this study are listed in Table 2. at its N terminus to a cleavage site (ENLYEQG) for tobacco etch virus (TEV) protease and to two immunoglobulin G (IgG) TheYRB1gene was isolated from a yeast genomic library
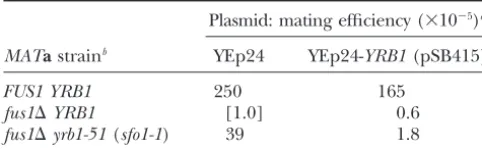
erated STE5 NcoI-BamHI fragment comprising the entire TABLE 3
ORF (using primers OSTE5-1, 5⬘-GGGGGGCCATGGGTAT Theyrb1-51(sfo1-1) mutation suppresses the cold-sensitive GGAAACTCCTACAGAC-3⬘, and OSTE5-2, 5⬘-GGGGGGAT mating defect of afus1⌬mutant CCCTATATATAATCCATATGG-3⬘) into pNOPPATA ( Hell-muthet al.1998) and subsequent recloning of the insert as PstI fragment into pNOPGFP2L. This vector is based on Plasmid: mating efficiency (⫻10⫺5)a
pRS425 and contains a 1.4-kbBamHI-PstINOP1p-GFPcassette MATastrainb YEp24 YEp24-YRB1(pSB415)
(K. HellmuthandE. Hurt,unpublished results).
Preparation of rabbit polyclonal anti-Yrb1 antiserum: To
FUS1 YRB1 250 165 generate a (His)6-Yrb1 fusion protein containing all but the
fus1⌬YRB1 [1.0] 0.6 first 10 residues of Yrb1, the correspondingYRB1coding
se-fus1⌬yrb1-51(sfo1-1) 39 1.8 quence was excised as a SalI fragment from pMK102 and
ligated into theXhoI site of pRSETA (Invitrogen), yielding aMating efficiency is defined as the number of diploids
pMK104. For expression in E. coli, strain BL21(DE3)/pLysS formed per number of input haploids of the strain tested.
(Studier1991) was transformed with pMK104 and the fusion The values given represent the average of three independent protein was induced by addition of
isopropyl--d -thio-galacto-trials, each performed in triplicate, and have been normalized
pyranoside (IPTG) to a final concentration of 0.4 mmfollowed to the mating efficiency of thefus1⌬mutant. by incubation at 37⬚
for 2 hr. Selection for pMK104 had to bThe indicated strains [JTY2023, MATa FUS1 YRB1;
be maintained by adding 50 mg/liter carbenicillin (Sigma, St. JTY2024, MATa fus1⌬ YRB1; JTY2025, MATa fus1⌬ yrb1-51
Louis), a more stable derivative of ampicillin, to the medium (sfo1-1)] were transformed with either YEp24 (aURA3-marked
because the fusion protein was relatively toxic to the cells. 2-m DNA vector) or pSB415 (YEp24-YRB1) and mated with
The fusion protein was purified fromE. coliusing Ni2⫹-chelate JY416 (MAT␣fus1⌬), as described inmaterials and methods.
affinity chromatography (Ni-NTA resin; QIAGEN, Chats-worth, CA), according to the manufacturer’s recommenda-tions. The purified protein was used to raise polyclonal anti-previously (Hellmuthet al.1998). Plasmids pMK294-51 and
sera in two adult female New Zealand White rabbits (nos. 1390 pMK294-52, and pMK291-wt, pMK291-51, and pMK291-52,
ex-and 1391), following stex-andard protocols (HarlowandLane
pressing Yrb1-green fluorescent protein (GFP) fusions under
1988), using 600g of protein in 50% Titermax (CytRx, Nor-control of the authentic YRB1 promoter, were constructed
cross, GA) for the first immunization and 400g of protein by replacing an internalBglII fragment in the YRB1coding
in 50% incomplete Freund’s adjuvant (Sigma) for each of two sequence in pMK284n, which expresses a functional
Yrb1-subsequent immunizations administered after 3 and 5 weeks, GFP chimera (Hellmuthet al.1998), with the corresponding
respectively. Bleeds were taken after 4 weeks (2 ml), 6 weeks fragments fromyrb1-51 and yrb1-52, followed by subsequent
(50 ml), 7 weeks (2 ml), and 8 weeks (terminal) and stored recloning of the respectiveYRB1-GFPgene fusions asEco
RI-in 0.02% sodium azide at ⫺70⬚. For detection of Yrb1 by NotI fragments into pRS424.
immunoblotting the resulting antisera (nos. 1390 and 1391) Fusions of full-length Yrb1 to the Gal4 transcriptional
activa-were used as primary antibodies at a dilution of 1:5000. tion domain (TAD) and full-length Gsp1(G21V) to theE. coli
Two-hybrid assay: To assess interactions between LexA LexA DNA-binding domain (DBD) were generated via PCR,
(DBD)-Gsp1(G21V) and Gal4(TAD)-Yrb1 fusion proteins, using the two-hybrid vectors pGAD424 and pBTM116,
respec-strain JTY2500 harboring theE. coli lacZgene under control tively. Fragments comprising the entireYRB1ORF were
syn-of eight LexA-binding sites was cotransformed with the appro-thesized using 5⬘-CCG AAT TCG GTC CAG GTG GTA GCG
priate pBTM116- and pGAD424-based plasmids. Transfor-AAG ATA AGA AAC CTG TCG-3⬘(OSFO15) and the M13/
mants were grown in SCGlc medium lacking leucine and tryp-pUC reverse sequencing primer (New England Biolabs) as
tophan to midexponential phase (A546 nm⫽ⵑ1) and assayed the primers and pUC19 carrying the chromosomalYRB1
-con-for-galactosidase acitivity as described previously (Ku¨ nzler
taining EcoRI-XbaI fragment (pMK102), or pUC19 carrying
andHurt1998). the corresponding fragments from theyrb1-51oryrb1-52ORFs,
Preparation of yeast cell extracts:Yeast cells were washed as templates. The PCR products were digested withEcoRI and
once with one culture volume of cold phosphate-buffered PstI and inserted into the corresponding sites in pGAD424,
saline (PBS), aliquoted into 1.5-ml microcentrifuge tubes yielding pMK199-wt, pMK199-51, and pMK199-52,
respec-(ⵑ20A546 nmunits per tube), and stored as pellets at ⫺70⬚. tively. Similarly, a fragment comprising the entire ORF coding
Frozen cell pellets were thawed by adding 0.2 ml cold lysis for Gsp1(G21V) was produced using 5⬘-GCG AGG CCT TGC
buffer (50 mmTris-HCl pH 7.5, 150 mmNaCl, 20 mmMgCl2, CCC AGC TGC TAA CGG TGA AG-3⬘ (OGSP7) and RSET
10% glycerol, 2 mm DTT, and 1 mm PMSF) and lysed by (5⬘-AAC TGC AGC CAA CTC AGC TTC C-3⬘) as the primers,
vigorous vortexing with 0.2 g of acid-washed glass beads (0.45– andE. coliexpression vector pRSETB (Invitrogen, Carlsbad,
0.6 mm diameter) for six 30-sec periods (separated by 1-min CA) carrying a PCR-mutated genomicPvuII-HindIII fragment
periods of cooling on ice). The lysate was clarified by centrifu-coding for Gsp1(G21V) as the template. The resulting PCR
gation for 5 min at 13,000⫻gat 4⬚and the protein concentra-product was cleaved withStuI andPstI and inserted into the
tion was determined by a dye-binding method (Bradford SmaI andPstI sites of pBTM116, yielding plasmid pMK195-GV.
1976) using commercially available reagents (Bio-Rad, Her-Plasmid YEplac195-AU-L25NLS-GFP was derived from
YE-cules, CA) and bovine serum albumin (BSA) as the standard. plac195-ADE2-URA3-L25-GFP (Hurtet al.1998) by removing
Purification of ProtA-TEV-Gsp1(G21V) from yeast: Trans-most of the RPL25coding sequence, except for the 5⬘-end
formants of wild-type strain CRY1, coexpressing ProtA-TEV-that encodes the first 52 residues of L25 (and contains an
Gsp1(G21V) from pNOPPATA-GSP1G21V and either Yrb1-intron), using a two-step PCR procedure (GiebelandSpritz
GFP, Yrb1(A91D)-GFP, or Yrb1(R127K)-GFP from plasmids 1990; mutagenic primer, 5⬘-GGG ACA ACT CCA GTG AAA
pMK284n, pMK294-51, or pMK294-52, respectively, were AGT CTT CTC TTT GCT CTC GAG TGG AAC AGC CTT
grown in selective medium at 26⬚to aA546 nm⫽ⵑ1.5. Purifica-GGA AGC-3⬘; O. GadalandE. Hurt,unpublished results).
tion of Gsp1(G21V) from these cells was performed essentially Plasmid pNOPGFP2L-STE5 expressing a GFP-Ste5 fusion
pro-as described (Hellmuthet al.1998), with the minor modifica-tein from a multicopy-plasmid under control of the
throughout the purification, including cell lysis, washing steps, recessive allele ofYRB1, which encodes the homolog of and elution. Elution by cleavage with TEV-protease (GIBCO- mammalian RanBP1 (Ouspenski et al.1995; Schlen-BRL, Gaithersburg, MD) was performed by incubation for
stedtet al.1995). Hence,sfo1-1was redesignatedyrb1-51.
1 hr at room temperature.
A recessive ts mutation, stp52 (sterile
pseudorever-Nuclear protein import and RNA export assays:
Tempera-ture-sensitive mutants, and their otherwise isogenic wild-type sion), closely linked to TRP1, was isolated as an
ex-strains, containing plasmids that express constitutively nuclear tragenic suppressor of the mating defect of a MATa transport substrates fused to GFP(S65T), namely SV40NLS-Gal4- ste5-3 ⫻ MAT␣ ste5-3 cross at restrictive temperature (TAD)-GFP (pGADGFP), GFP-Npl3 (pNOPGFPAU-NPL3),
(Katzet al.1987). Thestp52mutation also suppressed
and L25NLS-GFP (YEp195-AU-L25NLS-GFP), were cultivated to
other ste5,ste4, andste7 missense (ts) alleles (Katz et
early exponential phase (A546 nm⫽ ⵑ0.5) in selective SCGlc
medium at 23⬚, split into two equal portions, and incubated al. 1987). Although the linkage analysis reported by
at either 23⬚or 37⬚for various periods of time. Strains carrying Katz et al. (1987) assigned the stp52mutation to the plasmids expressing SV40NLS-GFP--galactosidase (pPS817) opposite side of theTRP1locus fromyrb1-51, we found under control of theGAL1promoter were pregrown to early
that ayrb1-51/stp52diploid strain was still ts, and that the
exponential phase (A546 nm⫽ⵑ0.5) in selective SCRaf medium
ts growth defect of thestp52mutant could be completely
at 23⬚ before Gal (2%) was added to the cultures and the
cells were incubated at 23⬚for another hour (to allow mRNA rescued by the clonedYRB1 gene on a plasmid (data
synthesis and export). The induced cultures were split into not shown). These results demonstrated that thestp52 two equal portions, and one portion was shifted to 37⬚ for mutation was another recessive allele of YRB1, as was 3 hr, while the other portion was maintained at 23⬚for the
confirmed by sequencing of the mutant DNA (see
be-same period. Fluorescence microscopy of living yeast cells
low). Hence,stp52was redesignatedyrb1-52.
expressing GFP fusion proteins was done according to
Hell-muthet al.(1998). Cells were concentrated by brief centrifuga- Phenotypic characterization of the yrb1-51 and yrb1-tion and resuspended in the residual growth medium without 52 mutations:To understand how alterations inYRB1 any washing steps. To assay mRNA export, cells were cultivated can suppress mating-defective mutants, we examined, in YPGlc medium as described above for strains harboring
first, the physiology of theyrb1mutants. At 23⬚,yrb1-51
constitutively expressed GFP fusion proteins. Poly(A)⫹RNA
mutant cells grew nearly as well as wild-type cells,
was localized byin situhybridization as described previously
(Segrefet al.1997). whereas the yrb1-52 mutant cells displayed impaired
Miscellaneous:SDS-PAGE and immunoblotting were con- growth already under these conditions; bothyrb1-51and ducted as described previously (Ku¨ nzler andHurt 1998). yrb1-52cells ceased growth and lost viability within 3–6 Multiple sequence alignment was done using the CLUSTALW
hr after shift to 37⬚ (data not shown). Similar results
1.7 (Thompsonet al.1994) and BOXSHADE 3.21
[Bioinfor-were observed for the corresponding homozygous
dip-matics group of the Swiss Institute for Experimental Cancer
Research (ISREC)] programs. Identities to the Yrb1 sequence loids (data not shown). As judged by immunoblotting
were calculated on the basis of pairwise alignments using the of cell lysates (Figure 1A), after shift to 37⬚ for 3 hr, ALIGN algorithm from the FASTA package (Pearson and the product of theyrb1-51allele was hardly detectable,
Lipman1988).
whereas the yrb1-52product remained relatively stable
even 6 hr after temperature shift. Thus, theyrb1-51
muta-tion appears to destabilize the gene product at higher
RESULTS
temperature, whereas the yrb1-52product is stable
un-Isolation ofyrb1ts
mutations as suppressors of mating der the same conditions. Upon prolonged incubation
defects: The mating deficiency of a fus1⌬ mutant is at 37⬚, an apparent degradation product of Yrb1
accu-much more pronounced at 14⬚than at 30⬚. At 14⬚, dip- mulated in yrb1-52 cells, but was also observed in the
loid formation in a MATa fus1⌬ ⫻ MAT␣fus1⌬cross wild-type control cells. Yrb1 was expressed at similar
is ⬍0.5% that of a MATa FUS1 ⫻ MAT␣ fus1⌬ cross levels in MATa, MAT␣, and MATa/MAT␣ cells (data
(Table 3). A screen for extragenic suppressors of this not shown) and its level inMATacells was not elevated in
mating defect (see materials and methods) yielded response to treatment with␣-factor mating pheromone
a single mutation,sfo1-1.This suppressor mutation re- (data not shown).
producibly enhanced mating competence of a fus1⌬ Examination of the cell morphology revealed that
mutant 30–50-fold (Table 3), but did not fully restore haploid yrb1-51 cells arrested mostly as enlarged cells
mating proficiency to the level of aFUS1cell. The sup- with a large bud or as large unbudded cells upon shift
pressor segregated 2:2 through two backcrosses against to 37⬚ (data not shown;Ba¨umer et al. 2000), which is
afus1⌬strain and cosegregated with a recessive ts growth reminiscent of cell cycle progression mutants. Similar
defect (in⬎15 tetrads analyzed per cross). Genetic map- results were previously observed forstp52/yrb1-52cells
ping of the mutation to the right arm of chromosome (Katzet al. 1987; Ouspenski 1998). Another striking
IV betweenCEN4and theTRP1gene (data not shown), phenotype of bothyrb1ts alleles was the appearance of
complementation of the ts growth defect by the wild- chains of elongated nonseparated cells, most evident in
typeYRB1gene on a plasmid (data not shown), elimina- homozygous diploids grown on plates at a
semipermis-tion of the suppression phenotype by plasmid-borne sive temperature (30⬚; Figure 1B). Such cell elongation
YRB1(Table 3), and nucleotide sequencing of the mu- is diagnostic of mutations that delay G2-M progression
(Lew2000). We observed a similar morphological
nonical cell division cycle (cdc) mutations, and might indicate a role of Yrb1 at multiple stages of the cell
cycle. Consistent with such a notion, theyrb1-51
muta-tion interferes with both the G1/S transimuta-tion and the
passage through mitosis (Ba¨umeret al.2000), and
dis-played synthetic growth defects when combined with
two differentcdc28 alleles (cdc28-4and cdc28-1N) that
are diagnostic for different cell cycle stages (G1 and G2/M, respectively; Table 4).
Yrb1-51 and Yrb1-52 are altered in conserved residues of the Ran-binding domain and defective for
Ran-bind-ingin vivo:To determine the nature of the alterations
in the mutant proteins, PCR was used to recover the
YRB1 coding sequences from the mutant strains (see
materials and methods). The DNA sequence of each mutant ORF contained a single point mutation, both of which alter a highly conserved residue in the RBD
(Figure 2A). Theyrb1-51mutation is a C-to-A
transver-sion on the coding strand at position 272 (where⫹1 is
the first base of the initiator ATG), which substitutes
Asp for Ala at codon 91 (A91D). Theyrb1-52allele is a
G-to-A transition on the coding strand at position 380, which substitutes Lys for Arg at codon 127 (R127K). On the basis of homology modeling of Yrb1 on the crystal structure of the first RBD (RanBD1) in mamma-lian Nup358 (RanBP2) complexed with Ran bound to Figure1.—Effects ofyrb1-51 and yrb1-52 mutations onin a nonhydrolyzable GTP analog (Vetter et al. 1999), vivo stability of Yrb1 and cell morphology. (A) Stability of A91D replaces a nonpolar residue in the hydrophobic normal and mutant Yrb1 at restrictive temperature. Haploid
core of Yrb1 with a bulkier, charged residue (Figure
strain YPH499 (YRB1) and its congenic derivatives, JTY2026
2B). This change should destabilize the global fold of
(yrb1-51) and JTY2483 (yrb1-52), were grown at 23⬚in SCGlc
Yrb1, consistent with the rapid degradation of this
mu-medium to midexponential phase, shifted to 37⬚, and samples
were withdrawn at the indicated times. Total protein was ex- tant protein observed at restrictive temperature (Figure
tracted from each sample and analyzed by SDS-PAGE and 1A). Two other existent yrb1 alleles, yrb1-1 and yrb1-2 immunoblotting using a rabbit polyclonal anti-Yrb1 antiserum
(Schlenstedtet al.1995), alter residues (F187 and L93,
(no. 1390). A band that cross-reacts nonspecifically with the
respectively) that project into the same hydrophobic
anti-Yrb1 antiserum served as a loading control. The asterisk
pocket as A91. In contrast, R127K makes a seemingly
indicates a major degradation product of Yrb1. (B) Morphol-ogy of homozygous diploidyrb1tscells. Strains YPH501 (YRB1/
modest change in a surface-exposed residue that forms
YRB1), JTY2482 (yrb1-51/yrb1-51), and JTY2485 (yrb1-52/yrb1- a bridge to residues in the long C-terminal “arm” of 52) were cultivated on YPGlc plates at 16⬚or 30⬚, as indicated,
Ran that embraces the RBD (Figure 2B), an alteration
and viewed by Nomarski optics.
unlikely to disrupt the overall structure, consistent with the observed stability of the mutant protein at restrictive temperature (Figure 1A). We confirmed that each
muta-fect in srp1-31ts/srp1-31ts diploids (data not shown).
Srp1/␣-importin is the adaptor necessary for recogni- tion was both necessary and sufficient to confer the ts
phenotype of the corresponding allele by inserting each tion and nuclear import of proteins that contain a
clas-sical nuclear localization signal (NLS) by the Kap95/ mutant DNA into a plasmid and introducing it into a
yrb1⌬background (seematerials and methods).
-importin receptor (Enenkelet al.1995). At 37⬚,
srp1-31cells are impaired in import of NLS-containing re- Two independent approaches demonstrated that the
yrb1-51and yrb1-52 mutations interfere with Yrb1-Ran porter proteins and arrest uniformly as large-budded
cells indicative of a defect in mitosis (Loebet al.1995). (Gsp1) interactionin vivo.The GTPase-deficient form
of Gsp1, Gsp1(G21V), binds more strongly to Yrb1 than Correspondingly, degradation of Clb2, whose
destruc-tion is required for exit from mitosis, is impaired in normal Gsp1 (Schlenstedtet al.1995); hence, we used
Gsp1(G21V) in our analyses. First, we applied the two-srp1-31cells (Loebet al.1995); likewise, degradation of
Clb2 and of two anaphase inhibitors, Pds1 and Sic1, is hybrid method using full-length wild-type Yrb1, Yrb1
(A91D), or Yrb1(R127K) fused to the Gal4
transcrip-also impaired inyrb1-51cells (Ba¨umeret al.2000).
De-spite these similarities, the absence of a uniform cell tional activation domain [Gal4(TAD)] and full-length
Gsp1(G21V) fused to the LexA DNA-binding domain
cycle arrest phenotype distinguishes theyrb1-51and
chromo-TABLE 4
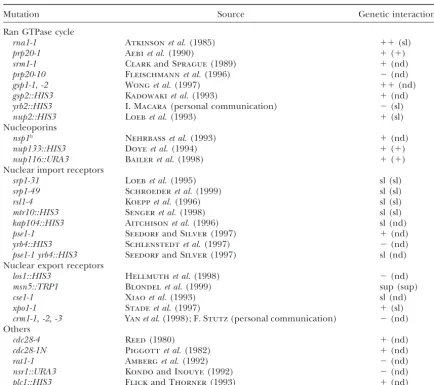
Summary of genetic interactions betweenyrb1-51(andyrb1-52) and nucleocytoplasmic transport factors
Mutation Source Genetic interactiona
Ran GTPase cycle
rna1-1 Atkinsonet al.(1985) ⫹⫹(sl)
prp20-1 Aebiet al.(1990) ⫹(⫹)
srm1-1 ClarkandSprague(1989) ⫹(nd)
prp20-10 Fleischmannet al.(1996) ⫺(nd)
gsp1-1, -2 Wonget al.(1997) ⫹⫹(nd)
gsp2::HIS3 Kadowakiet al.(1993) ⫹(nd)
yrb2::HIS3 I. Macara(personal communication) ⫺(sl)
nup2::HIS3 Loebet al.(1993) ⫹(sl)
Nucleoporins
nsp1ts Nehrbasset al.(1993) ⫹(nd)
nup133::HIS3 Doyeet al.(1994) ⫹(⫹)
nup116::URA3 Baileret al.(1998) ⫹(⫹)
Nuclear import receptors
srp1-31 Loebet al.(1995) sl (sl)
srp1-49 Schroederet al.(1999) sl (sl)
rsl1-4 Koeppet al.(1996) sl (sl)
mtr10::HIS3 Sengeret al.(1998) sl (sl)
kap104::HIS3 Aitchisonet al.(1996) sl (nd)
pse1-1 SeedorfandSilver(1997) ⫹(nd)
yrb4::HIS3 Schlenstedtet al.(1997) ⫺(nd)
pse1-1 yrb4::HIS3 SeedorfandSilver(1997) sl (nd)
Nuclear export receptors
los1::HIS3 Hellmuthet al.(1998) ⫺(nd)
msn5::TRP1 Blondelet al.(1999) sup (sup)
cse1-1 Xiaoet al.(1993) sl (nd)
xpo1-1 Stadeet al.(1997) ⫹(sl)
crm1-1, -2, -3 Yanet al.(1998);F. Stutz(personal communication) ⫺(nd)
Others
cdc28-4 Reed(1980) ⫹(nd)
cdc28-1N Piggottet al.(1982) ⫹(nd)
rat1-1 Amberget al.(1992) ⫺(nd)
nsr1::URA3 KondoandInouye(1992) ⫺(nd)
plc1::HIS3 FlickandThorner(1993) ⫹(nd)
Abbreviations in parentheses indicate phenotype observed with theyrb1-52allele.
a⫺, no synthetic growth phenotype;⫹, synthetic growth defect (see text for details); sl, synthetic lethality; nd, not determined; sup, no synthetic growth defect but extragenic suppression of mating defect (see Table 5).
somally inserted copy of E. coli lacZunder control of muthet al.(1998), eluted by cleavage with TEV-protease
(seematerials and methods), and analyzed by SDS-eight LexA-operator sites. In this system, Yrb1(A91D)
showed a reproducible reduction (⬎3-fold) and Yrb1 PAGE, Coomassie blue staining, and immunoblotting
using a polyclonal anti-Yrb1 antisera (no. 1391).
Exami-(R127K) showed a dramatic reduction (⬎50-fold) in
interaction with Gsp1(G21V) compared to wild-type nation of the input material (Figure 3B, load), and
flow-through fractions (data not shown), demonstrated that Yrb1 (Figure 3A). Immunoblotting with anti-Yrb1
antise-rum (seematerials and methods) and anti-Gsp1 anti- expression of normal and mutant Yrb1-GFP fusions was
comparable, as were their stabilities during purification. bodies (gift of P. Belhumeur) showed that all constructs
were expressed at equivalent levels (data not shown). No detectable Yrb1(R127K)-GFP copurified with Gsp1
(G21V), whereas significant amounts of both wild-type These results were confirmed by a biochemical
pro-cedure (Figure 3B). Yrb1-GFP, Yrb1(A91D)-GFP, or Yrb1-GFP and endogenous Yrb1 were retained by the
same beads (Figure 3B). A detectable amount of Yrb1
Yrb1(R127K)-GFP, produced from the authenticYRB1
promoter onCENplasmids, were expressed in wild-type (A91D)-GFP copurified with ProtA-Gsp1(G21V), but its
level was markedly less than the amount of wild-type cells (strain CRY1) also producing a ProtA-(TEV
site)-Gsp1(G21V) from the constitutiveNOP1promoter on Yrb1-GFP retained under the same conditions (Figure 3B).
yrb1-51 andyrb1-52 mutants are defective in nuclear
aCENplasmid. After growth at 26⬚, protein complexes
bound to bead-immobilized ProtA-(TEV site)-Gsp1(G21V) protein and RNA transport:To determine ifyrb1-51and
yrb1-52cause defects in nuclear protein import and RNA
Hell-Figure 2.—Positions of the altered residues in Yrb1 re-sulting from theyrb1-51andyrb1-52mutations. (A) Alignment of various Ran-binding domains (RBDs). Sequences shown are grouped into three subfamilies (RanBP1, RanBP2, and RanBP3), on the basis of certain shared sequence characteris-tics, and include the following (with GenBank accession num-bers): S. cerevisiae Yrb1 (L38489), Schizosaccharomyces pombe Sbp1 (D86381), mouse RanBP1 (X56045), human RanBP1 (X83617),Xenopus laevisRanBP1 (U09128),Arabidopsis thali-anaRanBP1 (U62742), mouse RanBP2 nucleoporin (X87337), human NUP358 (D38076), Bos taurus RanBP2 nucleoporin (L41691),Caenorhabditis elegansRanup96 (Z34801),S. cerevisiae Nup2 (X69964),S. pombeHba1 (U38783), and human RanBP3 (Y08697). Sequence ofS. cerevisiae Yrb2/Nup36 is from the Swiss Protein Database (accession no. P40517). The mouse and bovine RanBP2 are incomplete because they are derived from partial cDNA clones. An insert of 24 residues (possibly an intron) was omitted from the actualC. elegans Ranup96 sequence to optimize its alignment to the other RBDs. Identi-ties shared by 11 (or more) of the RBDs shown are indicated by white-on-black letters; chemically similar residues are shown as black-on-grey letters. The positions mutated in yrb1-51, A91A, and inyrb1-52, R127K, are indicated at the top. (B) Positions of the residues (A91 and R127) altered in the yrb1-51andyrb1-52mutants, respectively, have been modeled on the first RBD (RanBD1) in human NUP358 complexed with Ran bound to a nonhydrolyzable GTP analog (Vetteret al. 1999). Blue, Yrb1; purple, Ran; and red, GTP analog.
export, as observed before for theyrb1-1 andyrb1-2al- the cultures were then split into two equal portions, one
of which was maintained at 23⬚ and the other shifted
leles (Schlenstedtet al.1995), we first examined the
distribution of poly(A)⫹-RNA by in situ hybridization. to 37⬚. Samples were withdrawn at various times for
analysis. Results were more readily visualized in diploid Mutant or wild-type control cells were grown to
find-ings were made with haploid cells (data not shown). In
homozygousyrb1-51/yrb1-51diploids shifted to 37⬚for
2 hr, there was a rapid and clear-cut nuclear
accumula-tion of poly(A)⫹-RNA in every cell (Figure 4A); this
effect was readily apparent in ⵑ50% of the cells even
1 hr after the shift (data not shown). Poly(A)⫹ RNA
accumulated in the nucleus in a distinctly punctate pat-tern, a feature seen in mutants that have a strong RNA
export defect, such asmex67-5(Segrefet al.1997).
How-ever, onset of the RNA export defect in the yrb1-51/
yrb1-51 cells was slower than that in mex67-5 mutants and nuclear RNA accumulation was not as complete
(some cytosolic poly(A)⫹RNA signal remains even 2 hr
after shift to 37⬚). In striking contrast, yrb1-52/yrb1-52
diploids did not show any accumulation of poly(A)⫹
RNA (even 5 hr after shift to 37⬚), just like the wild-type
control (Figure 4A). Althoughyrb1-51cells manifested a
clear defect in RNA export, neitheryrb1-51mutants nor
yrb1-52mutants had any detectable effect on the nuclear export of proteins containing a leucine-rich NES, such
as SV40NLS-PKINES-GFP (Stadeet al.1997) or yAP1-GFP
(Yanet al.1998; data not shown).
To monitor the effect of theyrb1-51andyrb1-52
muta-tions on nuclear protein import, four different GFP fusions of nuclear proteins were examined. To assess
the␣-importin/Srp1 and
-importin/Kap95/Rsl1-depen-dent pathway, two chimeras containing the SV40 NLS
were used: a galactose-inducible SV40NLS-GFP-
-galactos-idase, which is so large it cannot diffuse out of the
nucleus after it has been delivered there (Lee et al.
1996), and a constitutively expressed SV40NLS-Gal4TAD
-GFP, which, due to its small size, can diffuse out of the nucleus unless ongoing import occurs continuously Figure3.—Yrb1(A91D) (yrb1-51) and Yrb1(R127K) (
yrb1-(Ku¨ nzler and Hurt1998). The third reporter was a
52) bind Gsp1(G21V) with reduced affinityin vivo.(A)
Interac-fusion of GFP to Npl3, an mRNA-binding protein, whose
tion between wild-type Yrb1, Yrb1(A91D), or Yrb1(R127K) and
Gsp1(G21V) was determined using the two-hybrid method as nuclear entry depends on the importin, Kap111/Mtr10 described inmaterials and methods.In brief, Yrb1 proteins (Sengeret al.1998). GFP-Npl3 accumulates rapidly in were fused to the Gal4(TAD), and Gsp1(G21V) was fused to
the cytoplasm if import is impaired because Npl3
contin-the LexA(DBD). In contin-the recipient strain ( JTY2500), contin-theE. coli
uously shuttles between the nucleus and the cytosol.
lacZgene is under control of eight LexA-operator elements.
The fourth transport substrate was constitutively
ex--Galactosidase activity is expressed in arbitrary units. Each
value represents the average of single measurements made on pressed and composed of GFP fused to the NLS of three independent transformants; error bars indicate standard ribosomal protein L25 (O. Gadal and E. Hurt, per-deviation of the mean. (B) Binding of Yrb1 to Gsp(G12V)
sonal communication). Transport of L25NLS-GFP into
was assessed by copurification. Cultures of strain CRY1 (YRB1
the nucleus utilizes two different import receptors,
GSP1) carrying CEN plasmids expressing a
ProtA-TEV-Kap121/Pse1 and Kap123/Yrb4 (Schlenstedt et al.
Gsp1(G21V) fusion from theNOP1promoter and either
wild-type Yrb1-GFP, Yrb1(A91D)-GFP, or Yrb1(R127K)-GFP ex- 1997). Like SV40NLS-Gal4TAD-GFP, L25NLS-GFP is small pressed from the authentic YRB1 promoter were grown in enough to diffuse out of the nucleus unless its import selective SCGlc medium at 26⬚. Extracts were prepared and
occurs continuously.
the ProtA-TEV-Gsp1(G21V) was purified on IgG-Sepharose
Control strains oryrb1-51andyrb1-52mutants carrying
(Pharmacia, Uppsala, Sweden) and eluted by digestion with
the reporter plasmids described above were cultivated
recombinant TEV protease (GIBCO-BRL, Gaithersburg, MD).
Equal fractions of the load and the eluate of each column in selective medium, shifted to restrictive temperature, were resolved by SDS-PAGE and analyzed, as indicated, by and examined by fluorescence microscopy. For the in-Coomassie blue staining and immunoblotting using a rabbit
ducible reporter, transformants were grown to
midexpo-polyclonal anti-Yrb1 antiserum (no. 1391). Endogenous Yrb1
nential phase in Raf-containing medium at permissive
served as a control to confirm equivalent loading and
function-temperature and induced for 1 hr by addition of 2%
ality of the immobilized Gsp1(G21V).
Gal (to allow for mRNA synthesis and export) before
Figure4.—Nuclear transport defects inyrb1tsmutants. (A) To examine poly(A)⫹RNA export, cultures of homozygous diploid strains YPH501 (YRB1/YRB1), JTY2482 (yrb1-51/ yrb1-51), and JTY2485 (yrb1-52/yrb1-52) were grown in YPGlc at 23⬚to early exponential phase, split into two equal portions, and incubated for another 2 hr either at 23⬚ or 37⬚. After fixation with formaldehyde, cells were stained with the DNA dye 4⬘,6-diamidino-2-phenylindole, analyzed byin situ hybrid-ization using a CY3-labeled oligo(dT) probe to visualize the subcellular distribution of poly(A)⫹RNA, and viewed by fluo-rescence microscopy using appropriate band-pass filters. (B and C) To examine nuclear protein import, the same strains as in A were transformed with multicopy plasmids expressing either SV40NLS-Gal4TAD-GFP (B) or L25NLS-GFP (C), respec-tively, cultivated and shifted as in A, and viewed directly by fluorescence microscopy and Nomarski optics.
significant cytoplasmic accumulation inyrb1-51andyrb1- in haploids and for the other two reporters (data not
shown). Inyrb1-52cells, the defect was noticeable even
52mutants after shift to 37⬚, compared to wild-type cells,
indicating a general defect in nuclear protein import. at permissive temperature. Our results showing a defect
in nuclear protein import inyrb1-52 cells are at odds
Results for SV40NLS-Gal4TAD-GFP (Figure 4B) and L25NLS
-GFP (Figure 4C) reporters in homozygous diploid with a previously published report on the same mutant
To exclude the possibility that cytoplasmic
localiza-tion of the reporter proteins inyrb-51andyrb1-52cells
was due to “leakiness” of the mutant nuclei,
accumula-tion of constitutively expressed SV40NLS-GFP-
-galactosi-dase (encoded by pPS815;Leeet al.1996) was examined
in the same strains before and after temperature shift. In contrast to the short-term assay with the inducible version of the same reporter protein (see above), no increased cytoplasmic GFP signal was observed (data
not shown), demonstrating that the nuclei in the
yrb1-51andyrb1-52mutants were not more “leaky” than wild-type nuclei.
Genetic interactions ofyrb1mutations with nucleocy-toplasmic transport factors:As an independent means
to confirm thatyrb1-51andyrb1-52compromise
nucleo-cytoplasmic trafficking even at permissive temperature,
Figure5.—Genetic interactions betweenyrb1tsalleles and
genetic interactions of these alleles with mutations in
components of the nucleocytoplasmic transport machinery.
genes encoding a variety of other factors involved in yrb1-51 (oryrb1-52) mutant strains were crossed with strains
nucleocytoplasmic transport were examined. Strains carrying a mutation in another gene of interest. The resulting
diploids were subjected to sporulation, and growth of
individ-carrying mutations of interest were crossed with strains
ual spores from tetratype asci was examined at various
temper-carrying the yrb1-51 or yrb1-52 mutation, and the
re-atures. Left, genetic interaction ofyrb1-51withnup2::HIS3is
sulting diploids were sporulated. Double mutant segre- manifested by the extremely poor growth (“⫹” in Table 4) of
gants from tetratype asci were compared to each single theyrb1-51 nup2::HIS3double mutant at 28⬚, a temperature
mutant segregant and to the wild-type segregant for clearly permissive for the congenic yrb1-51 and nup2::HIS3
single mutants (yrb1-51alone has a restrictive temperature of
their ability to grow at various temperatures (see Figure
31⬚under these conditions andnup2::HIS3alone has no
obvi-5). Mutations tested included alterations in genes
en-ous growth defect even at higher temperatures). Right, a
yrb1-coding components of the Ran GTPase cycle, nuclear 51 mutant carrying a URA3-marked multicopy plasmid
ex-import and export receptors, and nucleoporins (see Table pressing wild-typeYRB1(pMK103) was crossed with a strain
4). Theplc1⌬::HIS3mutation (FlickandThorner1993) carrying a ts mutation,srp1-31, in␣-importin. The resulting
diploid was sporulated and individual spores from a tetratype
was tested since there is evidence for a role of
PLC1-ascus were streaked on medium containing 5-FOA to
count-encoded phosphatidylinositol-specific phospholipase C
erselect against the plasmid. Genetic interaction of yrb1-51
in mRNA export (York et al. 1999; J. Flick,personal
withsrp1-31is manifested by the inviability or “synthetic
lethal-communication). Theyrb1-51mutation displayed dele- ity” (“sl” in Table 4) of theyrb1-51 srp1-31double mutant at
terious genetic interactions with many of these other any temperature, whereas the congenic yrb1-51 and srp1-31
single mutants are able to grow at permissive temperatures
classes of mutants that affect nucleocytoplasmic
trans-(here shown at 26⬚). Other double mutant combinations
port (Table 4). Combination of the yrb1-52 mutation
tested and their phenotype are listed in Table 4.
with at least 12 of these mutations revealed essentially
the same growth defects. For xpo1-1 yrb1-52 and
yrb2⌬::HIS3 yrb1-52, the growth defect was even more ing, presumably, the ability of these mutations to
sup-severe than for the corresponding double mutant with press mating defects. Far1 and Ste5 are currently the
yrb1-51 (Table 4). Equally pronounced growth defects only components of the mating pheromone response
were observed when yrb1-51was combined with muta- pathway known to shuttle between nucleus and
cyto-tions in genes encoding certain nuclear transport recep- plasm (Blondelet al.1999;Mahantyet al.1999) and
tors (Table 4). In all these cases, the double mutant was yrb1-52 was isolated as an extragenic suppressor of a
not viable at any temperature (synthetic lethality); for ste5 missense mutation (Katz et al. 1987). Hence, we
the other genetic interactions observed, double mutants constructed a functional GFP-Ste5 chimera and
exam-were viable at the permissive temperature (23⬚) but re- ined its subcellular localization in yrb1-51 and yrb1-52
vealed a restrictive temperature that was considerably mutants (and in control cells) in the absence of
phero-(⬎3⬚;⫹⫹) or slightly (ⱕ3⬚;⫹) lower than the one of mone to avoid the complications of signal-induced
any of the two single mutants (synthetic growth defect). changes. In wild-type cells at steady state, GFP-Ste5
accu-Thus, the functions of Yrb1(A91D) and Yrb1(R127K) mulated in the nucleus, even though cytoplasmic
stain-are at least partially defective even under permissive ing was also evident (Figure 6), as observed before
conditions. (Mahantyet al. 1999;Pryciak andHuntress1998).
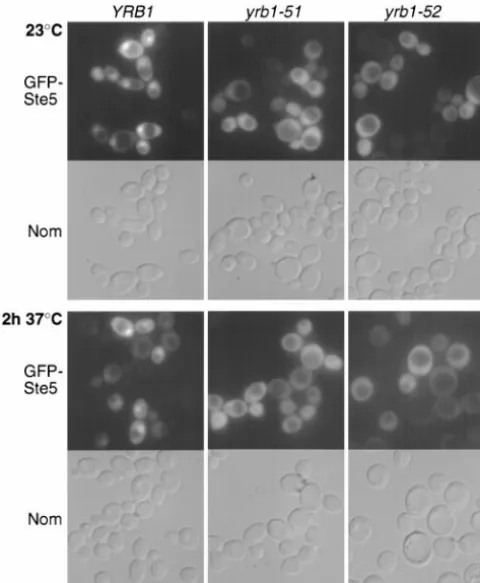
Nucleocytoplasmic trafficking of Ste5 is altered in In contrast, no nuclear accumulation was observed in
yrb1-51 and yrb1-52 cells: Our results indicate that im- either of the twoyrb1tsmutants, even at permissive
tem-paired nucleocytoplasmic transport is the primary cause perature (Figure 6). Since Ste5 shuttles continuously
between nucleus and cytoplasm (Mahantyet al.1999),
includ-TABLE 5
Suppression of the partial mating defect of a msn5⌬ null mutant by theyrb1-51andyrb1-52mutations
Genotypea Mating efficiency (%)b
MSN5 YRB1 92.3⫾10
msn5⌬YRB1 20⫾12
MSN5 yrb1-51 96.6⫾4.7
msn5⌬yrb1-51 89⫾12.2
MSN5 YRB1 67.3⫾18.7
msn5⌬YRB1 18.3⫾2.8
MSN5 yrb1-52 89.3⫾8.9
msn5⌬yrb1-52 70.6⫾24.5
aMATaspores of the indicated genotype from a cross be-tween strains HMK30 (msn5⌬), and JTY2027 (yrb1-51) and JTY2484 (yrb1-52), respectively, were assayed for quantitative mating with the MAT␣ tester strain DC17 as described in
materials and methods.
bMating efficiencies are the ratio of the number of diploids formed to the number of viable inputMATahaploids, are the average of three independent trials, and are given as percent-age together with the standard deviations of the mean.
DISCUSSION
Ran GTPase is one of the most highly conserved pro-Figure6.—Localization of GFP-Ste5 inyrb1tsmutants.
Hap-teins in nucleated cells (Macaraet al.2000;Sazerand
loid strains YPH499 (YRB1), JTY2026 (yrb1-51), and JTY2483 Dasso2000). Ran action has been implicated in nucleo-(yrb1-52) expressing a GFP-Ste5 fusion protein from a cytoplasmic transport, microtubule assembly, nuclear multicopy plasmid were cultivated, shifted, and viewed as
de-envelope formation, maintenance of chromatin
struc-scribed in the legend to Figure 4, B and C.
ture and nuclear (and nucleolar) organization, chromo-some segregation, DNA replication, RNA metabolism, and cell cycle progression. It is still a matter of some the observed mislocalization in cells with defective Yrb1
could be due, in principle, either to inhibition of Ste5 debate whether the pleiotropic phenotypes of
alter-ations in the Ran GTPase cycle are due solely to the import into the nucleus or enhancement of Ste5 export
from the nucleus. Since we have demonstrated thatyrb1- established role of Ran in nucleocytoplasmic transport
(Go¨ rlichandKutay1999) or whether Ran has addi-51andyrb1-52clearly impede nuclear import of various
reporter proteins, the former possibility seems more tional roles in the cell. Recent studies using in vitro
systems have implicated Ran function directly in micro-likely.
To obtain further evidence that suppression of mating tubule organization (Kahana and Cleveland 1999)
and nuclear envelope formation (Hetzer et al. 2000;
defects byyrb1-51 and yrb1-52arises from impairment
of nuclear import of Ste5, we tested whether theseyrb1ts ZhangandClarke2000). Our evidence indicates that
the Ran GTPase cycle is linked to the signaling events
mutations could suppress the mating defect of amsn5⌬
mutant. Msn5 is thought to be the nuclear receptor required for mating as a consequence of the role of
Ran in nucleocytoplasmic transport. required for pheromone-stimulated export of Ste5 from
the nucleus (Mahanty et al.1999). Eachyrb1tsmutant A decade ago, before the function of the essential Ran
regulator, RanGEF1, was fully appreciated, a ts mutation
was crossed against a msn5⌬ strain and the wild-type
single mutant, and double mutant spores from the re- (srm1-1) in its yeast homolog (SRM1/PRP20/MTR1) was
isolated as a suppressor of the mating defect of haploid sulting tetratype asci were tested for their relative mating
proficiency using a quantitative mating assay performed cells lacking pheromone receptors (ClarkandSprague
1989). Unlike other ts mutations in yeast RanGEF1
iden-at semipermissive temperiden-ature (30⬚) for theyrb1tsalleles.
Both theyrb1-51andyrb1-52mutations restored the mat- tified subsequently (Amberg et al. 1993), srm1-1 does
not cause dramatic nuclear accumulation of poly(A)⫹
ing efficiency of the msn5⌬ mutant to essentially the
wild-type level (Table 5). This suppression is consistent RNA at restrictive temperature (Kadowakiet al.1993;
and its effect on nuclear protein import was not exam-with the idea that a higher cytoplasmic pool of Ste5 (due
to its inefficient nuclear import in theyrb1ts mutants) ined), which left open the possibility that the Ran
GTPase cycle had some role in mating distinct from compensates for its inefficient pheromone-stimulated