organic papers
o2992
Silvaet al. C9H6N2O3 doi:10.1107/S1600536805025894 Acta Cryst.(2005). E61, o2992–o2993
Acta Crystallographica Section E Structure Reports Online
ISSN 1600-5368
6-Nitroquinolin-2(1
H
)-one
Luiz Everson da Silva,a,b Antonio C. Joussef,aSabine Forob* and Boris Schmidtb
aDepartamento de Quı´mica–UFSC, 88040-900
Floriano´polis, SC, Brazil, andb
Clemens–Scho¨pf-Institut fu¨r Organische Chemie und Biochemie, Technische Universita¨t Darmstadt,
Petersenstrasse 22, D-64287 Darmstadt, Germany
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study T= 299 K
Mean(C–C) = 0.004 A˚ Rfactor = 0.055 wRfactor = 0.149
Data-to-parameter ratio = 10.3
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2005 International Union of Crystallography
Printed in Great Britain – all rights reserved
The title compound, C9H6N2O3, is nearly planar, with maximum deviations from the mean plane of 0.024 (3) A˚ for the C atomparato the ring N atom and 0.048 (2) A˚ for one of the nitro O atoms. Three C—H O and one N—H O intermolecular hydrogen bonds stabilize the crystal structure.
Comment
As part of our ongoing studies on quinoline derivatives as fluorophores, we attempted to prepare, by treatment of 2-chloroquinoline with potassium nitrate and sulfuric acid, the corresponding nitroquinoline derivatives (I) and (II) (Kimber
et al., 2003). However, analytical data from the reaction mixture indicated, besides (I) and (II) as the main products, the presence of another compound whose structure could not be resolved unequivocally by spectroscopic methods. This minor product is the title compound, (III); in order to identify the structure of (III), and hence to help in clarifying the reaction mechanism, its crystal structure was determined by X-ray diffraction.
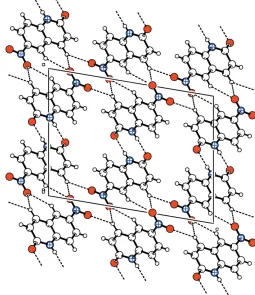
Four intermolecular hydrogen bonds, viz. one N—H O and three C—H O, form a three-dimensional network (Fig. 2). Details of the hydrogen-bonding geometry are given in Table 1. The title compound is nearly planar, with maximum deviations from the mean plane of 0.024 (3) A˚ for C3 and 0.048 (2) A˚ for O2.
Experimental
The title compound, (III), was obtained as a minor product from a mixture of 2-chloroquinoline, potassium nitrate and sulfuric acid, according to the literature procedure of Kimberet al.(2003). Crystals suitable for X-ray data collection were obtained by recrystallization from dichloromethane–hexane (1:1).
Crystal data
C9H6N2O3
Mr= 190.16 Monoclinic,P21=c a= 11.470 (2) A˚
b= 4.8880 (6) A˚
c= 15.065 (2) A˚
= 99.53 (1)
V= 833.0 (2) A˚3
Z= 4
Dx= 1.516 Mg m 3 CuKradiation Cell parameters from 25
reflections
= 6.6–26.7
= 1.00 mm1
T= 299 (2) K Prism, orange 0.180.100.05 mm
Data collection
Nonius CAD-4 diffractometer
!/2scans
Absorption correction: none 1941 measured reflections 1504 independent reflections 892 reflections withI> 2(I)
Rint= 0.069
max= 68.0
h=13!13
k= 0!5
l=18!18 3 standard reflections
frequency: 120 min intensity decay: 1.0%
Refinement
Refinement onF2
R[F2> 2(F2)] = 0.055
wR(F2) = 0.149
S= 1.01 1504 reflections 146 parameters
Only H-atom coordinates refined
w= 1/[2(F
o2) + (0.0734P)2] whereP= (Fo2+ 2Fc2)/3 (/)max< 0.001
max= 0.29 e A˚ 3 min=0.19 e A˚
3
Extinction correction:SHELXL97
Extinction coefficient: 0.0053 (11)
Table 1
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
C3—H3 O2i
1.04 (3) 2.48 (3) 3.377 (4) 145 (2) C3—H3 O3ii 1.04 (3) 2.50 (3) 3.230 (4) 127 (2) C9—H9 O2i
0.95 (3) 2.53 (3) 3.371 (4) 147 (2) N1—H1N O1iii
0.83 (3) 1.97 (3) 2.794 (3) 175 (3)
Symmetry codes: (i) xþ2;y;zþ1; (ii) x;y1 2;z
1 2; (iii)
xþ1;y2;zþ1.
The H atoms were located in a difference map and refined with
Uiso(H) = 1.2Ueq(parent atom).
Data collection:CAD-4/PC Software (Nonius, 1996); cell refine-ment: CAD-4/PC Software; data reduction: REDU4 (Stoe & Cie, 1987); program(s) used to solve structure: SHELXS97 (Sheldrick, 1997); program(s) used to refine structure:SHELXL97(Sheldrick, 1997); molecular graphics:PLATON(Spek, 2003); software used to prepare material for publication:SHELXL97.
The authors thank Professor Dr Hartmut Fuess, FG Strukturforschung, FB Material- und Geowissenschaften, Technische Universita¨t Darmstadt, Petersenstrasse 23, 64287 Darmstadt, for diffractometer time.
References
Kimber, M. C., Geue, J. P., Lincoln, S. F., Ward, A. D. & Tiekink, E. R. T. (2003).Aust. J. Chem.56, 39–44.
Nonius (1996).CAD-4/PC Software. Version 2.0. Nonius GmbH, Solingen, Germany.
Sheldrick, G. M. (1997). SHELXS97 and SHELXL97. University of Go¨ttingen, Germany.
Spek, A. L. (2003).J. Appl. Cryst.36, 7–13.
[image:2.610.61.278.71.192.2]Stoe & Cie (1987).REDU4. Version 6.2c. Stoe & Cie GmbH, Darmstadt, Germany.
Figure 1
[image:2.610.309.564.75.370.2]The molecular structure of (III), showing the atom labeling and displacement ellipsoids drawn at the 50% probability level.
Figure 2
supporting information
sup-1 Acta Cryst. (2005). E61, o2992–o2993
supporting information
Acta Cryst. (2005). E61, o2992–o2993 [https://doi.org/10.1107/S1600536805025894]
6-Nitroquinolin-2(1
H
)-one
Luiz Everson da Silva, Antonio C. Joussef, Sabine Foro and Boris Schmidt
6-Nitroquinolin-2(1H)-one
Crystal data
C9H6N2O3 Mr = 190.16 Monoclinic, P21/c
Hall symbol: -P 2ybc
a = 11.470 (2) Å
b = 4.8880 (6) Å
c = 15.065 (2) Å
β = 99.53 (1)°
V = 833.0 (2) Å3
Z = 4
F(000) = 392
Dx = 1.516 Mg m−3
Cu Kα radiation, λ = 1.54180 Å
Cell parameters from 25 reflections
θ = 6.6–26.7°
µ = 1.00 mm−1
T = 299 K
Prism, orange
0.18 × 0.10 × 0.05 mm
Data collection
Nonius CAD-4 diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
ω/2θ scans
1941 measured reflections 1504 independent reflections 892 reflections with I > 2σ(I)
Rint = 0.069
θmax = 68.0°, θmin = 3.9°
h = −13→13
k = 0→5
l = −18→18
3 standard reflections every 120 min intensity decay: 1.0%
Refinement
Refinement on F2
Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.055 wR(F2) = 0.149
S = 1.01
1504 reflections 146 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: difference Fourier map Only H-atom coordinates refined
w = 1/[σ2(Fo2) + (0.0734P)2]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001
Δρmax = 0.29 e Å−3
Δρmin = −0.19 e Å−3
Extinction correction: SHELXL97, Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
C1 0.6288 (3) −0.8802 (6) 0.4297 (2) 0.0474 (7)
C2 0.7255 (3) −0.7901 (7) 0.3868 (2) 0.0542 (8)
H2 0.737 (3) −0.891 (6) 0.332 (2) 0.065*
C3 0.8002 (3) −0.5927 (6) 0.4233 (2) 0.0490 (8)
H3 0.865 (3) −0.522 (7) 0.389 (2) 0.059*
C4 0.7856 (2) −0.4635 (6) 0.50588 (19) 0.0414 (7)
C5 0.6906 (2) −0.5469 (5) 0.54806 (19) 0.0408 (7)
C6 0.6712 (3) −0.4285 (6) 0.6279 (2) 0.0477 (8)
H6 0.611 (3) −0.475 (6) 0.659 (2) 0.057*
C7 0.7479 (3) −0.2273 (6) 0.6680 (2) 0.0469 (8)
H7 0.733 (3) −0.144 (6) 0.720 (2) 0.056*
C8 0.8416 (2) −0.1494 (5) 0.62635 (18) 0.0404 (7)
C9 0.8616 (2) −0.2619 (6) 0.5478 (2) 0.0437 (7)
H9 0.925 (3) −0.205 (6) 0.5181 (19) 0.052*
N1 0.6160 (2) −0.7475 (5) 0.50739 (17) 0.0451 (6)
H1N 0.566 (3) −0.799 (6) 0.538 (2) 0.054*
N2 0.9223 (2) 0.0666 (5) 0.66792 (16) 0.0460 (6)
O1 0.55994 (18) −1.0659 (4) 0.39957 (15) 0.0588 (7)
O2 1.0044 (2) 0.1299 (4) 0.63055 (15) 0.0616 (7)
O3 0.9011 (2) 0.1710 (4) 0.73709 (15) 0.0611 (7)
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
C1 0.0494 (17) 0.0431 (17) 0.0507 (18) −0.0013 (15) 0.0110 (14) 0.0024 (14)
C2 0.0585 (19) 0.0518 (18) 0.055 (2) −0.0083 (16) 0.0188 (16) −0.0052 (15)
C3 0.0476 (17) 0.0503 (18) 0.0527 (18) −0.0102 (15) 0.0192 (14) 0.0013 (14)
C4 0.0421 (15) 0.0367 (14) 0.0463 (16) −0.0019 (13) 0.0098 (12) 0.0048 (12)
C5 0.0398 (14) 0.0351 (15) 0.0483 (16) −0.0022 (13) 0.0099 (12) 0.0082 (12)
C6 0.0459 (17) 0.0478 (18) 0.0525 (18) −0.0021 (15) 0.0177 (14) 0.0043 (14)
C7 0.0503 (18) 0.0455 (16) 0.0466 (18) 0.0015 (15) 0.0127 (14) 0.0011 (14)
C8 0.0431 (15) 0.0354 (14) 0.0432 (16) 0.0010 (13) 0.0090 (12) 0.0092 (12)
C9 0.0419 (16) 0.0431 (16) 0.0486 (17) −0.0026 (13) 0.0150 (14) 0.0044 (13)
N1 0.0406 (13) 0.0469 (14) 0.0499 (15) −0.0084 (12) 0.0138 (11) 0.0031 (11)
supporting information
sup-3 Acta Cryst. (2005). E61, o2992–o2993
O2 0.0647 (14) 0.0614 (14) 0.0622 (14) −0.0222 (12) 0.0210 (11) 0.0003 (11)
O3 0.0682 (15) 0.0575 (14) 0.0590 (15) −0.0083 (12) 0.0151 (12) −0.0132 (11)
Geometric parameters (Å, º)
C1—O1 1.239 (3) C6—C7 1.389 (4)
C1—N1 1.366 (4) C6—H6 0.92 (3)
C1—C2 1.442 (4) C7—C8 1.385 (4)
C2—C3 1.347 (4) C7—H7 0.93 (3)
C2—H2 0.99 (3) C8—C9 1.358 (4)
C3—C4 1.429 (4) C8—N2 1.474 (3)
C3—H3 1.04 (3) C9—H9 0.95 (3)
C4—C9 1.396 (4) N1—H1N 0.83 (3)
C4—C5 1.409 (3) N2—O2 1.214 (3)
C5—N1 1.377 (4) N2—O3 1.220 (3)
C5—C6 1.386 (4)
O1—C1—N1 120.6 (3) C7—C6—H6 115 (2)
O1—C1—C2 123.3 (3) C8—C7—C6 118.8 (3)
N1—C1—C2 116.0 (3) C8—C7—H7 122.1 (18)
C3—C2—C1 121.3 (3) C6—C7—H7 119.1 (18)
C3—C2—H2 122.0 (18) C9—C8—C7 122.6 (3)
C1—C2—H2 116.5 (18) C9—C8—N2 118.4 (2)
C2—C3—C4 120.9 (3) C7—C8—N2 119.0 (3)
C2—C3—H3 119.8 (18) C8—C9—C4 119.8 (3)
C4—C3—H3 119.1 (18) C8—C9—H9 122.8 (18)
C9—C4—C5 118.1 (3) C4—C9—H9 117.3 (18)
C9—C4—C3 123.5 (3) C1—N1—C5 124.9 (2)
C5—C4—C3 118.4 (3) C1—N1—H1N 121 (2)
N1—C5—C6 120.4 (3) C5—N1—H1N 114 (2)
N1—C5—C4 118.4 (3) O2—N2—O3 124.7 (3)
C6—C5—C4 121.2 (3) O2—N2—C8 117.8 (2)
C5—C6—C7 119.5 (3) O3—N2—C8 117.5 (2)
C5—C6—H6 125 (2)
O1—C1—C2—C3 −178.1 (3) C6—C7—C8—N2 179.2 (3)
N1—C1—C2—C3 2.0 (5) C7—C8—C9—C4 0.3 (4)
C1—C2—C3—C4 −0.5 (5) N2—C8—C9—C4 −178.9 (2)
C2—C3—C4—C9 178.7 (3) C5—C4—C9—C8 −1.0 (4)
C2—C3—C4—C5 −0.3 (5) C3—C4—C9—C8 −180.0 (3)
C9—C4—C5—N1 −179.4 (2) O1—C1—N1—C5 177.2 (2)
C3—C4—C5—N1 −0.4 (4) C2—C1—N1—C5 −2.8 (4)
C9—C4—C5—C6 1.4 (4) C6—C5—N1—C1 −178.7 (3)
C3—C4—C5—C6 −179.6 (3) C4—C5—N1—C1 2.1 (4)
N1—C5—C6—C7 179.7 (3) C9—C8—N2—O2 −1.3 (4)
C4—C5—C6—C7 −1.1 (5) C7—C8—N2—O2 179.4 (3)
C5—C6—C7—C8 0.4 (4) C9—C8—N2—O3 178.0 (3)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
C3—H3···O2i 1.04 (3) 2.48 (3) 3.377 (4) 145 (2)
C3—H3···O3ii 1.04 (3) 2.50 (3) 3.230 (4) 127 (2)
C9—H9···O2i 0.95 (3) 2.53 (3) 3.371 (4) 147 (2)
N1—H1N···O1iii 0.83 (3) 1.97 (3) 2.794 (3) 175 (3)