This is a repository copy of
Assessment of density-functional approximations: Long-range
correlations and self-interaction effects
.
White Rose Research Online URL for this paper:
http://eprints.whiterose.ac.uk/4021/
Article:
Jung, J, Garcia-Gonzalez, P, Alvarellos, J E et al. (1 more author) (2004) Assessment of
density-functional approximations: Long-range correlations and self-interaction effects.
Physical Review A. 052501. -. ISSN 1094-1622
https://doi.org/10.1103/PhysRevA.69.052501
[email protected] https://eprints.whiterose.ac.uk/
Reuse
Items deposited in White Rose Research Online are protected by copyright, with all rights reserved unless indicated otherwise. They may be downloaded and/or printed for private study, or other acts as permitted by national copyright laws. The publisher or other rights holders may allow further reproduction and re-use of the full text version. This is indicated by the licence information on the White Rose Research Online record for the item.
Takedown
If you consider content in White Rose Research Online to be in breach of UK law, please notify us by
promoting access to White Rose research papers
Universities of Leeds, Sheffield and York
http://eprints.whiterose.ac.uk/
White Rose Research Online URL for this paper:
http://eprints.whiterose.ac.uk/4021
Published paper
Garc'ia-González, P, Jung, J, Alvarellos, JE, Godby, RW (2004)
Assessment of
Density Functional Approximations: Long-Range Correlations and
Self-Interaction Effects
Physical Review A 69 (052501 - 7 pages)
arXiv:cond-mat/0404559v1 [cond-mat.other] 23 Apr 2004
Assessment of density functional approximations: long-range correlations and
self-interaction effects
J. Jung,1 P. Garc´ıa-Gonz´alez,2 J. E. Alvarellos,1 and R. W. Godby3
1
Departamento de F´ısica Fundamental, Universidad Nacional de Educaci´on a Distancia, Apartado 60141, E-28080 Madrid, Spain
2
Departamento de F´ısica de la Materia Condensada, C-III, Universidad Aut´onoma de Madrid, E-28049 Madrid, Spain
3
Department of Physics, University of York, Heslington, York YO10 5DD, United Kingdom
The complex nature of electron-electron correlations is made manifest in the very simple but non-trivial problem of two electrons confined within a sphere. The description of highly non-local correlation and self-interaction effects by widely used local and semi-local exchange-correlation en-ergy density functionals is shown to be unsatisfactory in most cases. Even the best such functionals exhibit significant errors in the Kohn-Sham potentials and density profiles.
PACS numbers: 31.15.Ew,31.25.-v,71.15.Mb
I. INTRODUCTION
The Kohn-Sham (KS) [1] formulation of Density Func-tional Theory (DFT) [2] is in the present day the most popular method in electronic structure calculations. In this scheme, the exact ground state energy and electron density could be found self-consistently if the
exchange-correlation (XC) energy functional EXC[n] was known.
EXC[n] contains all the quantum many-body effects of
the electron system, but very simple mean-field prescrip-tions like the Local Density Approximation (LDA) often suffice to obtain accurate results for a wide variety of systems at an affordable computational cost.
However, there are several problems that are well be-yond the capabilities of the local approximation and any
semi-local extension thereof, because of the clear
mani-festation of the very non-local nature of electron-electron correlations. For example, the long-ranged van der Waals interactions, the image potential at metal surfaces and
clusters, or several pathological behaviors of the exact
XC potential cannot be described at all using simple XC functional models [3]. Nevertheless, these limita-tions should not be important in many other situalimita-tions, and new XC functionals are being proposed with the aim of reaching chemical accuracy while keeping the imple-mentation ease of the local density based approxima-tions [4, 5]. This could open the appealing possibility of making predictive studies of relevant aspects of Quantum Chemistry such as reaction paths, atomization energies, and bond lengths and energies.
The purpose of this paper is to bring further insight in the capabilities and limitations of local and semi-local XC functionals, showing that even in very simple prob-lems the complexity of the quantum electron-electron correlations might prevent any of these approaches from properly describing the ground state properties of such systems. We do not intend to make a comprehensive assessment of mean-field-like approximations, but just to provide a representative common picture of all them. Hence, among the realm of proposals existing in the
liter-ature, we have chosen the well-known LDA prescription by Perdew and Wang [6], the Generalized Gradient Ap-proximation (GGA) by Perdew-Burke-Erzenhof [7], and the very recent meta-GGA (MGGA) proposed by Tao
et al [8]. These three approaches have the virtue of
be-ing designed usbe-ing general considerations (i.e. they do not include empirical parameters). Furthermore, they can be seen as a coherent set of conceptual progressive improvements starting from the strictly local approxi-mation, then considering the dependence on the density variation, and finally including information from KS or-bitals through its associated kinetic energy density.
We will study a very simple but non-trivial system: two electrons confined within a sphere of hard walls, whose solution has been found through accurate Configuration Interaction calculations [9, 10]. This system is an inter-esting benchmark reference due to the following reasons. First, its simplicity: the density is isotropic, hence hav-ing a simple mathematical one-dimensional problem re-stricted to the radial coordinate. Second, its ground state has singlet spin configuration. As a consequence, Pauli’s correlation (exchange) between the electrons is absent
and the exchange energyEX[n] just corrects the spurious
electron self-interaction in the classical Hartree
electro-static energyWH[n]. That is, the Coulomb correlation is
actually the only source of quantum many-body effects in this system. Finally, different correlation regimes can
be easily achieved by varying the radiusRof the
confin-ing sphere. Thus, at smallR(high mean density) we are
in the low-correlation limit where the confinement by the sphere dominates over the electron-electron interaction,
so having a system with anatomic-like behavior. By
in-creasing the value ofR(decreasing the mean density) we
gradually enter into a highly correlated regime in which the correlation exhibits long-ranged and anisotropic ef-fects.
2
TABLE I: Comparison between the exact exchange and correlation energies for several sphere radiiRand the results given by the local and semi-local functionals evaluated on the exact density profile. The exact total energy is also included to illustrate the increasing importance of correlation for highR.
R Eex
tot E ex X E LDA X E GGA X E MGGA X E ex C E LDA C E GGA C E MGGA C 1 11.5910 -1.7581 -1.5230 -1.6843 -1.7805 -0.0507 -0.1424 -0.0775 -0.0647 5 0.7016 -0.3335 -0.2914 -0.3213 -0.3412 -0.0383 -0.0715 -0.0501 -0.0421 10 0.2381 -0.1592 -0.1407 -0.1550 -0.1651 -0.0288 -0.0481 -0.0362 -0.0301 25 0.0633 -0.0590 -0.0537 -0.0596 -0.0634 -0.0163 -0.0257 -0.0201 -0.0166 50 0.0249 -0.0278 -0.0262 -0.0296 -0.0311 -0.0093 -0.0151 -0.0116 -0.0095
to achieve the exact exchange energy for arbitrary
one-and two- electron systems (−WH[n] and−WH[n]/2
re-spectively), we can easily see how important is this limi-tation for two-electron densities with very distinct mean densities. On the other hand, LDA and GGA suffer from spurious correlation self-interaction, a limitation which is corrected by the MGGA [8, 11]. Nonetheless, the proper self-interaction correction does not guarantee the overall accuracy of the correlation functional, and the actual role played by non-local correlation effects has to be checked carefully.
As a first test, we will evaluate these functionals over the exact densities, i.e. in a non-self-consistent fashion, comparing the XC energies and potentials with the ex-act ones. Then, we will present the fully self-consistent solutions, in such a way that we will assess not only the self-consistentent energies but also the DFT densi-ties that minimizes the corresponding total energy
func-tionals. Atomic units (~ = me = e = 1) will be used
throughout the paper.
II. RESULTS ON THE EXACT DENSITY
PROFILE
The Hamiltonian of the two-electron model system is given by
ˆ
H =−1
2
2 X
i=1 ∇2i +
1
|~r1−~r2|+ 2 X
i=1
V (ri)
V(r) =
0 r < R
∞ r≥R.
The hard wall described byV(r) impose strict boundary
conditions atr =R, but does not have a direct
contri-bution to the total energy functionalE[n]. Therefore:
E[n] =TS[n] +WH[n] +EXC[n], (1)
whereTS[n] is the kinetic energy of the fictitious KS
non-interacting system. For a singlet state, the set of KS
equations is reduced to a single Schr¨odinger equation
−1
2∇
2+v S(r)
φ(r) =εφ(r), (2)
where vS = vH+vX+vC is the KS effective potential
which is the sum of the Hartree (vH), exchange (vX), and
correlation (vC) potentials. The density is related to the
ground-state orbital of (2) through the simple equality
φ(r) =pn(r)/2.
Under the exact DFT formulation, if nex(r) is the
ground-state density of the system, the corresponding
eigenvalueεexmust equal the ionization energyE[nex]−
E(1), where E[nex] = Eex
tot is the two-electron ground
state energy and E(1) =π2/ 2R2
is the energy of the one-electron system. Thus, the exact correlation poten-tial can be written explicitly as
vexC(r) =εex+
1 2
∇2p
nex(
r)
p
nex(
r)
−1
2
Z
dr′n
ex(
r)
|r−r′|, (3)
where we have used the exact relation EX[n] =
−WH[n]/2.[13] However, it is worth pointing out that (3)
is an expression only valid for the exact density profile. An exact functional expression for the correlation
poten-tialvC(r) of an arbitrary spin-unpolarized two-electron
densityn(r) would require to know the external potential
that defines the two interacting electron system whose
ground state isn(r) and then include such a potential.
Then, the simplicity suggested by (3) is just apparent. The performance of different local and semi-local pre-scriptions when evaluating the XC energy on the exact density profile is shown in Fig. 1 and Table I. The LDA systematically underestimates the absolute value of the exchange energy, whereas the GGA partially corrects this trend, although in the low density limit the GGA
over-estimates|EX|. The MGGA behaves reasonably well for
small radii, which is not a surprise since it reproduces exactly the exchange energy of the hydrogen atom and, as we said in the Introduction, in this range the model system behaves precisely like an atom. Thus, although the MGGA does not cancel exactly the spurious Hartree self-interactions, it fairly accounts for such a cancella-tion in atomic-like systems. When decreasing the mean electron density, the system cannot be considered like an atom any more and the MGGA greatly overestimates the
self-interaction corrections toWH, becoming even worse
than GGA.
Regarding the correlation energy EC[n], the LDA
3
5
DX
∆(
WRWS
HU
F
HQ
W
∆(
&S
HU
F
HQ
W
∆(
;S
HU
F
HQ
W
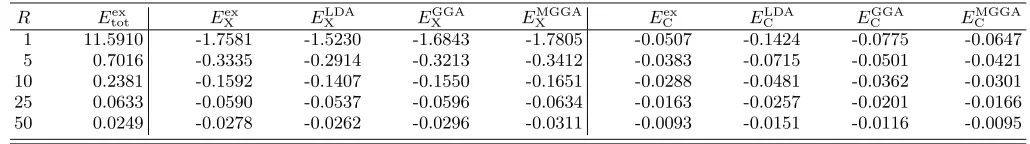
FIG. 1: Percent errors ∆E = 100× EDFT −Eex
/Eex for the exchange, correlation, and total energies as functions of the sphere radiusR. All DFT results are obtained non-self-consistently over the exact density. Solid line: LDA; dashed line: GGA; dotted line: MGGA; dash-dotted line: EXX+M.
too negative. On the contrary, the MGGA behaves ex-tremely well for all densities. Its relative error in the high density limit (around 20%) has a minor influence in the total energy, and such an error is less than 5% for lower densities. This excellent performance is in
agree-ment with the conclusions by Seidl et al [12] about the
essentially correct behavior of the meta-GGA correlation functional proposed in Ref. 11 (the basic ingredient of
the MGGA by Tao et al) under uniform scaling to the
low density limit. We have to bear in mind that in the
highly correlated regime, the two electrons are localized
in two different positions. That is, if one of the electrons
is at a distancerfrom the center, the probability to find
the second one is concentrated around a point on the op-posite side [9]. Thus, the LDA/GGA main source of error in this regime, corrected by the MGGA, is the absence of self-interaction corrections.
In the lower panel of Fig. 1 we present the accuracy of the corresponding non-self-consistent DFT total en-ergies. The well known compensation of errors between exchange and correlation in LDA and GGA is easy to observe in the atomic-like limit, but in the high
[image:5.612.60.297.49.379.2]UDX
Y
;&U
D
X
Y
&U
D
X
Y
;U
D
X
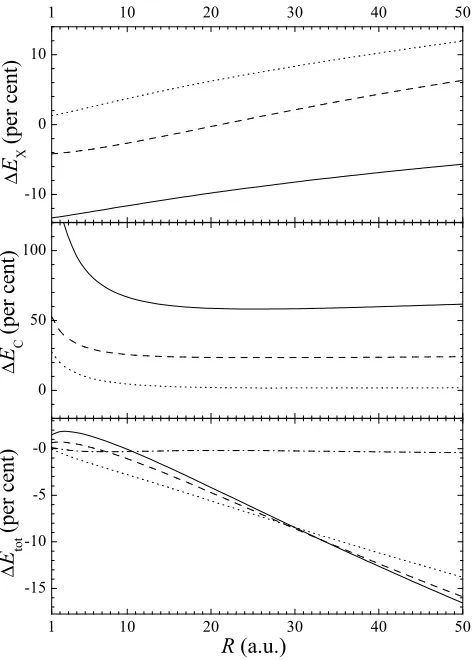
FIG. 2: Exchange and correlation potentials obtained from the exact density profile (R = 10). Thick solid line: exact results; thin solid line: LDA; dashed line: GGA; dotted line: MGGA; dash-dotted line: EXX+M. Note that the shape of the LDA XC potential reproduces fairly well the exact one excepting in the region around the center of the sphere. This error is amplified by the EXX+M prescription, where the ex-change part is exactly correct but the correlation potential is the same as the MGGA.
lation regime both functionals fail badly. The MGGA does not benefit from any cancellation of errors (it
over-estimates the absolute value of exchangeand correlation
energies). Hence, it slighty underestimates the total en-ergy for small radii, but the error on exchange dominates for lower densities where the MGGA perfoms poorly. A hyper-GGA [5], designed with the aim of being free of any self-interaction error, must yield very accurate total energies for all the ranges in our model system. Then, its performance should be similar to the presented in the lower panel of Fig. 1, where we plot the relative error on the total energy evaluated through a hybrid functional, EXX+M, where the exchange is calculated exactly and the correlation approximated by the MGGA. In this case, the error on the total energy is always less than 1 %, and
forR≃1 the absolute deviation is just 7 mHa/e, fairly
close to the chemical accuracy (around 2 mHa/e). From the shape of the potentials it is possible to see the underlying physics contained in the approximations.
4
highly correlated limit is an obvious consequence of the electrostatic repulsion, which tend to dominate over the confinement by the wall as we increase the radius of the system. Since the Hartree energy contains spurious self-interactions, the role of the exchange is to compensate
partially the effects due toWH. As we can see in the
up-per panel of Fig. 2, wherevX(r) is plotted for the model
system withR= 10, the exact exchange potential has a
mininum in the center of the sphere, favouring an atomic-like behavior. The corresponding LDA potential is not
able to account completely for this exchange attractive
feature, whereas the overall shift of vLDA
X is a
concomi-tant consequence of the local dependence on the density. The semi-local potentials exhibit a similar behavior but there are unphysical oscillations reflecting the presence of the gradient and the Laplacian of the density in the expression of the exchange potential.
On the contrary, the Coulomb correlation enhances the localization through a potential barrier located in the center of the sphere (see the middle panel of Fig. 2). This barrier reflects a truly non-local correlation effect.
In fact, forR= 10, the electron density is almost
homo-geneous around r = 0, and then vLDA
C (r) is practically
constant in this region. On the other hand, the LDA
fits reasonably well the exact potential ifr&5, but this
partial agreement is completely lost under the GGA and MGGA. This overall bad quality of the local and semi-local correlation potentials is a general feature in finite systems [14], although in some cases it could be masked if
we focus on the totalvXC(r). As we may see in the lower
panel of Fig. 2, the XC potentials given by LDA, GGA, and MGGA are rather similar (excepting the above men-tioned unphysical oscillations). Their shape is close to
the exact one forr&5 but, as expected, they do not
re-produce at all the correlation barrier atr= 0. Under the
EXX+M prescription, the situation is even worse, since
the XC potential reaches a minimum atr= 0 due to an
exact description of exchange which is not compensated by an accurate correlation potential. Therefore, although the correlation energies given by the MGGA are excel-lent, the potentials derived from it are not able to repro-duce the non-local effects that manifest themselves in the
shape of vC(r). As we will see in the next section, this
will lead to important deviations from the exact density profile when solving self-consistently the KS equation.
III. SELF CONSISTENT RESULTS
One of the major advantages of KS-DFT is its fully self-consistent character: any previous knowledge of the electron density profile is not required excepting for set-ting up an initial guess to start the iterative resolution of the KS equations. For many purposes, LDA and/or GGA give accurate self-consistent densities because the corre-sponding approximate potentials are very similar to the exact ones in those regions relevant for the calculation of the electron density. This justifies the use of more
Y
6U
ε
D
X
[image:6.612.322.558.49.198.2]UDX
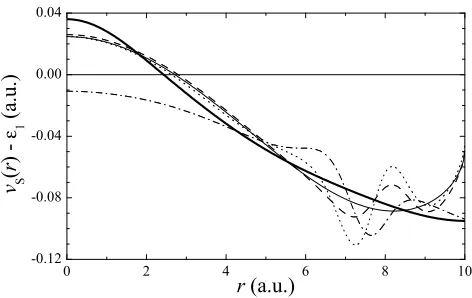
FIG. 3: Kohn-Sham potentialvS(r) for the exact density pro-file (R = 10). Thick solid line: exact result; thin solid line: LDA; dashed line: GGA; dotted line: MGGA; dash-dotted line: EXX+M. In order to allow an easier comparison, the potentials have been shifted in such a way that the corre-sponding first eigenvalue for each approximate potential is equal to zero. The behavior of the potentials near the sphere walls is less important due to the role played by the boundary conditionn(R) = 0.
ticated functional expressions as a mere correction over the self-consistent LDA/GGA densities. Consequently, the main effort carried out during the last years had been directed towards the improvement of the XC en-ergies, paying less attention to the characteristics of the
XC potentialvXC(r).
Nonetheless, in the previous section we have seen that all the functionals considered in this paper fail to re-produce the exact XC potential in a region where the electron density is far from being negligible. The con-sequences can be seen in Fig. 3, where we compare the
exact KS potential vS(r) with the DFT ones obtained
from the exact density profilenex(r). The LDA, GGA,
and MGGA do not reproduce the exact shape of vS(r)
and it is reflected by a classical forbidden region greater than the actual one: the self-consistent density will be pushed towards the walls. The EXX+M model, as co-mented, incorporates exactly the attractive character of
exchange, but the wrong description ofvC(r) makes the
effective potential less confining than the exact one: this hybrid approach tends to concentrate the density around
r= 0.
These deviations from the exact two-electron density profile, which can be quantified through the expression
δn= 1 2
Z
dr|nex(r)−n(r)|, (4)
5
5 DX
5 DX
Q U Â 5 U Q U Â5
5 DX
U5
[image:7.612.60.294.52.258.2]
U5
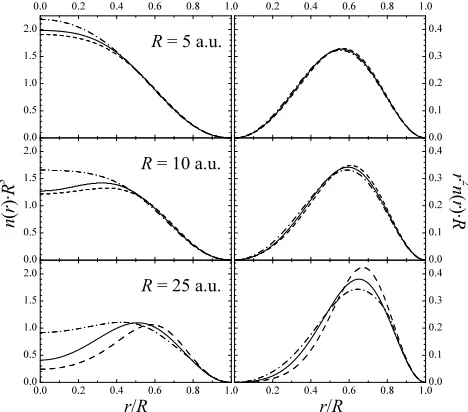
FIG. 4: Exact (solid line) and self-consistent GGA (dashes) and EXX+M (dash-dots) densities n(r) and reduced radial densities r2
n(r). All the quantities have been scaled with the radius of the confining sphere. The self-consistent LDA and MGGA densities are very similar to the GGA ones and have not been included in the figure. None of the approx-imate functionals is able to reproduce the correct behavior of the density in the center of the sphere, and their overall performance is very poor forR≫0.
compare the DFT densitiesn(r) as well as the the
cor-responding reduced radial densitiesr2n(r) with their
ex-act counterparts. In the atomic-like regime (R.5), we
can observe genuine errors on the DFT densities around the center of the sphere. However, this wrong behavior will lead to marginal errors on integrated quantities, as suggested by the overall good agreement shown by the reduced radial densities. At intermediate mean densities
(R ≃ 10), the differences on r2n(r) can be already
ob-served at a first glance. Moreover whereas the system shows an incipent localized behavior, which is
character-ized by a density reaching a local maximum at r 6= 0,
the EXX+M self-consistent density still has an atomic
behavior characterized by a maximum atr= 0. Finally,
in the low density limit, all the approximate functionals fail to describe the exact density profile with a minimum
accuracy. For instance, if R= 25 the error given by (4)
is 8.6 % using the EXX+M functional and 14.2 % using the GGA.
Then, the full minimizimation of E[n] adds a
fur-ther source of error due to the inaccuracies of the
self-consistent density. Nonetheless, these
self-consistency-inducederrors in the total energies are going to be less
im-portant because there is an overall trend to cancellation, see Table II. For instance, the EXX+M self-consistent density is smoother than the exact one, which lowers the kinetic and exchange energies while increasing the Hartree interaction energy. However, in spite of the dis-torted density profile, there is a fortunate cancellation of
TABLE II: Self-consistent DFT-KS results for several radii compared with the exact ones. The last file of each entry contains the DFT density errorδnas defined in Eq. 4.
Exact LDA GGA MGGA EXX+M
R= 1
Etot 11.5910 11.7338 11.6376 11.5540 11.5770
TS+WH 13.3999 13.3955 13.3974 13.3973 13.4000
EX -1.7581 -1.5193 -1.6820 -1.7785 -1.7582
EC -0.0507 -0.1423 -0.0778 -0.0648 -0.0647
δn 0.0070 0.0049 0.0056 0.0006
R= 5
Etot 0.7016 0.7104 0.7017 0.6897 0.6975
TS+WH 1.0734 1.0713 1.0716 1.0720 1.0747
EX -0.3335 -0.2897 -0.3196 -0.3401 -0.3348
EC -0.0383 -0.0713 -0.0504 -0.0421 -0.0423
δn 0.0210 0.0222 0.0196 0.0157
R= 10
Etot 0.2381 0.2371 0.2346 0.2305 0.2362
TS+WH 0.4261 0.4245 0.4246 0.4249 0.4275
EX -0.1592 -0.1395 -0.1537 -0.1643 -0.1608
EC -0.0288 -0.0479 -0.0363 -0.0301 -0.0306
δn 0.0383 0.0448 0.0371 0.0401
R= 25
Etot 0.0633 0.0587 0.0584 0.0582 0.0626
TS+WH 0.1387 0.1377 0.1376 0.1377 0.1395
EX -0.0590 -0.0533 -0.0597 -0.0632 -0.0600
EC -0.0163 -0.0256 -0.0195 -0.0163 -0.0170
δn 0.1352 0.1424 0.1300 0.0858
R= 50
Etot 0.0249 0.0197 0.0199 0.0204 0.0243
TS+WH 0.0620 0.0621 0.0622 0.0623 0.0624
EX -0.0278 -0.0270 -0.0318 -0.0330 -0.0282
EC -0.0093 -0.0154 -0.0105 -0.0088 -0.0099
δn 0.3317 0.3348 0.3343 0.1084
errors that makes the sum of these three terms practi-cally equal to the exact value. Thus, the minimization of the energy leads to changes on the density profile favoring lower correlation energies, which is done by increasing the density around the center of the sphere. As a result, self-consistent total energies are just slighty worse than the
non-self-consistent ones, the relative error ∆Etotranging
from -0.1% forR= 1 to -2.5% forR= 50. A similar
con-clusion reads for the remaining functionals (LDA, GGA,
MGGA), although in this case the self-consistent TS[n]
is greater than the exact one, whereas the classical inter-action energy is reduced after the minimization. Hence, self-consistency keeps the fairly good quality of the to-tal energies in the atomic limit, where there are minor changes in the density profile, and for intermediate and
low densities the dramatic changes onn(r) induce a few
[image:7.612.318.563.96.427.2]6
IV. CONCLUSIONS
In this work we have assessed the quality of some
of the most popular XC functionals used in ab-initio
electronic structure calculations in a simple system of electrons where several correlation regimes can be easily achieved. None of the exchange functionals is completely free of self-interaction errors, making imposible to obtain accurate energies for highly correlated electrons. Local and GGA correlation exhibit a similar drawback, but the MGGA correlation shows an excellent performance for all the regimes. Nonetheless, neither LDA/GGA nor MGGA can describe highly non-local correlation effects that lead to a non-trivial behavior of the correlation potential, so seriously affecting the quality of the self-consistent den-sities. Due to persistent cancellation of trends, the corre-sponding changes on the total energy after minimization are less important, but this uncontrolled source of error might prevent these approximate functionals from having full predictive accuracy.
The overall bad quality of the self-consistent KS po-tential also compromises the evaluation of post-self-consistency corrections based on more sophisticated methods, like Many-Body Perturbation Theory or time-dependent DFT [3], if the wrong effective potential is not corrected as well. On the other hand, an accurate description of the XC potential is required, for instance, when studying neutral excitations in finite systems
us-ing time-dependent DFT [15]. As shown recently by
Della Sala and G¨orling [16], for those systems having an HOMO orbital with nodal surfaces, the exact exchange potential tends to a constant if going to infinity over a set of zero measure directions. This leads to the appear-ance of potential barriers that, although could be of mi-nor importance when obtaining the self-consistent static results, might be essential if a proper description of all the unoccupied KS orbitals was required. Here, although in a very different context, potential barriers induced by the non-locality of the many-body effects in an electron system have been observed as well.
The limitations observed for this family of proto-type systems might have relevance for real molecular or condensed-matter systems in some cases. Prospective tests of the MGGA functional used in this work show an excellent performance for a wide variety of typical molec-ular and solid state systems which, moreover, seems to be kept if partial self-consistency is achieved [8]. How-ever, there is no guarantee that the energy minimization procedure may lead to small, but relevant changes on the local electron density in, for instance, the bonding region between two species, so giving a wrong account of the nature of such a chemical bond. Finally, for this model system we have seen that there are not substantial differ-ences between the fully self-consistent GGA and MGGA densities, although in this case the MGGA functionals take a simple GGA-like form. In spite of this simplifi-cation, the MGGA-XC potential amplifies the spurious oscillations already appearing under the GGA prescrip-tion. As a conclusion, the overall capability of MGGA and envisaged improvements thereof to yield accurate XC energies can hardly be questioned (specially for the cor-relation part), but further studies of the actual perfor-mance of the corresponding KS potentials are required. The latter point is relevant for those situations in which LDA/GGA are not able to reproduce the electron density with the requiered accuracy, and for other DFT-based
ap-plications needing an overall good description ofvS(r).
Acknowledgments
We wish to thank R. Almeida, T. Gould, J. M. Soler, D. Garc´ıa Aldea, J. Tao and D. C. Thompson for fruit-ful discussions, and K. Burke for providing the subrou-tine for the PBE GGA functional. This work has been funded in part by the EU through the NANOPHASE Research Training Network (Contract No. HPRN-CT-2000-00167), the Spanish Science and Technology Min-istry grant BFM2001-1679-C03-03 and by a grant from the UNED Fund for Research 2001.
[1] W. Kohn and L. Sham, Phys. Rev.140, A1133 (1965). [2] P. Hohenberg and W. Kohn, Phys. Rev. 136, B864
(1964).
[3] R. W. Godby and P. Garc´ia-Gonz´alez, in A primer in Density Functional Theory, edited by C. Fiolhais, F. Nogueira, and M. Marques (Springer, Berlin 2003), p. 185, and references therein.
[4] ´A. Nagy, Phys. Rep.298, 1 (1998).
[5] J. P. Perdew and S. Kurth, inA primer in Density Func-tional Theory, edited by C. Fiolhais, F. Nogueira, and M. Marques (Springer, Berlin 2003), p. 1.
[6] J. P. Perdew and Y. Wang, Phys. Rev. B 45, 13244 (1992).
[7] J. P. Perdew, K. Burke, and M. Ernzerhof, Phys. Rev. Lett.77, 3865 (1996).
[8] J. Tao, J. P. Perdew, V. N. Staroverov, and G. E. Scuse-ria, Phys. Rev. Lett.91, 146401 (2003).
[9] J. Jung and J. E. Alvarellos, J. Chem. Phys.118, 10825 (2003).
[10] D. C. Thompson and A. Alavi, Phys. Rev. B66, 235118 (2002);ibid.68, 039901 (2003) (E).
[11] J. P. Perdew, S. Kurth, A. Zupan, and P. Blaha, Phys. Rev. Lett.82, 2544 (1999).
[12] M. Seidl, J. P. Perdew, and S. Kurth, Phys. Rev. A62, 012502 (2000).
[13] Note that in this particular case with two electrons the exact exchange energy defined in the KS-DFT formalim is equivalent to the usual Restricted Hartree Fock ex-change energy.
7
(1994).
[15] G. Onida, L. Reining, and A. Rubio, Rev. Mod. Phys. 74, 601 (2002) and references therein.