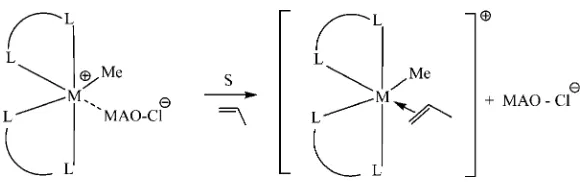Catalyzed by Octahedral Complexes
VICTORIA VOLKIS, ELENA SMOLENSKY, ANATOLI LISOVSKII, MORIS S. EISEN Department of Chemistry and Institute of Catalysis Science and Technology, Technion–Israel Institute of Technology, Haifa, 32000 Israel
Received 27 February 2005; accepted 4 May 2005 DOI: 10.1002/pola.20915
Published online in Wiley InterScience (www.interscience.wiley.com).
ABSTRACT: The effect of solvents (toluene, dichloromethane, and hexane) was studied on the polymerization of propylene with the octahedral complexes bis(trimethylsilyl) benzamidinate titanium dichloride(a), bis(acetylacetonate) titanium dichloride(b), and bis(diethylamino) titanium di-2-(diphenylphosphanylamino)pyridine as catalytic precursors and methylalumoxane as the cocatalyst. For comparison, the polymeriza-tion was also performed in plain liquid propylene without the addipolymeriza-tion of any solvent. The obtained polymers were fractionated by refluxing hexane. The activity of the complexes and the molecular weights and tacticities of the whole polymers and their different fractions were the studied parameters. VVC 2005 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 43: 4505–4516, 2005
Keywords: MAO; polymerization of propylene; solvent effect; titanium complexes
INTRODUCTION
During the last 2 decades, much academic and industrial interest has been focused on the poly-merization of a-olefins promoted by organome-tallic complexes. The understanding of the rela-tionships between the properties of the obtained polymers and the catalyst structure, activity, and selectivity has been the momentum of the research.1 For metallocenes of groups 3 and 4, which are activated by either methylalumoxane (MAO) or perfluoro aromatic boron cocatalysts, a high level of sophistication has been obtained with respect to the stereoregularities, polydis-persities of the polymers, and control over the polymeric architecture.2 In general, the struc-ture of the ancillary ligation affects electroni-cally and sterielectroni-cally the properties of the cata-lysts, influencing their activity and
stereoselec-tivity in the polymerization. Thus, complexes withC2 orC1 symmetry are expected to produce isotactic polyolefins, whereas complexes with
C2v symmetry are known to induce the forma-tion of atactic polymers.3 Although the micro-structure of the polypropylene obtained in the presence of a well-defined single-site catalyst has been correlated to the symmetry of the active organometallic complex, some complexes have been found not to follow such expectations and have produced distinctive polymeric materi-als, providing new, spectacular insights into the mechanism for the polymerization of propylene.4 In the polymerization ofa-olefins catalyzed by organometallic compounds, the activity of the complexes and the molecular weight and tacticity of the polymers are the most relevant parameters for describing the performance of the catalytic system. The activity of the complexes is charac-terized by the total amount of the polymer obtained per amount of the active catalyst species as a function of time. The molecular weight of the polymers is determined by the relative rates of
Correspondence to: M. S. Eisen (E-mail: chmoris@tx.
technion.ac.il)
Journal of Polymer Science: Part A: Polymer Chemistry, Vol. 43, 4505–4516 (2005) V
VC2005 Wiley Periodicals, Inc.
the propagation and termination processes, whereas the tacticity of the polymers is influenced by the stereochemistry of the olefin insertion and the relative position of the growing polymeric chain to the monomer substituent.5 The tacticity of the polymer is also affected by any misinser-tions (2,1- and 3,1-insermisinser-tions) and/or epimeriza-tion of the last inserted monomeric unit.6
Besides the listing parameters, one of the fac-tors strongly influencing the polymerization pro-cess is the nature of the solvents used to prepare the catalytic mixture. It has been shown that the nature of the solvent, particularly the solvent polarity, remarkably affects the activity of the metallocene systems and the properties of the poly-mers.7 For example, a linear increase in the rate of the syndiotactic propylene polymerization with thei-Pr(CpFluo)ZrCl2/MAO catalytic system was observed with an increase in the content of di-chloromethane in a mixture with toluene.7(a) However, in dichloromethane, the syndiospecific-ity of the catalyst was much lower than that observed in toluene. This result was assumed to be caused by an isomerization of the free zircono-cene species, separated by the polar solvent, via migration of the growing polymer chain before the next monomer insertion.7(a)A second example relates to the homopolymerization of 1-hexene in polar solvents (CH2Cl2 and o-dichlorobenzene) with the aforementioned catalytic system. The use of the polar solvents allowed a drastic reduc-tion of the cocatalyst MAO amount to achieve a maximum activity, but the stereoregularity of the polymer was much lower than those obtained in the less polar solvent toluene.7(b) The effect of solvents with increasing donor character and steric hindrance on the copolymerization of pro-pylene with higher a-olefins promoted by the Et(Ind)2ZrCl2/MAO system was also studied.7(c–f) The activity of the catalyst for the homopolymeri-zation and copolymerihomopolymeri-zation increased with the enhancement of the dielectric constant (e) of the solvents in the order of o-dichlorobenzene > dichloromethane > chlorobenzene > toluene > heptane, without changes in the stereoregularity of the copolymers.7(c)The lowest 1-hexene content in the copolymers observed in toluene was explained in terms of a competition between the nucleophilic medium and the monomer coordina-tion to the active site.7(e–f) In general, several plausible schemes have been suggested for the solvation of active species by nonpolar and non-coordinating (heptane), polar (CH2Cl2 and o -C6H5Cl), and coordinating (C6H5CH3) solvents,
regarding the copolymerization behavior of the catalyst.7(f)
In this communication, we report for the first time a comparison study of the effect of small amounts of solvents (toluene, dichloromethane, and hexane), differing in polarity and coordinat-ing ability, on the high-pressure homopolymeri-zation of propylene catalyzed by bis(trimethyl-silyl)benzamidinate titanium dichloride (com-plex A), bis(acetylacetonate) titanium dichloride (complex B), and bis(diethylamino) titanium di-2-(diphenylphosphanylamino)pyridine (complex
C) as catalytic precursors activated by MAO. The effect of the mixture of the solvents on the activity of the complexes and stereoregularity of the polymers has also been investigated. In addition and for comparison, the polymerization in neat liquid propylene was performed without the addition of the supplementary solvents. The obtained polymers were fractionated via reflux-ing in hexane. The activity of the complexes and the properties of the whole polymers and their different fractions were the studied parameters.
EXPERIMENTAL
Materials and Instruments
con-ducted in deuterated tetrachloroethane at 858C. The tacticities of the resultant polypropylene were determined according to the pentad analysis method.12The molecular weights of the polymers were determined by the GPC method with a Waters-Alliance 2000 instrument with 1,2,4-tri-chlorobenzene as a mobile phase at 1508C.
Synthesis of Bis(diethylamino) Titanium Di-2-(diphenylphosphanylamino)pyridine (Complex C)
Ti(NEt2)4(0.23 g, 6.85103mol)11was added via a syringe under an argon flow to a solution containing 0.34 g (1.22103mol) of the ligand 2-(diphenylphosphanylamino)pyridine10in 10 mL of dry toluene. After the mixture was stirred at 25
8C overnight, the solvent and diethylamine were removed by vacuum. The residue was washed with hexane through a fritted glass to remove the unreacted ligand, and the orange powder of the obtained product was recrystallized at 45 8C from a toluene solution; this yielded 0.45 g (88%) of orange crystals of complexC.
ELEM. ANAL. Calcd. for C42H48P2N6Ti: C, 67.58%; H, 6.48%; N, 11.25%. Found: C, 68.35%; H, 6.32%; N, 10.58%. 1H NMR (200 MHz, tol-uene-d8, d): 0.86 (t, 12H, 3JHH ¼ 7 Hz, CH3), 3.79 (br, 2H, CHHCH3), 4.78 (br, 2H,
CHHCH3), 5.95 [dd, 2H, 3JHH ¼ 4.0 Hz, 3
JHH ¼ 6 Hz, H(b)], 6.13 [dd, 2H, 3JHH ¼ 8.5 Hz, H(d)], 6.54 [dd, 2H,3JHH¼8.5 Hz,3JHH¼6 Hz, H(c)], 7.22 (br, 6H,m,p-aromatic rings), 7.56 (br, 2H,o-aromatic ring), 7.81 (br, 2H, o-aromatic ring), 7.97 [d, 2H,3JHH¼4 Hz, H(a)].
13
C NMR (100 MHz, toluene-d8, d): 13.0 (CH2CH3), 45.7 (CH2CH3), 111.9 (CH, Cb), 112.2 (CH,Cd), 128 (CH, m,p-aromatic rings), 131.8 (CH, C1), 132.0 (CH, C3), 137.5 (CH, Cc), 143.4 (CH, Ca), 168 (C, Ce). 31P NMR (81 MHz, toluene-d8, d): 35.12 (s, PPh2).
Propylene Polymerization
The polymerization experiments were carried out in a 100-mL, stainless steel reactor equipped
with a magnetic stirrer and a thermocouple. The reactor was charged inside a glovebox with 10 mg of the complex and the corresponding amount of MAO to keep the Al/Ti molar ratio at 800. After the introduction of the solvent (e.g., 6 mL of the solvent) via a syringe under an argon flow, the reactor was frozen in liquid nitrogen and pumped down. Liquid propylene (40 mL) was then vacuum transferred into a fro-zen reactor, the valve was closed, and the tem-perature was quickly (5 min) raised to 25 8C. During this heating time, no stirring was per-formed. At the desired temperature, the stirrer was operated, and the reaction was stirred for 4 h at a pressure of 10.2 atm. The reaction mix-ture was then quenched by the exhaustion of the unreacted propylene, followed by the intro-duction of 40 mL of a mixture of acetyl acetone and water. The polymer was filtered, washed with methanol and acetone, and dried in vacuo. The polymerizations were repeated to obtain at least three good reproducible results (<5%).
The possibility of polypropylene formation dur-ing the fast warmdur-ing of the reaction mixture from the temperature of liquid nitrogen to room tem-perature without stirring was found to be negli-gible. When the polymerization reaction was quenched immediately after room temperature (þ258C) was reached, the amount of the polypro-pylene formed was less than 0.3% of the total amount of the polymer obtained at room tempera-ture with normal stirring. In addition, the activ-ities of the studied complexes were not high enough to induce heating of the reaction mixture and, consequently, a change in pressure.
Fractionation of Polypropylene
The polymer samples were fractionated by extraction with refluxing hexane. The polymers (1 g) were packed into a cellulose thimble placed in a Soxhlet extractor. The latter was attached to a round-bottom flask (250 mL) filled with hexane (100 mL). After extraction for 24 h, the solvent was evaporated. Thus, for every polypropylene sample, two fractions [hexane-soluble (HS) and hexane-in[hexane-soluble (HI)] were obtained.
RESULTS AND DISCUSSION
COCH-COCH3)2(B) were prepared by the reaction in tol-uene of TiCl4 with 2 equiv of the bis(trimethyl-silyl)benzamidinate lithium tetramethylethylene-diamine (TMEDA) salt (eq 1)8or the bis(acetylaceto-nate) lithium salt (eq 2), respectively.9The complex Ti(Ph2PNpy)(NEt2)2(C) was prepared by the
reac-tion of 2 equiv of the neutral ligand Ph2PNHpy10 and 1 equiv of the homoleptic tetrakis(diethylami-no)titanium11(eq 3).13These complexes activated by MAO produced active catalytic systems for the poly-merization of propylene, yielding under high mono-mer pressure an elastomono-meric polypropylene:
To shed some light on the conceptual question regarding the effects of different solvents on the polymerization of propylene promoted by octahe-dral complexes, we present here the results ob-tained with three complexes (A, B, and C) dif-fering in their ancillary ligation. The effects of
chain termination, and the number of mono-meric units in each polymer are also presented in Table 1.
We begin the discussion of the polymerization results with a comparison of complexes A and
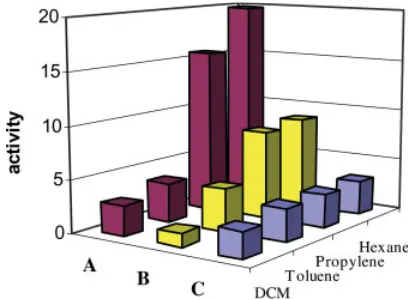
B. As can be seen in Table 1 and Figure 1, for these precatalysts, the polymerization rate in the presence of dichloromethane (e ¼ 8.93) is lower than that in toluene (e ¼ 2.38). At the same time, in both solvents, complexesA andB
are considerably less active in comparison with hexane (e¼1.90) and liquid propylene (e¼1.90). These results are opposite to those in the liter-ature data, according to which the polymerization activity of zirconocenes in a polar solvent (CH2Cl2) is higher than that in toluene, whereas the yields of the polymers in a nonpolar medium (heptane) are considerably lower in comparison with dichloromethane and toluene.7(a–f)
The different characters of the solvent effects on the polymerization of propylene, observed in our experiments and described in the literature, are due to the unique structures of the studied active titanium octahedral complexes, in com-parison with those of the metallocenes, and the polymerization conditions.3,8,9As for the reasons for the inferior polymerization rates observed in dichloromethane and toluene, in comparison
with hexane and neat propylene, let us examine likely interactions of the former solvents with the active cationic centers of the titanium dichloride benzamidinate (A) and acetylaceto-nate (B) complexes.
[image:5.594.67.546.98.269.2]One of the known mechanisms for the inter-action of polar solvents with the organometallic complexes activated by MAO has been proposed to be the separation of the ion couples between the cationic complex and the cocatalyst counter-ion forming a coordinative unsaturated catcounter-ionic
Table 1. Effects of Different Solvents on the Polymerization of Propylene with ComplexesA,B, andCActivated by MAOa
Complexi
A B C
dcm tol hex prop dcm tol hex prop dcm tol hex prop
PPb 1.75 2.31 12.4 9.75 1.64 5.08 11.1 10.6 1.32 1.67 1.74 1.69
Ri1021c 6:27 8:28 44:96 34:95 5:88 18:21 39:79 38:00 4:73 5:98 6:24 0:65
Ri106d 4:31 3:00 22:90 16:97 1:39 1:71 4:40 6:02 0:69 0:87 0:67 0:80
ne 124 198 440 442 331 2244 1281 1231 744 830 694 1154
A104f 2.82 3.72 19.9 15.7 1.30 4.03 8.97 8.42 2.46 3.11 3.25 3.15
Mw103 23 40 50 52 64 264 183 150 122 129 137 160
MWDg 4.4 4.8 2.7 2.8 4.6 2.8 3.4 2.9 3.9 3.7 4.7 3.3
mmmmh 12.3 10.2 9.2 8.1 24.5 20.5 16.0 12.2 9.7 9.3 9.0 8.9
aAmount of the complex
¼10 mg; Al/Ti molar ratio¼ 800; volume of the solvent ¼6 mL; volume of liquid propylene ¼ 40 mL; temperature¼258C; pressure¼10 atm; reaction time¼4 h.
bYield of polypropylene (g). c
Rate of monomer insertion (molecules/h).
dRate of chain termination (mol of polymer/h). e
Number of monomeric units in the polymer.
fActivity (g of PP
mol of Ti1
h1). g
Molecular weight distribution (Mw/Mn). h
mmmmpentad content (%).
idcm
¼CH2Cl2; tol¼toluene; hex¼hexane; prop¼neat liquid propylene.
Figure 1. Effect of the solvent on the activity of
[image:5.594.328.532.531.681.2]complex, which does not strongly interact with the solvent (Scheme 1).7(f),8(a,b)This effect makes the separated cationic species more accessible for the incoming monomer and, in principle, must result in the augmentation of the polymer-ization rate as a result of the better charge sep-aration caused by the solvents with higher polarity. However, as noted, both complexes A
and B were unexpectedly found to be more active in the less polar solvents (Table 1 and Fig. 1).
Hence, another possible effect responsible for the polymerization activity of the complexes may be the coordination of CH2Cl2 and toluene, due to their donor abilities, to the free orbital of the metal obtained in the formation of the cati-onic complex by the transfer of the methide group (CH3) to MAO (Scheme 2).7(f),8(a,b) Such coordination would stabilize the active cationic center, thus decreasing its accessibility and rap-idity for the insertion of the monomer molecules. In comparison with toluene, this effect must be
larger for the more polar dichloromethane, as follows from the data of Table 1.
It can be deduced that the summarized effects of the solvents are determined by both kinds of interactions (Schemes 1 and 2). Apparently, under the studied conditions, precatalystsA and
B are more sensitive to the stabilization action of the polar solvents (dichloromethane and tol-uene) than to the charge separation effect, that resulting in a decrease in the polymerization activity of the complexes. This effect found for the polar solvents may be corroborated by the fact that all the studied complexes were found to be inactive when the polymerizations were performed with the more polar solvent tetrahydrofuran, having a higher coordination capability.
The polymerization of propylene in dichloro-methane, toluene, or hexane was performed with a large excess of liquid monomer with respect to the amounts of the solvents. There-fore, we can assume the coexistence of
solvent-Scheme 1. Plausible separation of the cationic center and the MAO counterion by
the polar solvent (M¼metal; L–L¼ancillary ligation; S¼solvent).
[image:6.594.161.453.70.161.2]separated and propylene-separated ion pairs, in accordance with the literature data.1(a),2(c),14
Noticeably, in the polymerization of propy-lene, larger rates were observed when the reac-tion was carried out in neat monomer (Table 1 and Fig. 1). Clearly, in this case, liquid propy-lene also acts as a solvent, ensuring the forma-tion of the active species without other external interactions hindering the insertion of the in-coming monomer molecules. The high polypropy-lene yields in hexane, similar to those observed in liquid propylene (Table 1 and Fig. 1), are indi-cative of the lack of interactions of the former solvent with the cationic complexes.
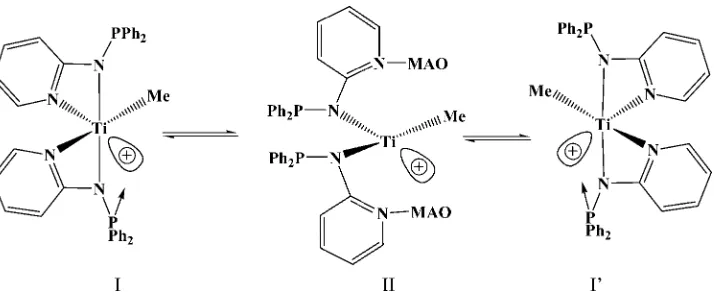
A similar but less pronounced effect of the solvents on the polymerization of propylene was also observed for complexC(Table 1 and Fig. 1). The small effect of the solvents is the result of a plausible existence of complex C in two equili-brium forms with both a closed pseudo C2 sym-metry (I and I0) and an opened pseudo C2v configurations (II), in addition to the internal coordination of the same ancillary ligations (Scheme 3).13
The variation of the molecular weight of the polymers as a function of the nature of the sol-vent is presented in Table 1 and Figure 2. For complexesA and C, the molecular weight of the polymers slightly decreased with increasing sol-vent polarity. For complex B, the lowest molecu-lar weight was obtained when the reaction was carried out in CH2Cl2, whereas in the less polar solvents, polymers with much higher molecular weights were obtained.
To rationalize these results, we must take into account the relationships of the rates for chain propagation and termination for each complex.
The mechanism for the termination of the polymerization corresponding to each of the
complexes has been previously studied. We have found that b-methyl elimination, b-H elimina-tion, and aluminum-transfer mechanisms were the major operative termination pathways for complexes A, B, and C, respectively.8,9,13 The data in Figure 2 allow us to assume that for complex A the effect of the solvent is primarily related to the rate of termination rather than to the rate of propagation. From the rate of propa-gation, we should expect higher molecular weights for the less polar solvents (Fig. 1). How-ever, because the obtained molecular weights are similar for all solvents, the rate of termina-tion must follow the trend hexane > propylene
>toluene dichloromethane to compensate the propagation rate yielding the tendency observed in Figure 2. This assumption is corroborated by the calculated rate of chain termination pre-sented in Table 1. For complex B, a similar
[image:7.594.130.487.72.220.2]Scheme 3. Dynamic behavior of cationic complexCin the presence of MAO.
Figure 2. Dependence of the molecular weight of
trend for the rate of termination also must be operative, though much stronger (for hexane and propylene), besides the more polar CH2Cl2, which induces a low number of insertions. For complex C, similar catalytic activities and weight-average molecular weight (Mw) of the polymers were observed in all solvents; this indicated comparable rates for monomer inser-tion and chain terminainser-tions, as corroborated by the calculated rates presented in Table 1.
Scheme 4 shows the possible interactions of the cationic center with the monomer, polymer chain, and solvents. By comparing plausible rates of solvation of the active complexes with the rate of termination and taking into account that the rate of solvation affects both the rates of propagation and termination, we can explain the dependences shown in Figure 2. For complex
A, the rate of rehybridization of the methyl group (k1 ¼ rate of chain termination) must be faster than the rate of solvation (k2) of the cati-onic center (k1 >k2). This effect increases the b -CH3-elimination rate and, as a result, decreases the molecular weight of the polymers. The net effect of the different solvents can be estimated relatively by the amount of the monomeric units inserted into the polymer chain (Table 1). For complex B, the interaction of the b-hydrogen (centrosymmetric orbital interaction) with the metal (k3) must be much weaker than the inter-action of the strong polar solvent (k3 <k2), thus impeding fast monomer insertions. If this is the only operative mechanism, it will lead to a
de-crease in the molecular weight by increasing the solvent polarity. In addition, the rate of termina-tion will be increased by the reductermina-tion of the polarity of the solvent. Therefore, the higherMw obtained in toluene, in comparison with that obtained in hexane or plain propylene, is the net effect obtained from the competition between the insertion of propylene (high concentration of the monomer-intermolecular process) and chain termination (low concentration of the catalyst-intramolecular process), allowing a large amount of monomer insertions but a small amount of chain eliminations. The same effect is also observed for CH2Cl2, although this solvent impedes much more strongly the monomer insertion, producing low activity as well as low molecular weights. The b-hydrogen interactions have been proved to be extremely important, inducing the first step for the epimerization of the last inserted monomer in the polymerization of olefins by metallocenes, but not causing a major reduction in the molecular weight of the polymers.15 The use of hexane as a solvent with complex Bunexpectedly promotes the formation of a polymer with a slightly higher molecular weight in comparison with plain propylene, and this indicates that the b-H elimination in the former is much slower than that in the plain monomer. This result is related to the lack of stabilization of the cationic center by the hexane solvent, which allows a higher chain of propaga-tion (stabilizapropaga-tion via the monomer) in compari-son with the chain of termination (see Table 1).
For complexC, the rate of the interaction of the polar solvent with the metal (k2) must be compa-rable to or slightly larger than the rate for alu-minum transfer (k4; k2 ˜ k4). For complex C, the molecular weight of the polymer decreased with an increasing amount of MAO (beyond Al/ Ti ¼ 800) when the reaction was carried out in plain propylene, hexane, or toluene. Evidently, the rate of termination is a combined effect of the MAO concentration and its competition toward the stabilization of the cationic center by the solvents.16 Such an effect was not observed for complexesAandB.
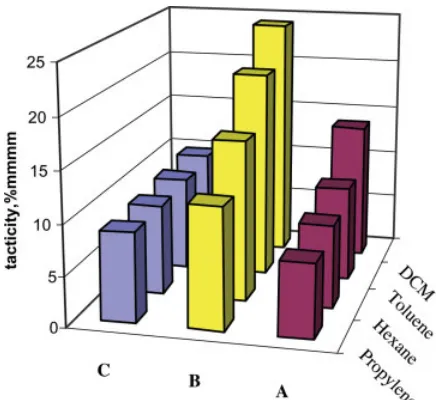
For complexes A and C, the effect of the sol-vents on the tacticity of the polymers seems to be negligible, whereas for precatalystB, themmmm
pentad contents in the polymers increased as the polarity of the solvents increased (Fig. 3). The last result suggests that during the reaction in the presence of CH2Cl2, which is in close interaction with the empty d-orbital of the active cationic complex, the stereoregular dis-crimination of the two olefinic enantiofaces is larger, allowing an easier insertion of the pre-ferred face leading to higher isotacticities, as corroborated by the lowest activity of the system
B/MAO observed in dichloromethane (Table 1 and Fig. 1).17 The isospecificity of complex A is, in general, lower than that for Bbecause of the high amount of misinsertions.12(d) An
independ-ence of the mmmm content in the polymers, obtained with the systemC/MAO, of the polarity of the solvents presumably may be a result of the dynamic behavior of this complex and inter-nal ligand coordination.13
[image:9.594.70.288.72.272.2]The most important question concerns the ability of small amounts of the polar solvents (in the presence of an excess of liquid propylene) to influence drastically the polymerization activity of the complexes and properties of the polymers (vide infra). This remarkable fact can be illus-trated by the results obtained when the poly-merization reaction was performed with the cat-alytic system B/MAO in binary mixtures of CH2Cl2 with toluene or with hexane (Table 2), followed by fractionation of the polymers in the refluxing hexane. The polypropylene obtained in dichloromethane was more soluble in hot hexane than the polymer formed in toluene (entries 1 and 2). In addition, for the polymer obtained in toluene, the isotacticity of the HS fraction was higher than that for the polymer obtained in dichloromethane, whereas the opposite effect was observed for the isotacticity of the HI frac-tion. Increasing the amount of dichloromethane, while the total volume (solvent and liquid propy-lene) was kept constant, did not practically influence the yield of the polypropylene, the polymerization rate, and the molecular weight and isotacticity of the whole polymer (before frac-tionation) and both HS and HI fractions (entries 2 and 3). However, when the reaction was carried out in a mixture of CH2Cl2 and toluene, the activity of the catalytic system and the proper-ties of the polymers were mainly affected by the presence of the polar solvent (CH2Cl2), as ob-served for both soluble and insoluble fractions. Remarkably, this effect is independent of the concentration of the polar solvent in the reaction mixture (cf. entries 5 and 6). These results shed light on the large ability of the polar solvent, even in small amounts in a mixture of solvents, to control the activity of the organometallic com-plex in the polymerization of propylene as well as the properties of the obtained polypropylene. As already mentioned, the highest polypropy-lene yield was reached in hexane (entry 4). Nevertheless, the data obtained in mixtures of hexane and dichloromethane (entry 7) are also comparable to those obtained in dichlorome-thane (entry 2). Hence, the polymerization activ-ity of the complexes is mostly operative by the interaction of minute amounts of the polar sol-vent (dichloromethane) with the active cationic
Figure 3. Dependence of the tacticity of the
center, via their electron-donor/coordinating abilities, almost independently of the presence of additional nonpolar solvent in the reaction mixture.
The results of the propylene polymerization with catalytic system B/MAO, carried out in a mixture of solvents (Table 2), corroborate the aforementioned effect of the solvents on the molecular weight of the polymers. When equal amounts of CH2Cl2 and toluene (3 mL of each) were used, Mw of the polymer (66,000) was not distinct from that of the polymer obtained in CH2Cl2alone (Mw¼ 64,000; entries 2 and 5). In addition, when the polymerization was carried out in a mixture of CH2Cl2 and hexane (3 mL of each), a similar result was obtained (Mw
¼ 68,000), indicating again the large effect caused by the more polar solvent.
The separation of the polypropylene into two fractions, soluble and insoluble in hot hexane, correlates with the wide polydispersity of the polymers obtained. Taking into account that these fractions are produced, at least, by the two different active sites, we can propose that the initial active complexes undergo a specific transformation under the influence of solvents. Because the separated fractions of the polymers strongly differ in isotacticity (Table 2), it is very plausible that the active sites responsible for their formation also differ by their stereospeci-ficity, depending on the kinds of interactions with the solvent. We can propose the active intermediates1and 2responsible for the forma-tion of the heterotactic and isotactic polymeric fractions, respectively. Active site 1 has an empty d-orbital (in addition to the cationic orbi-tal) that can be coordinated by the nonpolar sol-vent, propylene, at the expense of an a-agostic interaction from the polymer chain. This kind of interaction will not avoid any rotation of the polymer, and therefore, the insertion of the monomer molecules will not be maintained trans between the methyl group of the monomer and the growing polymer chain, a requirement for stereoregular insertions.5
[image:10.594.76.248.72.726.2]With active site 2, the polar solvent (toluene or dichloromethane) is strongly attached to the empty d-orbital, impeding the free rotation of the growing polymer chain, thus inducing a higher isotacticity of the polymer. These plausi-ble kinds of interactions can be corroborated by the trend observed for the HI fractions produced by the system B/MAO. It may be seen that the higher the polarity of the solvent, the larger the isotacticity of the polymers obtained. When mix-tures of the solvents are used, the more polar solvent mainly induces the isospecificity (except the mixture of a large amount of toluene and a small amount of dichloromethane, when a ki-netic competition of the solvents is operative). An additional corroboration can be seen from the data obtained in the polymerization of propylene in the absence of a polar solvent (Table 1). Performing the reaction in hexane or in the neat propylene resulted in the formation of only heterotactic polymer. These results are also confirmed by the fact that the more polar the solvent is, the less active the catalytic sys-tems are, but the better the stereoselectivities of the polymerization are.
CONCLUSIONS
Solvents of various polarities and coordination abilities (dichloromethane, toluene, and hexane) influence the high-pressure propylene polymer-ization catalyzed by the octahedral titanium dichloride complexesA,B, andC activated with MAO. In comparison with CH2Cl2 and toluene, all the complexes are considerably more active in the nonpolar hexane and in liquid propylene (bulk polymerization). The different effects of the solvents were assumed to be determined by two effects: the separation of the ion couples between the cationic complex and cocatalyst counterion and the stabilization of the metal via the coordination of the solvents. Increasing the solvent polarity resulted in a decrease in the molecular weight of the polymers produced by complexes A and B (only for CH2Cl2), whereas for complexC, a less drastic effect was observed. We have rationalized the variation of the poly-mer molecular weight as a function of the sol-vent nature, taking into account the relation-ships of the rates for the chain propagation and termination for each complex. For complex B, the isotacticity of the polymers increased with
an augmentation of the solvent polarity, whereas the stereoregularities of the polymers produced by complexes A and C were nearly independent of the solvent nature.
The authors thank the German Israel Foundation (contract I-621-27.5/1999) and the Technion VPR Fund for the Promotion of Research at the Technion for their support of this work. V. Volkis thanks the Technion Institute of Catalysis Science and Technol-ogy for a postdoctoral fellowship. A. Lisovskii thanks the KAMEA program for its financial support.
REFERENCES AND NOTES
1. (a) Kaminsky, W. InAdvances in Catalysis; Gates, B. C.; Kno¨zinger, H., Eds.; Academic:San Diego, 2002; Vol. 46, p 89; (b) Kaminsky, W.; Arndt, M. Adv Polym Sci 1997, 127, 143 and references therein; (c) Brintzinger, H. H.; Fisher, D.; Mu¨l-haupt, R.; Rieger, B.; Waymouth, R. M. Angew Chem Int Ed Engl 1995, 34, 1143; (d) Alt, H. G.; Ko¨pll, A. Chem Rev 2000, 100, 1205 and referen-ces therein; (e) Coates, G. W. Chem Rev 2000, 100, 1223; (f) Ittel, S. D.; Johnson, L. K.; Brook-hardt, M. Chem Rev 2000, 100, 1169; (g) Chen, Y.-X.; Marks, T. J. Chem Rev 2000, 100, 1391; (h) Britovsek, G. J. P.; Gibson, V. C.; Wass, D. F. Angew Chem Int Ed 1999, 38, 428 and references therein; (i) Kaminsky, W. J Polym Sci Part A: Polym Chem 2004, 42, 3911.
2. (a) Mu¨lhaupt, R. In Polypropylene: An A–Z Refer-ence; Karger-Kocsis, J., Ed.; Kluwer Academic: Dordrecht, 1999; p 454; (b) Metz, M. V.; Schwartz, D. J.; Stem, C. L.; Nickias, P. N.; Marks, T. J. Angew Chem Int Ed 2000, 39, 1312; (c) Lanza, G.; Fragala, I. L.; Marks, T. J. J Am Chem Soc 2000, 122, 12764; (d) Williams, V. C.; Piers, W. E.; Clegg, W.; Elsegood, M. R. I.; Collins, S.; Marder, T. B. J Am Chem Soc 1999, 121, 3244; (e) Temme, B.; Erker, G.; Karl, J.; Luftmann, H.; Frohlich, R.; Kotila, S. Angew Chem Int Ed Engl 1995, 34, 1755.
3. (a) Averbuj, C.; Tish, E.; Eisen, M. S. J Am Chem Soc 1998, 120, 8640; (b) Even, J. A.; Jones, R. L.; Razavi, A.; Ferrara, J. D. J Am Chem Soc 1988, 110, 6255; (c) Mo¨hring, P. C.; Coville, N. J. J Organomet Chem 1994, 479, 1; (d) Even, L. A. J Am Chem Soc 1984, 106, 6355; (e) Catalyst Design for Tailor-Made Polyolefins; Soga, K.; Ter-rano, M., Eds.; Elsevier: Tokyo, 1994.
5. Miller, A. A.; Bercaw, J. E. Organometallics 2002, 21, 934 and references therein.
6. For misinsertions, see ref. 1(c). For epimerization mechanisms, see (a) Busico, V.; Cipullo, R. J Am Chem Soc 1994, 116, 9329; (b) Busico, V.; Capor-aso, L.; Cipullo, R.; Landriani, L. J Am Chem Soc 1996, 118, 2105; (c) Resconi, L. J Mol Catal A 1999, 146, 167.
7. (a) Herfert, N.; Fink, G. Macromol Chem Phys 1992, 193, 773; (b) Coevoet, D.; Cramail, H.; Def-fieux, A. Macromol Chem Phys 1999, 200, 1208; (c) Forlini, F.; Tritto, I.; Locatelli, P.; Sacchi, M.; Piemontesi, F. Macromol Chem Phys 2000, 210, 401; (d) Forlini, F.; Fan, Z.-Q.; Tritto, I.; Locatelli, P.; Sacchi, M. C. Macromol Chem Phys 1997, 198, 2397; (e) Fortini, F.; Princi, E.; Tritto, I.; Sacchi, M. C.; Piemontesi, F. Macromol Chem Phys 2002, 203, 645; (f) Sacchi, M. C.; Fortini, F.; Losio, S.; Tritto, I. Macromol Chem Phys 2003, 193, 45; (g) Makio, H.; Fujita, T. Macromol Symp 2004, 213, 221. 8. (a) Volkis, V.; Shmulinson, M.; Averbuj, C.;
Lisov-skii, A.; Edelmann, F. T.; Eisen, M. S. Organome-tallics 1998, 17, 3155; (b) Volkis, V.; Nelkembaum, E.; Lisovskii, A.; Hasson, G.; Semiat, R.; Kapon, M.; Botoshansky, M.; Eishen, Y.; Eisen, M. S. J Am Chem Soc 2003, 125, 2179.
9. Shmulinson, M.; Galan-Fereres, M.; Lisovskii, A.; Nelkenbaum, E.; Semiat, R.; Eisen, M. S. Organo-metallics 2000, 19, 1208.
10. (a) Xavier, K. O.; Smolensky, E.; Kapon, M.; Aucott, S. M.; Woollings, J. D.; Eisen, M. S. Eur J Inorg Chem 2004, 4795; (b) Aucott, S. M.; Slawin, A. M. Z.; Woollins, J. D. Organomet Chem 1999, 582, 83. 11. Bradelly, D. C.; Thomas, I. M. J Chem Soc 1959,
340.
12. (a) Stehling, F. S.; Knox, J. R. Macromolecules 1975, 8, 595; (b) Song, W.; Yu, Z.; Chien, J. C. W. J Organomet Chem 1996, 512, 131; (c) Busico, V.; Cipullo, R.; Caporaso, L.; Angelini, G.; Serge, A. L. J Mol Catal A 1998, 128, 53; (d) NMR of
Poly-mers; Bovey, F. A.; Miray, P. A., Eds.; Academic: San Diego, 1996.
13. In the reaction of complex Cwith MAO, bridging CH3 or Al moieties were not trapped on NMR tube experiments; see (a) Smolensky, E. M.Sc. Thesis, Technion–Israel Institute of Technology, 2003; (b) Smolensky, E.; Kapon, M.; Woollings, D.; Eisen, M. S. Submitted for publication, 2005.
14. (a) Busico, V.; Castelli, V. V. A.; Aprea, P.; Cipullo, R.; Segre, A.; Talarico, G.; Vacatello, M. J Am Chem Soc 2003, 125, 5451; (b) Beck, S.; Lieber, S.; Schaper, F.; Geyer, A.; Brintzinger, H. H. J Am Chem Soc 2001, 123, 1483; (c) Zhou, J.; Lan-caster, S. J.; Walker, D. A.; Beck, S.; Thomton-Pett, M.; Bochmann, M. J Am Chem Soc 2001, 123, 223; (d) Xu, Z.; Vanka, K.; Firman, T.; Michalak, A.; Zurek, E.; Zhu, C.; Ziegler, T. Orga-nometallics 2002, 21, 2444.
15. (a) Busico, V.; Castelli, V. V. A.; Aprea, P.; Cipullo, R.; Segre, A.; Talarico, G.; Vacatello, M. J Am Chem Soc 2003, 125, 5451; (b) Beck, S.; Lieber, S.; Schaper, F.; Geyer, A.; Brintzinger, H. H. J Am Chem Soc 2001, 123, 1483; (c) Zhou, J.; Lan-caster, S. J.; Walker, D. A.; Beck, S.; Thomton-Pett, M.; Bochmann, M. J Am Chem Soc 2001, 123, 223; (d) Xu, Z.; Vanka, K.; Firman, T.; Michalak, A.; Zurek, E.; Zhu, C.; Ziegler, T. Orga-nometallics 2002, 21, 2444.
16. The larger the amount of MAO was, the lower Mw of the polymers was. When the reaction was quenched after a short period, no olefin chain end groups were observed.