AbstrAct
Vascular Endothelial Growth factor (VEGF), a term coined by Napolean Ferrera in 1989, is a molecule of great impact in the physiological event of angiogenesis. VEGF shot to prominence in research in the past score of years but there have been earlier reports that have had a seminal impact on the run-up to its discovery and extensive study. Among the various studies, the works of Warren Lewis (1927), J.C. Sandison and Gordon Ide (1939) lead to the identification of a potential tumour derived growth factor. Glenn Algire and team in 1945 then put forward the proposal that neovascularization was essential to tumorigenesis. These works were followed by a slew of experimental research trying to identify or purify mitogenic factors involved in neovascularization. In 1971, Folkman. J in a novel attempt to isolate a specific proangiogenic factor, identified a soluble endothelial mitogen terming it as “tumour angiogenic factor”. Independent research groups led by Donald senger and Harold dvorak further identified and purified what they termed the “Vascular Permeability factor” in 1983. Eventually, Napolean Ferrera et.al. isolated and cloned the endothelial mitogenic protein naming it as the Vascular Endothelial Growth factor. Two decades later, we are yet exploring the genetic and molecular events occurring in the VEGF signal transduction and its pathogenesis. This review makes a modest attempt to put in a nutshell the vast research done in the turf of VEGF and its implications in angiogenesis.
I. INtrODUctION
Vascular Endothelial Growth factor (VEGF), a term coined by Napolean Ferrera in 1989, is a molecule of great impact in the physiological event of angiogenesis. beginning in the late 1800s with the observations of rudolf c. Virchow about highly vascularised tumours, the discovery, identification and christening of the VEGF has taken an extensive century of research by the scientific fraternity on the mechanisms of angiogenesis. relentless studies have been pursued ever since towards the VEGF family of proteins for plausible prognostic and as well targeted drug therapy applications in medicine.
Although VEGF shot to prominence in research only in the past score of years, there have been earlier reports that have had a seminal impact on the run-up to its discovery and extensive study. Among the various studies, the works of Warren Lewis (1927), J.c. sandison and Gordon Ide (1939) lead to the identification of a potential tumour derived growth factor. Glenn Algire and team in 1945 then put forward the proposal that neovascularization was essential to tumorigenesis. these works were followed by a slew of experimental research trying to identify or purify mitogenic factors involved in neovascularization. In 1971, Folkman. J in a novel attempt to isolate a specific proangiogenic factor, identified a soluble
VEGF - A Precis’
1Bhaskari Janardhanan, M.Sc. Biotechnology., 2Dr. Lakshmi Krishnamoorthy 1Kidwai Memorial Institute of Oncology, Bangalore, Karnataka, India.
(Currently pursuing PhD in medical biochemistry at KMIO, Bangalore, India after working as JRF for 2 years in IITM)
endothelial mitogen terming it as “tumour angiogenic factor”. Independent research groups led by Donald senger and Harold dvorak further identified and purified what they termed the “Vascular Permeability factor” in 1983. Eventually, Napolean Ferrera et.al. isolated and cloned the endothelial mitogenic protein naming it as the Vascular Endothelial Growth factor. two decades later, we are yet exploring the genetic and molecular events occurring in the VEGF signal transduction and its pathogenesis. this review makes a modest attempt to put in a nutshell the vast research done in the turf of VEGF and its implications in angiogenesis.
II. VEGF strUctUrE
VEGF is a glycosylated, disulphide-linked homodimer and expressed in different isoforms ranging in size from 121 to 206 residues in humans with a molecular weight range of 32 - 45kDa. the isoforms result from different splicing events and all variants share the same 115 N-terminal as well as 6 c-terminal residues.1,2 A crystal structure analysis at 2.5Å resolution of a truncated construct of residues 8-109 showed that VEGF is a member of the cystine knot growth factor superfamily.
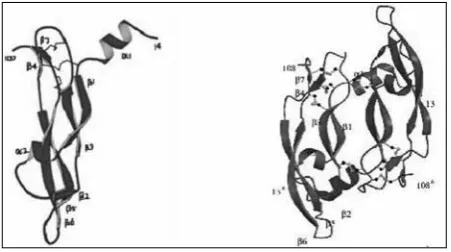
the overall structure of the VEGF monomer (figure 1) consists of a central antiparallel four-stranded β sheet with the characteristic cystine knot at one end and a short three stranded β sheet at the other end. the cystine knot consists of two disulphide bonds forming a covalently linked ring structure between two adjacent β strands [β3- β7], together with a third disulphide bond penetrating this
ring and connecting the beginning of strands β1 and β4. A detailed analysis reveals two α helical and 7 β strand segments. Helix α1 consists of residues 16-24 and the central four-stranded β sheet is formed by residues 27-34 (β1), 51-58 (β3), 73-83 (β5) and 89-99 (β6). the cystine knot motif at one end consists of residues 67-69 (β4) and 103-105 (β5) and the short three-stranded β sheet consists of residues 46-49 (β2), β5 and β6 at the other end. the central four stranded β sheet can be divided into two separate pairs of β strands; β1- β3 and β5- β6. there is a lack of hydrogen bonding at the centre of the sheet barring a single main-chain hydrogen bonding and is solvent exposed from either sides. the cystine knot is hence significant for fold stabilization.
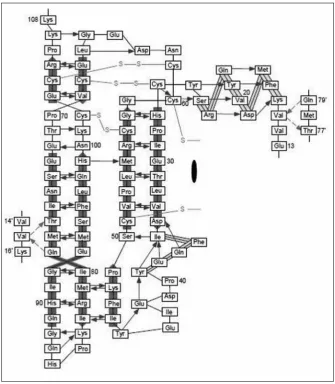
the hydrogen bonds formed in the monomer surrounding the centre are shown in figure 2. the hydrogen bonds are highly conserved in the molecule and only two main-chain hydrogen bonds are observed between the monomers in a dimer. the single N-glycosylation site in VEGF is at Asn-75 near the beginning of the strand β5 and this is known to have no functional impact but is required only for efficient secretion.3 Each VEGF monomer contains three solvent-accessible loop regions, namely the loops connecting strands β1 to β3, β3 to β4 and β5 to β6. the segment connecting strands β1 and β3 (residues 35-50) contains a single turn of α helix (α2) followed by residues in an irregular conformation and a short segment of β strand (β2). this short β strand together with neighbouring strands β5 and β6 form a short, three-stranded antiparallel β sheet at the opposite end of the monomer to the cystine knot and forms part of the receptor-binding face of VEGF, which packs with the N-terminal α helix from the other subunit. this is the main structural feature of the receptor binding face and accounts for 65% of the total surface buried within the dimer and is important for the stability of the receptor binding face. this also forms the second hydrophobic core of the dimer structure apart from the monomer centre. Loops β3 to β4 is adjacent to the cystine knot and differs greatly in length amongst cystine knot proteins. conformational variability of VEGF is facilitated by the loop connecting strand β5 to β6 that undergoes a concerted movement and is important for binding to both KDr and flt-1. Hence, this loop confers flexibility and is functionally important in accessing different conformations for binding to different receptors.
this in addition to the lack of an extended hydrophobic core at the center of the molecule in both the monomer and dimer contribute to the high chain mobility of the molecule. there are only two main chain hydrogen
bonds between the monomers at the outer strand β5. these bonds are formed between the N-terminal residue Val-15 of one monomer and thr-77 and Gln-79 of the second monomer (shown in figure 2 in broken lines) At the location of the dimer axis, four water molecules are conserved in all independent dimers. the total area buried between the two interfaces of the monomers is accounted for by the hydrophobic core containing the most extensive contacts within the dimer and being a part of the receptor-binding site, extends across the subunit interface.4 the positioning of the receptor-binding interfaces at each pole of VEGF seems to facilitate receptor dimerization, which is essential for transphosphorylation and signaling. both the receptor binding sites at each pole are equally significant for the pro-angiogeneic activity of VEGF
since it has been observed that mutant dimers with only one receptor-binding site antagonize native VEGF activity.5 the monomers initially come together through hydrophobic interactions and eventually stabilize by disulphide bonds between cys51 of one chain and cys61 of another6. the signal peptide containing an amphipathic α helix, essential for dimerisation, is eventually cleaved off during secretion.7,8,9
III. VEGF GENE AND sPLIcE VArIANts
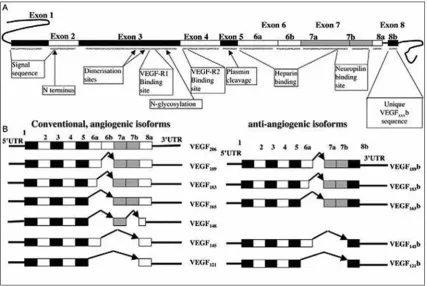
the VEGF gene spans about 16,272 bp with a coding region of ~14Kbp in chromosome 6p21.3.10,11,12 there are a total of 8 exons as shown in figure 3. Exon 1 and
and four residues of exon 2 code for the signal peptide that includes the α helix essential for dimerization of the monomers. Exons 2-5 code for the main receptor binding moieties of which three acidic (Asp63, Glu64 and Glu67) and three basic (Arg82, Lys84 and His86) residues essential for binding receptors VEGFr1 and VEGFr2 are in exons 3 and 4 respectively. the signal peptide is about 26 amino acids in length. Exons 2 to 5 code for about 115 amino acid residues of the protein. Exon 6a codes for 24 amino acids and 6b for 17 amino acids. Exon 7 codes for 44 amino acids and exon 8 codes for about 6 amino acid residues.13 the significance of each of the exon and the different splice variants, both angiogenic and anti-angiogenic is shown in figure 2. there are several cleavage sites coded for across these exons, namely for signal peptidases, plasmin and the urokinase type plasminogen activator. the target site for the signal peptidase lies just beyond exon 1, for plasmin in exon 5 and for uPA in exon 6a. the interaction sites for VEGFrs lie in the region coded by exon 3 and 4. the heparin binding domains are coded for by exon 6 and 7. Exons 6 and 7 encode for 12 and 15 basic amino acids within their coded residues that is responsible for the strong binding with heparan sulphate proteoglycans (HsPG) and the extracellular matrix. these basic sequences are termed cell surface retention consensus sequences. VEGFs also
bind to neuropilin (NrP) which is said to be mediated by two distinct domains; the basic heparin binding domain encoded by exon 6 and the NrP/heparin binding domain encoded by exons 7 and 8. the 55 carboxy terminal residues of VEGF165 containing the NrP/heparin binding domain encoded by exons 7 and 8 has two sub-domains each containing two disulphide bridges and a short two stranded antiparallel β sheet with the carboxy terminal sub-domain additionally containing a short α helix. VEGF165 is also known to bind to NrP through a domain encoded by an alternative exon 6 and part of exon 7 and this has a strong similarity with the domain coded for by exons 7 and 8. VEGF145 that lacks exon 7 also binds NrP though the domain encoded by exon 6.
table 1 summarizes the different VEGF isoforms, their coding exons and features.
VEGF is a molecule subject to intense scrutiny and regulation since it plays a pivotal role in angiogenesis across different stages of development and in grave maladies. the molecule has several variants to its credit, the reason for it being largely regulatory. there are 12 known variants arising from the splicing of the gene of which 7 are pro-angiogenic and the rest are known to be anti-angiogenic. Variants that are angiogenic
Figure 3: the figure represents the entire span of ~16kbp of VEGF gene with the number of base pairs in each exon. the transcriptional start site is tss and translational start site is AtG while the two alternative stop codons include tGA1 and 2 in exon 8. (reprinted from reference 55)
Table 1: Isoforms of human VEGF-A
Isoform Size (aminoacids) Coding exons Features
VEGF A 121 121 1-5, 8 secreted
VEGF-A145 145 1-6, 8 binds NrP2 not NrP1; secreted
VEGF-A165 165 1-5, 7, 8 binds NrP1 & 2, most abundant and active form
VEGF-A165b 165 1-5, 7, 8b secreted, antagonist to VEGF-A165
VEGF-A183 183 1-5, 6a, 7, 8 sequestered in EcM, released on cleavage
VEGF-A189 189 1-8 except 6b sequestered in EcM, released on cleavage
are VEGF121, VEGF145, VEGF148, VEGF165, VEGF183, VEGF186, VEGF206.
the variants are diagrammatically shown in figure 4. It is evident from the figurative representation that exon 8 exerts a distinctive effect of conferring pro or anti-angiogenecity. Exon 8a confers pro-angiogenecity while exon 8b confers anti-angiogenecity. both subsets of exon 8 code for 6 aminoacids at the c-terminus end. While exon 8a encodes cys-Asp-Lys-Pro-Arg-Arg, exon 8b codes for ser-Leu-thr-Arg-Lys-Asp. Alternative splicing that produces mrNA with 8b codons produce an entire set of isoforms of VEGF that are completely contradicting the effects of the mrNA 8a subsets. Perrin et.al. have reported the expression of VEGF121b, VEGF145b, VEGF165b and VEGF189b in humans. the family is termed VEGFxxxb to denote variants encoded by exon 8b. the neutralizing effect of VEGFxxxb isoforms is contributed to competitive inhibition that binds to the receptors but does not stimulate the tyrosine phosphorylation essential for downstream angiogenic activity as is caused by VEGF isoforms. the VEGFxxxb isoforms are hence an endogenous anti-angiogenic agent formed by alternative splicing and is said to be influenced by other growth factor conditions
and splice factor availabilities.14 the VEGF
165b isoform is reported to be down-regulated in various cancers such as renal cell carcinoma, prostate and colon carcinoma15,16,17 and is worth further detailed study. the alternative splicing of VEGF to generate angiogenic isoforms is fundamental to its bioavailability, the inclusion and exclusion of exons 6 and 7 that code for the heparin and neuropilin binding domains and affect generation of diffusible proteins. the heparin-binding domain encoded by exon 6 determines heparin-binding to the extracellular matrix and therefore the isoforms with this domain (VEGF145, VEGF189, VEGF206) are bound tightly to the cell surface heparin containing proteoglycans in the extracellular matrix and those lacking are diffusible. VEGF165 that has only one heparin binding region encoded by 7 is moderately diffusible. VEGF121 that has none of the domains coded by exon 6 or 7 is highly diffusible. the bound isoforms can be liberated by the action of plasmin on activation since plasmin degrades the extracellular matrix and cleaves the VEGF iosoforms into a VEGF110 that has the amino terminal 110 amino acids and is very highly diffusible.18 Urokinase type plasminogen activator directly cleaves VEGF189 and VEGF206 homodimers that have weaker circulation but have higher mitogenic activity on cleavage which is otherwise obstructed due to heavy
protein folding. this demonstrates that exons 6, 7 and 8 do more than just regulate VEGF expression, they define the bioavailability of VEGF isoforms through HsPG binding and also define the production of enhanced mitogenic or anti-angiogenic isoforms.
IV
.VEGF
rEcEPtOrs AND sIGNAL
trANsDUctION
there are five main known receptors for the VEGF family of proteins. the receptors known till date are the
• VEGF r-1 or flt-1,
• VEGF r-2 or KDr or FlK-1 • VEGF r-3 or flt-4
• Neuropilin 1 • Neuropilin 2
two of the VEGF receptors, neuropilin1 and 2, belong to the members of the collapsing/semophorin family and have no kinase activity of their own.
table number 2 shows a bird’s eye view of the receptors, their protein family, their binding targets and functions.
Table 2: Summary of VEGF receptor families
receptor VEGF r-1 VEGF r-2 VEGF r-3 Neuropilins Hspg
Protein family Flt family Flt family Flt family semaphorin
binding receptors
EcM associated proteoglycans
chromo-some 13q12,
30 exons 4q12 5q35 10p12 8q23
Molecular
weight ~180 kDa 200-230kDa 170kDa 120-140kDa Variable High molecular
weight binds to VEGF-A 110,
VEGF-A121, VEGF-A165, VEGF-b,
PIGF
VEGF-A110, VEGF-A121, VEGF-A145, VEGF-A165, VEGF-c, VEGF-D,
VEGF-E
VEGF-c
VEGF-D VEGF-AVEGF-b165, VEGF-AVEGF-A145165, , VEGF-A189, VEGF-A206, VEGF-b167
binding affinity to VEGF (Kd)
~10-100 pM 75-750 pM --- 200-300 pM ~165 pM
Expressed in Endothelium of adults, in healing wounds, vascular smooth muscles,
monocytes, trophoblast cells, monocytes and renal
mesangial cells.
Endothelial and hematopoietic progenitors and
proliferating endothelial cells
in the embryo, megakaryocytes and
retinal progenitor cells.
Adult lymphatic endothelium
Normal endothelial
cells, embryonic
vascular endothelial
cells
basement membrane
and components
of the cell surface
signaling
impact Proteases, growth factors Ec proliferation, migration and survival
LEc proliferation,
migration and survival
Enhanced VEGF r-2 signalling
storage of VEGF, Enhanced
tyrOsINE KINAsE rEcEPtOrs OF tHE
VEGF
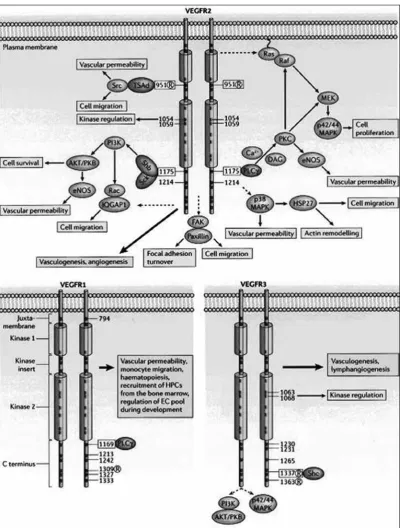
VEGF receptors belong to the “7-Ig” or flt gene family of tyrosine kinases characterized by seven extracellular immunoglobulin like domains, one membrane-spanning segment and a conserved intracellular tyrosine kinase domain interrupted by a kinase insert domain. specific roles of individual Ig-like domains in VEGFr functions have been uncovered as well. the second and third domains define high-affinity ligand binding in VEGF r1 and r2 respectively. the fourth domain is believed to be required for receptor dimerization. the fifth and sixth domains are understood to be required for VEGF retention post binding. the first domain is said to be regulatory in function for ligand binding. both receptors are glycosylated (not essential as in VEGFr1) but only the mature glycosylated forms (as in VEGFr2) can efficiently autophosphorylate on activation.19-25
VEGF r-1
the VEGF r-1 is also known as Flt-1 (Fms like tyrosine kinase 1; fms refers to feline mcdonough sarcoma virus). the VEGF r-1 binds to PIGF, VEGF-b, VEGF-A.26,27
VEGF r-1 has a strong binding affinity towards VEGF than VEGF r-2 but very weak kinase activity and has no direct mitogenic activity or cytoskeletal effects. It is therefore assumed to be a decoy receptor that negatively regulates VEGFr-2 downstream signaling. However, activation of VEGFr-1 is implicated in the increased expression of urokinase type of plasminogen activator and plasminogen activator inhibitor-1.28 these molecules play a role in extracellular matrix degradation and cell migration. VEGFr-1 also plays a role in monocyte chemotaxis. the human VEGFr-1 gene, containing 30 exons, produces a pre-mrNA that undergoes alternative splicing to produce a soluble receptor isoform (sVEGFr-1) that can bind to and inhibit the action of VEGF. sVEGFr-1 on cleavage of its signaling peptide, contains 661 aminoacids corresponding to the first 6 of the 7 extracellular immunoglobulin domains;29 thereby making it feasible for it to act as a functional analogue. the capacity to autophosphorylate in response to activation by VEGF is minimal since VEGFr1 fails to undergo threshold levels of activation loop phosphorylation which is important for further downstream signal transduction by the tyrosine kinase.30 On the contrary, the signal transduction induced by PIGF on VEGFr1 is significant and this variation is said to be due to the differences in the
ability of the ligands to induce different conformational changes.
there are six different phosphorylation sites for VEGFr1 and and they are tyr 1169, 1213, 1242, 1309, 1327 and 1333. While PIGF stimulates tyr 1309, VEGF165 stimulates tyr1213 phosphorylation, thereby possibly causing differential conformational change and further downstream signaling.31 It is also considered that the low kinase activity is owing to a small number of tightly knit and regulated phosphorylation sites. A repressor sequence has been identified in the VEGFr1 juxtamembrane domain, the replacement of which confers equal kinase activity as on par with VEGFr2. Hence this repressor sequence is considered to be highly regulated and influences folding in such a manner as to modulate access of the kinase activity site and may also regulate interaction with phosphotases.31 the phosphorylation of tyr 1333 allows binding of PLcγ and adaptor molecules crk and Nck, while the phosphorylation site tyr 1213 binds to 3 src-homology 2 domain containing proteins, namely, sHP2, PLcγ1 and Grb2. tyr 1242 and 1327 are sites with poor phosphorylation.
VEGFr2
by its ligand, can transactivate VEGFr1 and vice versa. this transactivation by homodimers represent a novel mechanism to be researched on and as well indicates that cross-talk between the two receptors is feasible and in some cases essential for manifold amplification of a signal32. VEGFr2 activates a number of downstream signals that ultimately cause endothelial cell proliferation, migration and survival. the PLcγ activation stimulates protein kinase c which in turn activates ras. this pathway induces the activation of the Erk pathway, MAPK pathway. the Erk pathway eventually results in activation of transcription factors including c-Jun, ternary complex factor and c-fos genes for survival. VEGFr2 also activates PI3K which in turn activates multiple cell permeability causal factors such as eNOs, PKb/Akt, rac. Akt/PKb inhibits bcl2 associated proteins and caspase 9 that in turn promotes cell survival. VEGFr2, on activation, interacts with MAP38K, focal adhesion kinase and substrate paxilin to induce cytoskeletal re-organization and migration as well.
VEGFr3
VEGFr3 is the key molecule in remodeling the primary capillary plexus in embryo and in adult lymphangiogenesis. this receptor occurs in embryonic vascular endothelial cells where its production decreases during development and is subsequently limited to the lymphatic vessels after their formation.33,34 It differs from the other two VEGFrs in undergoing proteolytic cleavage in the extracellular domain (6th Ig domain) into two disulphide-linked polypeptides. the 4.5kb and 5.8kb mrNA code for peptides varying in their c-termini and in their signaling properties due to an alternative 3` exon splicing.35,36 Five tyrosine residues in the c-termini end have been recognized as autophosphorylation sites for the receptor and them being residues 1230, 1231, 1265, 1337 and 1363. Phosphotyrosine 1337 serves to bind shc and Grb2 which occur at the beginning of the MAP kinase pathway. On stimulation by its ligand VEGF-c, VEGFr3 transactivates VEGFr2 and the interaction of the two receptors influences the pattern of transphosphorylation and signal transduction by preventing the phosphorylation of the shc and Grb2 binding sites. VEGFr3 activation also results in PI3K and Akt/PKb stimulation that is important for cell survival. Alam, et al in 2004 also reported the feasibility that VEGFr3 must interact with VEGFr2 in order to catalyze substrate phosphorylation.
NEUrOPILINs AND HsPG
INtErActION WItH VEGF
Works by sorker et.al and chan et.al. identified and discovered an additional receptor for VEGF that was known to be a neuronal recognition molecule and a neuronal cell adhesion molecule, namely the neuropilins. Neuropilins are transmembrane non-protein tyrosine kinase co-receptors for both the semaphoring and VEGF family proteins. Of the seven classes, neuropilins bind to select members of class III semaphorins. VEGF165 and VEGFb bind to neuropilin 1 and VEGF145, VEGF165 and VEGFc bind to neuropilin 2. Neuropilins act as co-receptors with VEGFrs and have no tyrosine kinase function of their own. the neuropilins are unusual since they act as co-receptors for two entirely distinct class of proteins, the VEGFs and semaphorins, and in combination with two different classes of co-receptors (VEFrs and plexins). there is evidence that shows that the semaphorins compete with the VEGF for binding neuropilins as co-receptors, thereby showing a plausible mode of regulation. Neuropilins also function as receptors for the VEGF isoforms independent of the VEGFrs. the neuropilins contain a large extracellular component, a transmembrane segment and a short intracellular portion that serves as a docking site. Neuropilin-1 has five distinct extracellular domains and the diversity of protein modules is consistent with the possibility of multiple binding ligands. NrP1 enhances binding of VEGF165 6 times when co-expressed with VEGFr2. NrP1 also potentiates the effects of VEGFb in cells where VEGFr1 is expressed. NrP1 induced sustained PAF synthesis when co-expressed with VEGFrs, thereby causing sustained vascular permeability, inflammation and endothelial cell migration. the neuropilins contain domains a1, a2, b1 and b2 where ligand binding happens (Figure 7). Of these there is competition at domains b1 and b2 for VEGF binding since semaphorins bind to all 4 domains. the c domain is essential for dimerization followed by the very short transmembrane and intracellular domains. the HsPGs unlike any of the receptors help in signal amplification and sustenance. transactivation of VEGFr-2 leads to prolonged and enhanced signal transduction due to Hs-dependent trapping of the active VEGFr-2 signaling complex. Hence HsPGs not only enhance receptor binding but also prolong the signal transduction.
Figure 6: the figure represents the VEGf receptor family. three signaling kinases (VEGFrs), accessory receptors: Neuropilins 1 and 2 and Heparan sulphate proteoglycans that serve as reserves of VEGf and also enhance VEGF signaling. (reprinted from reference 53)
Figure 7: the figure represents the structural design of neuropilins 1 and 2. Ic, intracellular segment. (reprinted from reference 27)
V. VEGF ExPrEssION PAttErNs,
PHysIOLOGy AND rEGULAtION
VEGF localization and expression information is derived mostly from studies on VEGF-A in mouse. It is detected from the embryonic stage day 7 in the extra-embryonic and embryonic endoderm. It is present in high levels in the trophoblast surrounding the embryo, in the embryonic myocardium, gut endoderm, embryonic mesenchyme and amniotic ectoderm. During development, it is found in the mesenchyme and neuroectoderm of the head. the expression rapidly declines in most tissues weeks after birth and is relatively low in adults except in few vascular beds, including those of the brain, choroids plexus, lung alveoli, kidney glomerulii and heart. VEGFA expression is also upregulated during specific physiological processes such as development of the endocrine in corpus luteum during pregnancy, wound healing and tissue repair. It is now strongly associated with pathogenesis as well such as neovascularization in cancers.
plasma fibronectin, victronectrin and thrombospondin. the activated platelets release a plethora of cytokines and growth factors to restore health in the area. One of them is VEGFA that attracts circulating neutrophils and monocytes. this chemotactic response is mediated by VEGFr1 expressed on inflammatory cells.37,38 the VEGFA expression in wounds is highly induced by the local hypoxia created. HIF-1 regulates VEGFA expression in wound margin keratinocytes. During re-epithelialization in wounds, plasmin secreted by activated keratinocytes is also known to mobilize bound forms of VEGF into diffusible forms and thereby results in increased tissue vascular permeability and repair.
VEGFA is also physiologically important during luteal angiogenesis.39 VEGFA mrNA and protein is detectable in the granulose cells of primordial and primary follicles. After ovulation, VEGFA expression is observed in granulosa derived luteal cells. VEGFA expression in the corpus luteum is highest in the early luteal phase and declines after mid-luteal phase. Neutralization of VEGFA expression in early luteal phase inhibits the development of normally extensive capillary bed and luteal function reflected by secretion of progesterone which is reduced by 60%. Leuteinizing hormone is the major regulatory factor involved in VEGF A expression than oxygen tension as in the case of wound repair.
VEGF being a major angiogenic factor is subject to intense multi-level regulation.
Hypoxia or low oxygen tension and hormones are two major regulatory factors physiologically but an understanding of the basic molecular events is necessary for indepth studies of VEGF expression and functioning and as to whether that knowledge can get us to therapies against pathogenic angiogenesis in maladies as cancer.
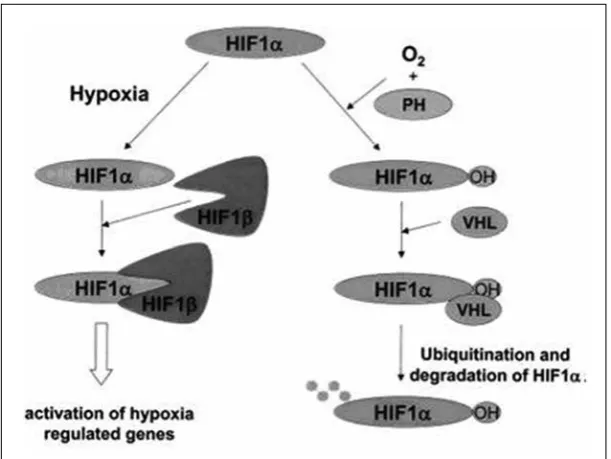
VEGF expression is highly induced by hypoxia. the VEGF A gene contains hypoxia responsive elements (HrEs) in its 5` and 3` Utr region. transcriptional regulation of VEGFA expression by hypoxia is mediated by binding of the transcription factor HIF-1 (hypoxia inducible factor 1) to the HrE. HIF-1 is a heterodimer of HIF-1α and HIF-1β sub-units. HIF-1α is very labile and under hypoxic conditions, it accumulates since proteosomal degradation is inhibited (figure 8). At normal oxygen tension, proline hydroxylation targets HIF-1α for preoteosomal degradation, but is inhibited under hypoxia because of the requirement of the responsible prolyl hydroxylases for molecular dioxygen. the product of the VHL tumor suppressor gene is also required for proteosomal proteolysis. In addition, VEGF mrNA is stabilized under conditions of hypoxia as a result of binding of unidentified factors to its 3` Utr. VEGFA expression is also upregulated by a variety of growth factors and cytokines including PDGF-bb, tGF-β, FGF-2, interleukin1β and interleukin-6, some of which act synergistically with hypoxia.
Dekel et.al have showed that L-VEGF (long-VEGF) is associated with hypoxia and tumor angiogenesis.40 the otherwise short lived VEGF mrNA is both induced and stabilized on exposure to hypoxia. It has a very long 1038bp 5`Utr that contains 2 IrEs elements (A&b). these two regulatory elements ensure efficient translocation under hypoxia where normal translation via ribosomal scanning mechanism is severely impared. the 5` Utr of VEGF harbours a conserved OrF which can add a 180 aminoacid stretch in front of VEGF. this OrF is initiated by a cUG codon followed by additional 3 conserved cUG codons. this lengthened isoforms is called Long VEGF (L-VEGF). Effective cleavage of L-VEGF results in the detachment of the mature VEGF from the 180 aminoacid OrF extension initiated from the upstream in-frame cUG codon. Expression of just the 180 amino acid extension of VEGF in itself leads to cytoplasmic staining. However, upon hypoxia the N-terminal segment is translocated to the nucleus via the nuclear localization signal (NLs) confined to the amino-terminus. Further, strong association between the level of VEGF expression and nuclear localization of the 180 amino acid OrF extension was observed in various normal angiogenic tissues that was hightened in corresponding tumours. Hence the nuclear localization of the 180 amino acid extension plays a significant role in VEGF-mediated angiogenesis and its regulation, both physiologically and pathologically.
VI. VEGF PAtHOGENEsIs
VEGF pathogenesis is a hot topic of research and debate worldwide in the research fraternity. VEGF pathogenesis is implicated in several maladies including diapetic retinopathy, rheumatoid arthiritis, cardiovascular ischemia, diabetis mellitus, peripheral vascular diseases, endometriosis, pre-eclampsia, psoriatic skin disease and the king of maladies, cancer. In this section, we shall have a bird’s eye view on the role of VEGF in various diseases which here I choose to section as malignant and non-malignant diseases.
NON MALIGNANt DIsEAsEs
rheumatoid arthritis is an autoimmune, polyarticular disease that eventually erodes joints and osteoarthritis is a non-inflammatory, degenerative disease affecting the hyaline cartilage. In due course of time, in either diseases, there is an onset of hypoxia and thereby VEGF upregulation. VEGFA121 and VEGF165 are known to be
upregulated in both theses disease and the microvessel density is increased significantly in both.41 It is considered that VEGF is not only responsible for lesion formation but also in recruitment of monocytes to the affected area.42 It also activates plasmin and PAI-1 which in turn activates matrixmetalloproteinases that are main enzymes involved in destruction of joints. Higher levels of VEGFA, in serum of patients has been correlated with worse prognosis and hence useful in predicting the vigourousness of the treatment to be given to the patient. thus serum VEGFA levels could act as biomarkers for prognosis in this case.
In cardiovascular ischemia there is an increased steady state levels of HIF-1α mrNA which in turn upregulates and as well induces expression of VEGFA. VEGFA expression is shown to be persistant longer than that of HIF-1α.43 Hence though the expression and action of HIF-1α is early in the onset of myocardial infarct formation, the effect and expression fo VEGFA is of longer duration and is probably necessary for the preservation of the myocardium and limitation of hypoxic cellular destruction.
Diabetes mellitus is an endocrinological disease characterized by general microangiopathy, especially affecting the kidney, retina, nervous and vascular systems. Proliferative diabetic retinopathy is a major cause of adult blindness. VEGF has emerged as the key mediator of intraocular neovascularization. It stimulates microaneurysm formation and capillary occlusion with ischemia, promotes vascular permeability and as well stimulates development of new retinal cells. It has been shown by Duh and Aiello et.al, that blocking VEGF signaling is sufficient to completely inhibit retinal neovascularization.44,45 VEGFA has also been suspected having a potential role in diabetic nephropathy but not as extensive as in diabetic retinopathy.
VEGFA hyperexpression is also proven in endometriosis, pre - eclampsia and ovarian hyperstimulation concentration of VEGFA have been found to be significantly higher in women with moderate to severe endometriosis in comparison to those with minimal to mild or no disease. VEGFA is localized predominantly in the glandular epithelium of enodmetrosis lesions.46
MALIGNANt DIsEAsEs
and is highly upregulated in most human cancers.1 the expression of VEGF correlates with intratumoral vessel density and poor prognosis in cancer patients2 and inhibition is known to decrease tumour vessel density and tumour growth.3 the main inducing factor in tumours is the growing hypoxic conditions in the centre of the tumour that has no initial vascularization. the low oxygen tension in the tumour induces VEGF expression and related molecules that initiate vascularization. both VEGF and its receptors are shown to be highly expressed in metastatic carcinomas and the expression of these proteins directly correlates with the degree of neovascularization. the role of VEGF and its receptors is studies widely based on inhibition experiments. retrovirus mediated expression of dominant negative VEGF receptor flk-1 dramatically inhibits the growth of a variety of tumours, including mammary, ovarian, lung carcinomas and c6 glioblastomas. Histological examination of these tumours demonstrated that the growth of blood vessels in the tumours is severely reduced. Intraperitoneal administration of anti-VEGF antibody in nude mice harbouring tumours significantly decreased tumour vessel density and suppresses tumour growth. these observations indicate that a general inhibition of VEGF activity in vivo results in reduced tumour angiogenesis and tumour growth. VEGF, in addition to stimulating blood vessel growth, also increases vascular permeability. In such a situation, VEGF can induce the formation of leaky blood vessels with fragmented membranes that can easily be penetrated by neoplastic cells to disseminate the primary tumour and favouring metastasis. thus VEGF, in conjunction with several other factors, has multiple roles in tumour angiogenesis.
cUrrENt trENDs IN VEGF rEsEArcH
VEGF sNPs and prognosis
the VEGF gene is located on chromosome 6p12 and includes a 14-kb coding region with eight exons and seven introns. At least 30 single nucleotide poly morphisms (sNP) in VEGF gene have been described in the literature. Polymorphisms of VEGF gene have been associ-ated with susceptibility to several types of cancer. some of these polymorphisms (+936c>t rs3025039, -2578c>A rs699947, -1154G>A rs1570360, -634G>c rs2010963, -460c>t rs833061, +405c>G rs2010963) have been related to protein expression of VEGF in cancers. since VEGF is significant in the tumour angiogenesis, it is reasonable to hypothesize that VEGF gene polymorphisms are good candidates for predicting the risk of developing
cancers. Many studies have investigated the role of VEGF polymorphisms as a genetic determinant for susceptibility and outcome of breast, prostate, NscL, and colorectal cancer. several polymorphisms have been reported within the promoter (-2578c > A, -2489c > t, -1498c > t, and -1154G > A), 5′-Utr (-634G > c and -7c > t), and 3′-Utr (936c > t and 1612G > A) for the VEGF gene47. current scenario is such that studies across different races have given unsettling results and data. Although certain sNPs are implicated as risk factors and some as protective, the sNPs and their allelic variations have not so far come out as a strong predictive factor in any of the cancers. Hence extensive genotyping and linkage studies are needed globally to arrive at an answer to the question as to whether VEGF sNPs can be used as a biomarker for cancer or atleast as a prognostic marker?
VEGF targeted therapies
VEGF targeted therapies are the most logical of conclusions in tackling cancer. VEGF-induced angiogenesis may be inhibited by targeting either the VEGF ligand itself or VEGFrs. bevacizumab is an IV administered humanized monoclonal antibody directed against VEGF-A, and this drug acts by binding and neutralizing all VEGF-A isoforms. In contrast, small-molecule tyrosine kinase inhibitors (tKIs) do not bind to VEGF directly; they inhibit the activity of VEGFrs and therefore block downstream signaling pathways activated by VEGF-A and other members of the VEGF family. Many tKIs have the advantage of oral bioavailability, but the serum half-lives of these agents are considerably shorter than that of bevacizumab, although this can be compensated for by daily administration of tKIs. small-molecule inhibitors commonly have additional activity against other tyrosine kinases, and such “multitargeting” activity may contribute to their antitumor effects. For example, sorafenib inhibits raf-1, VEGFr-2, VEGFr-3, and platelet-derived growth factor receptor β, potentially targeting proliferation via the ras–extracellular signal– related kinase pathway in addition to angiogenesis. Other VEGFr tKIs include vandetanib, sunitinib, bIbF 1120, and pazopanib
binds to VEGF-A but also binds to VEGF-b and placental growth factor. VEGF trap has a greater affinity for the VEGF ligand than VEGFrs and anti-VEGF monoclonal antibodies such as bevacizumab. the precise mechanisms by which angiogenesis inhibition might improve clinical outcome are not fully understood. Antiangiogenic agents may work by suppressing tumor vasculature formation, thus depriving the tumor from nutrients and limiting tumor growth. Another possibility is that antiangiogenic agents transiently normalize the tumor vasculature, allowing more efficient oxygen and chemotherapy delivery. targeting the VEGF pathway has proven to be a successful strategy in a variety of cancers. randomized phase III clinical trials of bevacizumab in combination with chemotherapy in patients with colorectal, lung, and breast cancer have all demonstrated a benefit for VEGF inhibition in terms of either the progression-free survival (PFs) or overall survival duration. In renal cancer, VEGF pathway inhibition, either with bevacizumab in combination with interferon or single-agent VEGFr inhibitors (sunitinib or sorafenib), has shown dramatic benefits in terms of the PFs time.48
there is a flip side of toxicity that does trouble VEGF targeted therapies. the major toxicities seen in the phase II trials of bevacizumab include hypertension, thrombosis,
proteinuria, hemorrhage, and GI perforation, some that do turn fatal. the therapies and the response to it from patients depends on several clinico-pathological status of the patient as well. Hence further studies on this line can be extremely useful.
turunen M.P. et.al. showed epigenetherapy, a gene therapy based on epigenetic mechanism at a promoter level as a new approach of therapy for treatment of several pathogeneic angiogenesis.49 they used a lentivirus mediated delivery of shrNA molecules targeted to specific regions in the mVEGFA promoter either inducing or repressing VEGFA expression via epigenetic modulation.
VEGF epigenetics
Kim et.al. have recently shown that VEGFr1 and VEGFr2 are regulated by epigenetic mechanism in stomach cancer, colon cancer and hepatocellular carcinoma.50 Quentmeier H.et.al., have shown methylated status of VEGFr2 and VEGFr3 across thirty cell lines representing lymphoma subtypes.51 these recent studies have included a new dimension for research and therapy based on VEGF epigenetics. the VEGF A promoter can
be manipulated using promoter-targeted small rNAs and this results in modulation of VEGFA expression. the epigenetic regulation of VEGF is shown to be more through histone codes than through DNA methylation. Hinton D et.al. studied the role of histone deacetylase inhibitor trichostatin A (tsA) for its ability to inhibit experimental choroidal neovascularization (cNV) and found that tsA inhibits cNV in vivo. Invitro studies have suggested that this is due to the inhibition of VEGF and VEGFr2 and their further therapeutical potential in angiogenesis. studies by Kim et.el. (as mentioned earlier) although suggested that VEGFr1 and VEGFr2 showed variable hypermethylation also showed no methylation in VEGF gene in tested cancer cell lines (colon, stomach, lung, melanoma, breast and thyroid cancers). yamada et.al. demonstrated that promoter methylation of VEGFr1 plays a key role in silencing of the gene in prostate cancer cells but the same was not elucidated in other cancer types. VEGF in turn is also known to epigenetically modulate the expression of other genes such as rex1 and Oct 4 genes. Upon treatment with VEGF A methylation patterns in promoters of both genes were diminished in endothelial progenitor cells. Interestingly a converse effect has been observed where expression of pro-angiogenic gene is stimulated by epigenetic mechanism. the lymphangiogenic gene VEGFc is known to be over-expressed in gastric cell line due to demethylation . sundrani D.P. et.al. observed changes in placental global DNA methylation levels in preeclampsia and examined gene promoter cpG methylation and expression of several angiogeneic genes to find differentially methylated cpG sites in the promoter regions of VEGF, Flt0-1 and KDr between control and preeclampsia groups.52 Hypomethylation and consequent upregulation of VEGF mrNA levels were observed between term and preterm preeclampsia and this could be a compensatory mechanism to restore normal angiogenesis and blood flow in preterm preeclampsia. Hence an altered DNA methylation in placental angiogenesis is strongly suggested in preeclampsia. currently there is very little understanding of the epigenetics of VEGF and plethora of research to gain insight is necessary.
rEFErENcEs
1. Houck K.A., Ferrara N., Winer J., cachianes G., Li b. & Leung D.W. (1991). the vascular endothelial growth factor family: identification of a fourth molecular species and characterization of alternative splicing of rNA. Mol. Endocrinol. 5, 1806–1814. 2. tischler E. et al., & Abraham J.A. (1991). the human gene for
encoded through alternative splicing of rNA. J. biol. chem. 266, 11947–11954.
3. Peretz, D., Gitay-Goren, H., safran, M., Kimmel, N., Gospodarowicz, D. and Neufeld, G. (1992). Glycosylation of vascular endothelial growth factor is not required for its mitogenic activity. biochem. biophys. res. commun.182, 1340-1347.
4. Muller. y.A. et.al., (1997). the crystal structure of Vascular Endothelial Growth Factor (VEGF) refined to 1.93 Å resolution: multiple copy flexibility and receptor binding. structure, 19997, 5:1325-1338.
5. siemeister, G., schirner, M., reusch, P., barleon, b., Marme, D. and Martiny-baron, G. (1998a). An antagonistic vascular endothelial growth factor (VEGF) variant inhibits VEGF-stimulated receptor autophosphorylation and proliferation of human endothelial cells. Proc. Nat.Acad. sci. UsA 95, 4625-4629.
6. Potgens, A. J., Lubsen, N. H., van-Altena, M. c., Vermeulen, r., bakker, A., schoenmakers, J. G., ruiter, D. J. and de-Waal, r. M. (1994). covalent dimerization of vascular permeability factor/ vascular endothelial growth factor is essential for its biological activity. Evidence from cys to ser mutations. J. biol. chem. 269, 32879-32885.
7. Leung, D. W., cachianes, G., Kuang, W. J., Goeddel, D. V. and Ferrara, N. (1989). Vascular endothelial growth factor is a secreted angiogenic mitogen. science 246, 1306-1309.
8. Keck, P. J., Hauser, s. D., Krivi, G., sanzo, K., Warren, t., Feder, J. and connolly, D. t. (1989). Vascular permeability factor, an endothelial cell mitogen related to PDGF. science 246, 1309-1312. 9. siemeister, G., Marme, D. and Martiny-baron, G. (1998b). the
alphahelical domain near the amino terminus is essential for dimerization of vascular endothelial growth factor. J. biol. chem. 273, 11115-11120.
10. Vincenti, V., cassano, c., rocchi, M. and Persico, G. (1996). Assignment of the vascular endothelial growth factor gene to human chromosome 6p21.3. circulation 93, 1493-1495. 11. Houck, K. A., Ferrara, N., Winer, J., cachianes, G., Li, b. and
Leung, D. W. (1991). the vascular endothelial growth factor family: identification of a fourth molecular species and characterization of alternative splicing of rNA. Mol. Endocrinol. 5, 1806-1814. 12. tischer, E., Mitchell, r., Hartman, t., silva, M., Gospodarowicz,
D., Fiddes, J. c. and Abraham, J. A. (1991). the human gene for vascular endothelial growth factor. Multiple protein forms are encoded through alternative exon splicing. J. biol. chem. 266, 11947-11954.
13. robinson c.J and stringer s.E. the splice variants of Vascular Endothelial Growth Factor and its receptors. Journal of cell science, 114, 853-865.
14. Nowak D.G., Woolard J., Amin E.M., Konopatskaya O., saleem M.A., churchill A.J., Ladomery M.r., Harper s.J., bates D.O. (2008). Expression of pro- and anti-angiogenic isoforms of VEGF is differentially regulated by splicing and growth factors. Journal of cell science, 121, 3487-3495.
15. bates D. O., cui t. G., Doughty J. M., Winkler M., sugiono M., shields J. D., Peat D, Gillatt D. and Harper s. J. (2002). VEGF1 65b, an inhibitory splice variant of vascular endothelial growth factor, is down-regulated in renal cell carcinoma. cancerres. 62, 4123-4131.
16. Woolard J., Wang W. y., bevan H. s., Qiu y., Morbidelli L., Pritchard-Jones r. O., cui t. G., sugiono M., Waine E., Perrin r. et al. (2004). VEGF165b, an inhibitory vascular endothelial growth factor splice variant: mechanism of action, in vivo effect on angiogenesis and endogenous protein expression. cancer res. 64, 7822-7835.
17. Varey A. H., rennel E. s., Qiu y., bevan H. s., Perrin r. M., raffy s., Dixon A. r., Paraskeva c., Zaccheo O., Hassan A. b. et al. (2008). VEGF 165 b, an antiangiogenic VEGF-A isoform, binds and inhibits bevacizumab treatment in experimental colorectal
carcinoma: balance of pro- and antiangiogenic VEGF-A isoforms has implications for therapy. br. J. cancer 98, 1366-1379. 18. Houck KA, Leung DW, rowland AM, Winer J, and Ferrara N
(1992) Dual regulation of vascular endothelial growth factor bioavailability by genetic and proteolytic mechanisms. J biol chem 267:26031–26037.
19. Davis-smyth t., chen H., Park J., Presta L. G. and Ferrara N. (1996). the second immunoglobulin-like domain of the VEGF tyrosine kinase receptor Flt-1 determines ligand binding and may initiate a signal transduction cascade. EMbO J. 15, 4919-4927. 20. barleon b., totzke F., Herzog c., blanke s., Kremmer E., siemeister
G., Marme D. and Martiny-baron G. (1997a). Mapping of the sites for ligand binding and receptor dimerization at the extracellular domain of the vascular endothelial growth factor receptor FLt-1. J. biol. chem. 272, 10382-10388.
21. Wiesmann c., Fuh G., christinger H. W., Eigenbrot c., Wells J. A. and de Vos A. M. (1997). crystal structure at 1.7 Å resolution of VEGF in complex with domain 2 of the Flt-1 receptor. cell 91, 695-704.
22. Fuh G., Li b., crowley c., cunningham b. and Wells J. A. (1998). requirements for binding and signaling of the kinase domain receptor for vascular endothelial growth factor. J. biol. chem. 273, 11197-11204.
23. shinkai A., Ito M., Anazawa H., yamaguchi s., shitara K. and shibuya M.(1998). Mapping of the sites involved in ligand association and dissociation at the extracellular domain of the kinase insert domaincontaining receptor for vascular endothelial growth factor. J. biol. chem. 273, 31283-31288.
24. Piossek c., schneider-Mergener J., schirner M., Vakalopoulou E., Germeroth L. and thierauch K. H. (1999). Vascular endothelial growth factor (VEGF) receptor II-derived peptides inhibit VEGF. J. biol. chem. 274, 5612-5619.
25. robinson c.J. and stringer s.E., splice variants of VEGF and their receptors. J. of cell sciences., 114, 853-865.
26. Waltenberger J, claesson-Welsh L, siegbahn A, shibuya M, Heldin cH. Different signal transduction properties of KDr and Flt1, two receptors for vascular endothelial growth factor. J biol chem 1994;269:26988–95.
27. VEGF signaling in tumour progression – robert roskoski Jr 28. Olofsson b, Korpelainen E, Pepper Ms, et al. Vascular endothelial
growth factor b (VEGF-b) binds to VEGF receptor-1 and regulates plasminogen activator activity in endothelial cells. Proc Natl Acad sci UsA 1998;95:11709–14.
29. Kendall rL, thomas KA. Inhibition of vascular endothelial cell growth factor activity by an endogenously encoded soluble receptor. Proc Natl Acad sci UsA 1993;90:10705–9
30. Ito N,Wernstedt c, Engstrom U, claesson-Welsh L. Identification of vascular endothelial growth factor receptor-1 tyrosine phosphorylation sites and binding of sH2 domain-containing molecules. J biol chem 1998;273:23410–8.
31. claesson-Welsh L (2003) signal transduction by vascular endothelial growth factor receptors. biochem soc trans 31:20–24. 32. Autiero M, Waltenberger J, communi D, et al. role of PlGF in the intra- and intermolecular cross talk between the VEGF receptors Flt1 and Flk1. Nat Med 2003;9:936–43.
33. Galland F, Karamysheva A, Pebusque MJ, et al. the FLt4 gene encodes a transmembrane tyrosine kinase related to the vascular endothelial growth factor receptor. Oncogene 1993;8:1233–40. 34. Kaipainen A, Korhonen J, Mustonen t, et al. Expression of the
fms-like tyrosine kinase 4 gene becomes restricted to lymphatic endothelium during development. Proc Natl Acad sci UsA 1995;92:3566–70.
35. Ortega N, Hutchings H, and Plouet J (1999) signal relays in the VEGF system. Front biosci 4:D141–D152.
VEGF-D expression in neuroendocrine cells and their receptor, VEGFr-3, in fenestrated blood vessels in human tissues. FAsEb J 14: 2087–2096.
37. schaffer cJ and Nanney Lb (1996) cell biology of wound healing. Int rev cytol 169:151–181.
38. Martin P (1997) Wound healing—aiming for perfect skin regeneration. science (Wash Dc) 276:75–81.
39. Fraser HM and Lunn sF (2000) Angiogenesis and its control in the female reproductive system. br Med bull 56:787–797. 40. Dekel. y.r., Fuchs A., yakirevich E., Azriel A., Mazareb s., resnick
M.b., Levi b.Z., (2005) Nuclear localization of long-VEGF is associated with hypoxia and tumour angiogenesis. bbrc 332: 271-278.
41. Walsh DA (1999) Angiogenesis and arthritis. rheumatology (Oxford) 38:103–112.
42. Ikeda M, Hosoda y, Hirose s, Okada y, and Ikeda E (2000) Expression of vascular endothelial growth factor isoforms and their receptors Flt-1, KDr and neuropilin-1 in synovial tissues of rheumatoid arthritis. J Pathol 191:426–433.
43. Lee sH, Wolf PL, Escudero r, Deutsch r, Jamieson sW, and thistlethwaite PA (2000) Early expression of angiogenesis factors in acute myocardial ischemia and infarction. N Engl J Med 342:626–633.
44. Duh E and Aiello LP (1999) Vascular endothelial growth factor and diabetes: the agonist versus antagonist paradox. Diabetes 48:1899–1906.
45. chakrabarti s, cukiernik M, Hileeto D, Evans t, and chen s (2000) role of vasoactive factors in the pathogenesis of early changes in diabetic retinopathy. Diabetes Metab res rev 16:393–407. 46. shifren JL, tseng JF, Zaloudek cJ, ryan IP, Meng yG, Ferrara N, Jaffe
rb, and taylor rN (1996) Ovarian steroid regulation of vascular endothelial growth factor in the human endometrium: implications
for angiogenesis during the menstrual cycle and in the pathogenesis of endometriosis. J clin Endocrinol Metab 81:3112–3118. 47. Jain L., Vargo c.A., Danesi r. (2009) the role of vascular
endothelial growth factor sNPs as predictive and prognostic markers for major solid tumours. Mcr, 10: 1158-1535
48. banerjee s., Gore M., (2009) the future of targeted therapies in ovarian cancer. the Oncologist, 14: 706-716
49. turunen M.P., Lehtola t., Heinonen s.E., Assefa G.s., Korpisalo P., Girnary r., et.al., (2009), circ res., 105: 604-609.
50. Kim J.y., Hwang J.H., Zhou W., shin J., Noh s.M, song I.s., et.al. (2009) the expression of VEGF receptors genes is concurrently influenced by epigenetic gene silencing of the genes and VEGF activation Landes biosci 4:5, 313-321.
51. Quentmeier H., Eberth s., romani J., Wiech H.A., Zaborski M., Drexler H.G., (2012) DNA methylation regulates expression of VEGF-r2 (KDr) and VEGF-r3 (FLt4). bMc cancer 12:19. 52. sundrani D.P., reddy U.s., Joshi A.A., Mehendale s.s.,
chavangautam P.M., Hardikar A.A., et.al. (2013) Differential placental methylation and expression of VEGF, FLt-1 and KDr genes in human term and preterm preeclampsia. clin Epi 5:6 53. Hoeben A., Landuyt b., Highley M.s., Wilders H.,
VanOosterom A.t., bruijn E.A., (2004), Vascular endothelial growth factor and angiogenesis, Pharmacol rev 56: 549-580.
54. Muller y.A., christinger H.W., Keyt b.A., deVos A.M., (1997), the crystal structure of vascular endothelial growth factor (VEGF) refined to 1.93Å resolution: multiple copy flexibility and receptor binding, structure 5:10: 1325-1338