0022-538X/94/$04.00+0
Copyright (C 1994,American Society for Microbiology
Multiple Functions of Capsid Protein Phosphorylation
in Duck Hepatitis B Virus Replication
MINSHU YU ANDJESSE SUMMERS*
Departmentof Cell Biology, University of New Mexico School of Medicine, Albuquerque, New Mexico 87131
Received 15 February 1994/Accepted 12 April 1994
We have investigated the role of phosphorylation of the capsid protein of the avian hepadnavirus duck hepatitis B virus in viral replication. We found previously that three serines and one threonine in the C-terminal 24 amino acids of the capsid protein serve as phosphorylation sites and that the pattern of
phosphorylationatthese sites in intracellular viral capsids is complex. In this study, we present evidence that the phosphorylation state of three of these residues affects distinct steps in viral replication. By substituting theseresidues with alanineinordertomimic serine, or with asparticacidin order to mimic phosphoserine, andassaying the effects of these substitutionson various steps in virusreplication,we were able to make the
following inferences. (i) The presence of phosphoserines at residues245 and259 stimulates DNA synthesis within viral nucleocapsids. (ii) The absence ofphosphoserine at residue 257 and at residues 257 and 259 stimulates covalentlyclosed circularDNA synthesis andvirus production,
respectively.
(iii) The presence of phosphoserine at position 259 is required for initiation of infection. The results implied that both phosphorylated and nonphosphorylated capsid proteins werenecessaryfor a nucleocapsid particle tocarryout all itsfunctions in virus replication, explaining why differential phosphorylation ofthecapsid protein occurs inhepadnaviruses.Whetherthesedifferentiallyphosphorylated proteins coexist on the same nucleocapsid, or whether thenucleocapsid acquires sequential functions throughselectivephosphorylation anddephosphory-lation, is discussed.
Hepadnavirusesare afamilyof small enveloped viruses with a partially double-stranded circular DNA genome that repli-catesthroughreversetranscription. During initiation of infec-tion, virus particles deliver the DNAgenome to the nucleus, where itis converted to acovalently closed circular (ccc)DNA thatservesas atranscriptional template for the production of RNAgenomes(pregenome) (8, 13,15, 17).RNApregenomes are encapsidated in the cytoplasm by viral capsid proteins to formimmature nucleocapsid particles within which viralDNA
synthesis occurs. Initially, aminus-strand DNAis synthesized by reverse transcription of the pregenome (7, 12). Minus-strand DNA is subsequently used as the template for
plus-strand DNAsynthesis.Matureintracellularnucleocapsids
con-taining double-stranded DNA proceed along one of two
alternatepathways. Earlyduring the infectious cycle, the DNA inmaturecapsidparticles is deliveredtothenucleus, resulting inan amplificationof the copy number of cccDNA(15).Late
during infection, productionof thelarge viral envelopeprotein
inhibits this amplification, redirectingviral nucleocapsids into
envelopedvirusparticles,whichareexportedfrom the cell(13, 14). Thus, nucleocapsidsareinvolved inanumber of sequen-tialfunctions, namely, RNApackaging, DNA synthesis, deliv-ery of viral DNA to the nucleus, and recognition of viral
envelopeproteins.Thesequential
expression
ofsomeof thesefunctionsmaybe
regulated by
protein
modification.The capsid protein of the duck hepatitis Bvirus
(DHBV)
consists of 262 amino acid residues. Theproteincanassemble into a capsid in the absence of other viral proteins. The C terminus of the capsid protein contains one threonine and threeserines that serve aspotentialphosphorylation
sites on the surface of immaturenucleocapsids (10,
11,20).
Ininfected*Corresponding author. Phone: (505) 277-7979. Fax: (505)
277-9494.Electronic mail address:[email protected].
cells, these four sites arephosphorylated in various
combina-tions,resulting inelectrophoretic heterogeneityof theprotein
insodium dodecyl sulfate(SDS) gels. While the population of intracellular viralcapsids contains capsid proteins phosphory-lated at zero to four of these sites, capsid proteins isolated from extracellular viruses are electrophoretically homoge-neous in SDS gels and comigrate with unphosphorylated capsid protein. This difference in the phosphorylationstatesof intracellular andmatureviralcapsids suggested that phosphor-ylationmayplayarole in intracellular capsid functionorviral morphogenesis (10).
Asafirststepin understandingthe role of phosphorylation invirusreplication,weexaminedthephenotypes ofaseries of serineorthreonineaminoacid substitutions in the C terminus of capsid protein. We assayed for RNA packaging, DNA
synthesis, intracellularlocalization of the protein, production
ofenvelopedviruses, and viralinfectivity. The resultssuggest that each of three of these four residues participates in a distinct manner incapsid function, dependingon its stateof
phosphorylation.
MATERUILSAND METHODS
Plasmid and mutant construction. Wild-type and mutant capsid
proteins
wereexpressed
inpermissive
cells from a plasmid in which the DHBVpregenome encoding sequenceslacking the precore region was cloned
immediately
down-stream of the immediate-earlycytomegalovirus (CMV)
pro-moter, aspreviouslydescribed (19). Stopcodons in the enve-lope gene (T->A at nucleotides 1327, 1346, and1349)
prevented the
expression
of eitherenvelope
protein.
Thesemutations, designated
collectively
as 1S, werepreviously
de-scribed (13).Inordertoalter the
potential
phosphorylation
sitesatthe C terminus ofcapsid protein,
a series ofpoint
mutationswere 4341on November 9, 2019 by guest
http://jvi.asm.org/
4342 YU AND SUMMERS
introduced
by
oligonucleotide-directed
mutagenesis
into codonscoding
for serine and threonine in the 3' end of thecapsid
gene openreading frame, resulting
in individual re-placementofserine and threonineby
alanineoraspartic acid,
as we describedpreviously. Fully sequenced
restrictionfrag-mentsthat contained themutation of interestwere
individually
subcloned into thecapsid protein expression plasmid.
Nomen-clature for thereplacement
mutants followed the convention ofaletterdesignation
of thewild-type
aminoacid,
followedby
the residuenumber,
followedby
the substituted amino acid(e.g., S245A).
Theplasmid pSPDHBV RV2650,
used intesting
the function of alteredcapsid protein by complementation,
consisted ofpSP65
containing
an EcoRI dimer of a DHBV genome in which a frameshift mutation atposition
2560destroyed
theproduction
of anycapsid protein (3).
We used twoversions of thisplasmid:
one in which theenvelope
genes wereintact,
and one in which stop codons in thep17
geneprevented expression
of bothpre-S
and Sproteins (13).
An infectious
plasmid
used fortesting
the effect of the mutation S259Aoninfectivity
wasconstructedby
substitution ofthe serine 259codonby
analaninecodonatthe3' end of thecapsid protein
gene. The mutant genome was cloned down-stream of theimmediate-early
CMV promoter in the vectorpUCl
19 such thattranscription began
at the authentic viralcap site
(14).
Cell culture and transfection. Transfection of
plasmid
DNAswascarriedoutby
the calciumphosphate
coprecipita-tion method(13)
inthe chickenhepatoma
celllineLMH(1, 4),
maintained in F-12-Dulbecco modifiedEaglemediumsupple-mented with 10%fetal bovineserum. Cellswereincubated at
37°C
for 4days
after addition of DNAs(10 ,ug/60-mm dish).
Cotransfection ofpSPDHBV
RV2650 andcapsid protein
expression plasmids
wasperformed
with 5 ,ug of each DNA.Primary
duckhepatocyte
cultureswereused for thegrowth
of DHBV.Primary hepatocytes
wereprepared
from 5-to 10-day-oldducklings by perfusion
of the liver withcollagenase
aspreviously
described(16).
Duckhepatocytes
were maintained in Leibowitz medium(L-15)
supplemented
withglucose (1
mg/ml),
insulin(1
jxg/ml),
hydrocortisone
hemisuccinate(10
,uM),
anddimethyl
sulfoxide(1%) (9).
Aliquots
of virusconcentrated from theculturefluidsof transfected LMH cells
by polyethylene glycol precipitation
were used to infect each 60-mm dish of duckhepatocytes
aspreviously
described(14).
Extractionandassay of intact viralcapsids containingRNA and DNA.TransfectedLMHcells in 60-mmdisheswerelysedby
the addition of 0.5 ml oflysis
buffer(50
mmTris-HCl [pH8.0],
1 mMEDTA,
1% NonidetP-40).
The platewasrockedgently
todistribute the buffer andwaskeptat37°Cfor 10 min.The
lysate,
containing capsids,
wassubjectedtomicrocentrifu-gation
to removenuclei and cell membrane debris.To removeDNA not present in
nucleocapsids,
we added 6 mM Mg-acetate and 100 ,ugofDNase Ipermltothe supernatant and incubated it at37°Cfor30 min.A
portion (10 ,ul)
of thelysatewasmixed with2[lI
ofsample buffer(50% glycerol,
0.1%bromphenol blue) andloaded ontoa 1% agarose gel prepared in 10 Mm Na-phosphate
electro-phoresis buffer,
pH 7.5. The capsids were electrophoresed toward the anode at 50 V with recirculation of the buffer.Capsids
were transferred directly to nylon or nitrocellulose filters with TNE buffer (10 mMTris-HCl
[pH 7.4], 1 mM EDTA, 150 mMNaCl)
by blotting,and thefilter was washed for 10 min indistilledwateranddried. At this point, the filterwas
probed
forcoreantigens (nitrocellulose)byimmunostain-ing
aspreviously
described (13), or the nucleic acid was released in situ forhybridization
analysisby wetting the filter(nylon)
for 10to20 s in 0.2 N NaOH-1.5 M NaCl, followedimmediately by neutralization in 0.2 N Trizma-HCI-1.5 M NaCl for 5 min, and washed again with distilled water for 10 min. The dried filter was probed for either total RNA and plus-strand DNA, or for minus-strand DNA with strand-specific 32P-labelled RNAs.
Extraction and analysis of DNA replicative intermediates from cultured cells. DNA replicative intermediateswere ex-tracted either from transfected LMH cells or from infected primary duck hepatocytes as previously described (13). Four days posttransfection, cells were lysed and DNA in the super-natantswasremovedby DNase I digestion. Nucleic acids were purified by protease digestion and phenol extraction and collectedbyethanolprecipitation. Viral DNAwasassayed in eachsampleby 1% agarose gel electrophoresis and Southern blot hybridization.
Analysis of cccDNA in transfected cells. Transfected cells in 60-mm plates were washed once with HBS buffer
(N-2-hy-droxyethylpiperazine-N'-2-ethanesulfonic acid [pH 7.45], 150 mM NaCl), and 1 ml of cccDNA isolation buffer (10 mM
Tris-HCl[pH 7.5], 10 mM EDTA,1% SDS)wasaddedtoeach
plateand incubated for 5 minat37°C. A total of 0.25 ml of 2.5 M KCl was added to the lysate, and the lysate was briefly
vortexed and chilled on ice for 5 min. After removal of the
detergent-protein complexes by centrifugation, the superna-tant was extracted with phenol, and the nucleic acids were
precipitatedwithethanol. The dried DNApelletwasdissolved in20 ,lIof TE (10mMTris-HCl [pH 7.4], 1 mM EDTA).
Toeliminate transfectedplasmidsthatusuallycontaminated the viral cccDNAfraction,the nucleic acidsweredigestedwith
DpnI, which carries out methylation-dependent cleavage at manysites in theplasmid.Residualfragmentsofplasmid DNA werefurtherdigestedwith exonuclease III.Briefly,5 ,ul of the cccDNAsolutionwasincubated with 5 U ofDpnI and 25 U of exonuclease III in restriction buffer containing 10 mM Tris
(pH 7.5), 10 mM magnesium acetate, 50 mM NaCl, 1 mM
dithiothreitol,and0.01% NonidetP-40and incubatedat37°C
for2 h. The viral cccDNA in thedigestwasassayeddirectly by agarosegel
electrophoresis
andSouthern blothybridization.Assay of virus particles from supernatants of transfected cells.Virusparticleswereprecipitatedfromthesupernatants of transfectedcellsbytheadditionof10% (wt/vol)
polyethyl-ene glycol (molecular weight, 7,000 to 9,000) as previously described (14). After centrifugation, the tube was carefully
drained and the insidewaswipedfree ofexcess polyethylene glycol. The pellet was dissolved in 1/50 volume of 2 mM HEPES(pH 7.4)-150mMNaCl-2 mMCaCl2.Atotal of 5
RI
of the dissolvedpelletwasaddedto15pAlof TEcontaining 750 ,ugof pronase per ml andincubated for 60 minat37°C. Thisdigestion was sufficient to disrupt viral cores that were not bound inalipid envelope,butenvelopedviruswascompletely
resistanttopronase (Sa). Free viral DNA was removed from thesuspension bythe addition of 10 mM Mg-acetate and 500 ,ug ofDNase Iperml (type I, Sigma) andincubation at 37°C for 30 min. The sample was subjected to electrophoresis
through a 1% agarose gel with DNA electrode buffer with bufferrecirculation. Thevirusparticles migrated with an Rf of about 0.15with respecttothe bromphenol blue tracking dye. Virus particles were transferred to a nylon filter by blotting with TNE. The filter was thoroughly dried, and the
DNA-containing particlesweredenaturedby soaking the filter in 0.2 N NaOH containing 1.5 M NaCl. The filter was neutralized with 0.2 M Trizma-HCl containing 1.5 M NaCl, washed in TNE, and dried. Viral DNA was detected by hybridization of thefilterwitha
32P-riboprobe
specific for detection of the viral minus strand.Immunofluorescent staining of capsid protein in cultured J. VIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
Capsid expression vector
CM
I)rR1 L iS-I
R1
229-rSkSrerrapTpqragSplprSSSShhrSpSprk-262
Capsid
defective genome
Rl
1S/1St
DHBV
Rl
r
S/wt
Rl
v IF AL v
_
_~1~capsid
-~M
pol
- ~ ~ ~
envelope
RV2650
I . I
3021/1 3021
FIG. 1. Expression plasmids used in the study. Capsid proteins
wereproduced from the expression vectordepicted on thetop.The immediate-earlyCMVpromoterwasusedtodriveexpression of viral RNA. The DHBVsequencescontaining the 5' packaging signalwere
deleted(I), andstopcodonswereintroduced into the envelopeopen
reading frame (iS). The EcoRI sitesdelimiting the cloned monomer
viral DNA (Ri)areshown. Wild-type andmutantproteinsweretested
for their ability to complement a capsid-defective pregenome. The
pregenome wastranscribed from a viral DNA dimer, shown at the bottom. The viral DNA sustained a2-bp deletion (RV2650) in the
capsidopenreading frame. The viralpregenomepromoter(DHBV) is indicated.wt,wildtype.
cells. Transfected LMH cells and infected hepatocytes were
fixed and stainedwith rabbitantibody specificfortheDHBV capsid protein aspreviously described (3).
RESULTS
We determined the phenotypes of various altered capsid proteins after their expression in the chicken hepatoma cell
lineLMH.Tomeasuretheeffects of mutationsonstepsin viral
DNA synthesis and virus production, we coexpressed the
capsid proteinswithaDHBVgenomethat couldnotproduce
any capsid protein and measured the ability of the altered proteins tocomplement the defect in the DHBV genome,as wehave previously described (19). Using this assay, we were
able to avoid effectsofchanges inthe Popen reading frame,
whichoverlaps the regionofthecapsidgenethatwemutated.
Thevectorsweused forexpression ofthe capsid proteinand
thecapsid-defective genome areshown inFig. 1.
Stop codons (1S mutation) introduced into the envelope
gene thatwaspresent in thecapsid protein expressionvector preventedthe expressionofanyintact envelope proteinfrom this plasmid. In experiments in which the effect ofenvelope protein expressionwasstudied,wecomparedthe complemen-tation ofa capsid-defective genome that contained an intact envelope region with that of one that contained the 1S
mutation.
A series of point mutations that we introduced into the capsidgene resultedin the substitution of alanine codons for nine individual serine codons andonethreoninecodonatthe 3' end of the core gene open reading frame. Using these mutants,wepreviouslyshowed that threeserines,S245, S257,
FIG. 2. Immunostaining ofDHBV capsid protein in transfected
LMHcells. LMHcellswerestainedfor DHBVcapsid proteinat48h
posttransfectionwith thewild-type capsid protein expressionvector.
Capsid proteindistributionwasalwayspredominantly cytoplasmic (as
shown)in allsubstitutionmutantsdescribedin thisstudy regardless of thepresence ofenvelopeproteinsorviral DNAsynthesis.
andS259, and onethreonine, T239, functionas
phosphoryla-tion sites in thewild-type protein. Since an alanine in one of
these positions might functionally mimic the nonphosphory-lated wild-type amino acid residue serine, we believed that
replication defects associated with these mutations might reveal the requirementsforphosphorylation atthese sites.
We also constructeda series ofmutants in which the four previously identified phosphoacceptor residues were
substi-tuted withaspartic acid,whichwebelievedmight functionally
mimicphosphoserine.Defectsassociatedwith these mutations mightrevealanyrequirementfornonphosphorylatedserineat
aparticularsite.
Intracellular distribution ofcapsid proteins in transfected cells. Immunofluorescent staining of cells transfected with either alanine or aspartic acid substitution mutants in each
case showed the capsid antigendistributed in cytoplasm in a
manner similar to that observed with the wild-type capsid
protein (showninFig. 2), suggestingthatcytoplasmic localiza-tion of the capsid protein ofDHBV was not affected by the phosphorylationstate ofanyindividualresidue.
Capsid assembly and pregenomeencapsidation in replace-ment mutants.Todeterminewhetherpreventionof serineor
threonine phosphorylation at specific residues exerted any
effect on viral capsid assembly, we assayed the formation of
capsids byourmutantsbyelectrophoresisofparticles through nondenaturing agarose gels and Western blot (immunoblot) 1
on November 9, 2019 by guest
http://jvi.asm.org/
[image:3.612.319.560.83.392.2] [image:3.612.61.298.190.300.2]4344 YU AND SUMMERS
6<< < < <:<:< << <
NN N N N N c, N 0s
> X X wen vo co u Uz <a) a U)< coQD < coLO <:a) 00
" C"4 NC N N
5 e
l-CLo Qo
(5
CD
oa a) 0U)e r C) U)
tt " N " N N" CN
w
s uz toLI)c>Gw w w
- - + _ + _ + - + - t + - -
[image:4.612.320.566.79.234.2]-presence of envelope protein
FIG. 4. Effectsofamino acidreplacementsonviralDNAsynthesis. LMH cellswerecotransfectedwithplasmids expressingthe indicated capsid protein and a capsid-defective genome in the presence or
absence ofenvelope protein expressionfrom thecomplementedviral
genome.Replicative intermediateswereassayed byagarosegel elec-trophoresisand blot hybridization. WT,wild type.
X,*..~~~~
FIG. 3. Effects of serine-to-alaninereplacementsoncapsid
forma-tion, RNApackaging, and DNA synthesis. LMHcells cotransfected withplasmids expressing the indicated capsid proteinsand a
capsid-defectivegenomewerelysed, andcytoplasmic capsidswere analyzed
byagarose gel electrophoreses and transfertoafilterfor(a) immu-nostainingor (b)detection oftotalplus-strandviral nucleic acid. In panelc,total viralreplicativeintermediateswere extracted from the capsidsandassayed byagarosegel electrophoresisandblot hybridiza-tion.WT,wildtype.
analysis.Theelectrophoreticpatternsproduced bythe altered proteinsweresimilartothoseproduced bythewild-type capsid protein (Fig. 3a), indicating that replacement mutants were
able todirecttheproductionof stable capsids.
We measured the viral specific nucleic acid content of intracellularcapsids bytransferof thecapsidstoanylon filter,
denaturation, and hybridization to a riboprobe specific for
detection of viral plus strands (Fig. 3b). The results showed thatindividual substitution ofthevariouspotential phosphor-ylation sites by alanine, including threonine 239, serine 257, and serine 259, failed to affect the levels of totalviral plus-strandnucleic acid (ameasureoftotal RNA plusmatureviral DNA) encapsidated compared with the wild-type protein. Alanine substitution of serine 245 caused a reduction in
plus-strand nucleic acid associated with nucleocapsids;
how-ever, thisreduced signal could be accounted for bya specific
defect in plus-strand DNA synthesis (see below) rather than reduced packaging of RNA. The results indicated that RNA packaging into nucleocapsids did not require any specific
individual phosphorylation state at these four amino acid residues.
Effect ofaminoacid substitutions on viral DNAsynthesis.
When individual serine and threonine mutants were used to supply capsid proteins to acapsid-defective genome intrans, replicative intermediates could be observed in all cases (Fig.
3c). However, alanine substitution of serine 245 resulted in a
specific failure to accumulate mature relaxed circular DNA (S245A). The phenotype was similar to that of mutants we
have previously described (class II phenotype) that resulted from truncations of 19 to 25 amino acids atthe C terminus (19). The region that is missing from the C terminus of all deletion mutantshavingthe class II phenotypewould include the serine 245 phosphorylation site, and the absence of this potentialphosphorylation site inthese deletionmutantscould accountfor theclass IIphenotype (19).
We showedpreviouslythat thedownstreamadjacent proline of each phosphoacceptor serine and threonine was essential
for the mobilityshift in SDS gels associatedwith phosphory-lation.Whethertheprolineactedas asignal for recognition by aprotein kinase, orwhetherreplacement ofthe prolinewith glycine rendered themobilityin SDS gelsinsensitive to phos-phorylation,wasnotdetermined. Replacement of proline 246 withglycinealsoresultedinafailuretosynthesizematureviral DNA (Fig. 4, P246G). To test further whether the specific S245A and P246G phenotypes were a result of a failure to
phosphorylate residue 245 or were due to the conservative missense mutationsthemselves,wesubstituted serine 245 with
aspartic acid. The capsid protein of this mutantwas able to support normal viral DNA maturation, as shown in Fig. 4
(S245D). It was likely, therefore, that phosphoserine rather
thanserine itselfwasfunctional inthe synthesis ofmatureviral DNA.
We also tested whether the presence of aspartic acid at
position245 couldrestore function tothe mutantP246G. As shown in Fig. 4, substitution of aspartic acid for serine in
mutantP246G(Fig.4,S245D/P246G)didnot restorewild-type
function. The result suggests that the defect caused by the proline-to-glycine substitution was not due to prevention of phosphorylation of serine245,butthat proline, along with its adjacentupstreamphosphoserineoraspartic acid,maybepart
ofasingle functional unit.
Substitutionof serine 259 withalanine resultedinathree-to fivefold defect intotal DNAsynthesis (Fig. 3c and4,S259A).
a
b
'41
.4
J. VIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:4.612.62.299.85.419.2]ca cn ul
qt
s < N N4 C%l CM
j~~~~~~
4
_
..l
~~~~
I1-tCl 0)
LO LO
LO
Lo
U) U) U) U)
[image:5.612.62.295.78.352.2] [image:5.612.319.553.83.184.2]N Cl CM CN
...
:w_.0
<
<
<
<
0D
C
0D
CM) LO 1LC CD L LO LD O
CO N' U' Ln L qt LO U)
, N C N N N CN N CN
5 w (1w 5 wO(: J
6.
cs m LO LO P rl- m O
m) In qI.. LOULO LO LO
N CN CM CM N N N N
5~~~5 CO 0 m CJ CJ
l_
4I1
FIG. 5. Effects of amino acid replacements oncccDNAsynthesis.
LMH cellswerecotransfected with plasmids expressing the indicated
capsid protein and a capsid-defective genome in the presence or
absence ofenvelope protein expression from the complemented viral
genome.ViralcccDNAwasselectively purified and assayed byagarose
gel electrophoresis and blot hybridization.WT,wildtype.
We interpret this defect as an inhibition of an earlystep in minus-strand DNAsynthesis, since RNA packagingwas
nor-mal, thepatternofnascentminus strandswasnormal, and the
minus strandswereusedastemplates for plus-strand synthesis
in a seemingly normal manner. Substitution of the adjacent
downstreamproline produced thesamedefect (Fig. 4, P260G),
while aspartic acid replacement resulted in normal levels of viral DNA (Fig. 4, S259D). These results suggested that the inhibitionofminus-strandsynthesis by alanine substitutionwas
duetolackofphosphorylation atresidue 259.
Effect of alanine substitutions on synthesis and regulation
of cccDNA. We analyzed the levels of cccDNA in cells trans-fected with the various alanine substitutionmutants.Since the maintenance of normal levels of cccDNA in the nucleus depends on the regulation by the 36-kDa pre-S envelope protein, we assayed cccDNA levels in cells expressing the altered capsid proteins in the absence andpresence ofpre-S envelope proteins.Theresults showed that withoneexception
(S245A), cccDNAsynthesis reachedrelatively equivalent lev-els in the cells transfected with the alanine substitution
mu-tants and that cccDNA levelswere reducedasexpectedin the
presenceof thepre-S envelope proteins (Fig. 5).
ThemutantS245Ashowed reducedproductionof cccDNA in the absence of the envelope protein, consistent with its defect in the production of mature DNA. However, this mutant produced nearlythe same cccDNAlevel in the
pres-ence of thepre-S envelope protein. This result indicated that the cccDNA level in this mutant was not regulated by pre-S envelope protein. Alteration of cccDNAregulation by a
mu-tation in the capsid protein suggests an involvement of the
FIG. 6. Effects of amino acid replacements on the production of enveloped virusparticles. LMH cells were cotransfected with plasmids expressing the indicated capsid proteins and a capsid-defective ge-nome expressingthe viral envelope proteins. Virus particles concen-trated from theculturefluidwere assayed by selective resistance to pronase-DNase I digestion, agarose gel electrophoresis, and blot hybridization. WT,wild type.
capsid proteinaswell as the pre-S protein in cccDNA
regula-tion, consistent with earlier models that pre-S binding of the nucleocapsid prevents its use for cccDNA synthesis (13, 14). A similarresultwas observed withthe class II C-terminal dele-tionmutantsthat weoriginallydescribed (21).
Effect of aspartic acid substitutions on cccDNA synthesis. Aspartic acid substitution of serine 257 produced a reduction in the levelsof cccDNA produced in the absence of envelope proteins (Fig. 5, S257D). Smaller reductions were seen in mutants S245D and S259D. Since all of these mutants pro-duced levels ofreplicative intermediates equivalent to levels
produced bythewild-type protein(Fig. 4),this result suggested that phosphorylation at position 257 would be expected to inhibit cccDNA DNA synthesis. This conclusion would be consistent with models that havebeen proposed in the litera-ture for control of nuclear localization of the capsid protein and cccDNA synthesis by phosphorylation (2, 18), provided that nuclearlocalization isastepin cccDNAsynthesis. In these
experiments, however, we never observed nuclear localization ofamajor portionof the capsid protein.
The level of cccDNA in the presence of envelope protein in allasparticacid substitutionmutantswasreduced below that in theabsence ofenvelopeprotein. We infer from this result that phosphorylation would not be expected to interfere with control of cccDNAamplification by the pre-S envelope pro-tein.
Influenceof aminoacid substitutionsonviralassemblyand
infectivity.
Wemeasured theamountofenvelopedvirus in the culturefluidsof transfected cellsby nondenaturingagarosegel electrophoresis.The results showed thatmutantS245A,which was defective in viral DNA maturation, failed to produceenvelopedvirus(Fig. 6).This result may beaconsequence of the failure of thismutant tosynthesizematurerelaxed circular DNA(see Discussion).Levels ofvirusproduced bythemutant
S259Awere depressedthree- to
fivefold,
commensuratewith thereductionin total viral DNAsynthesis
attributable tothis mutation.On the otherhand, asparticacid substitutionof serines 257 and 259causedspecificreductions inenvelopedvirus produc-tion, suggesting thatphosphorylation at these sites would be
expectedtoinhibitassemblyofmature
nucleocapsids
into virusparticles. These results were confirmed
by using
isopycnic
cesium chloridegradientcentrifugation
toassayforenveloped
virus particles (14). No alanine oraspartic
acid substitution altered the patternof viral DNA found in extracellular virus:i.e., only mature double-stranded DNA was present in the
so :1
on November 9, 2019 by guest
http://jvi.asm.org/
4346 YU AND SUMMERS
Duck hCDaton vtes RI
Duck henatocvtes
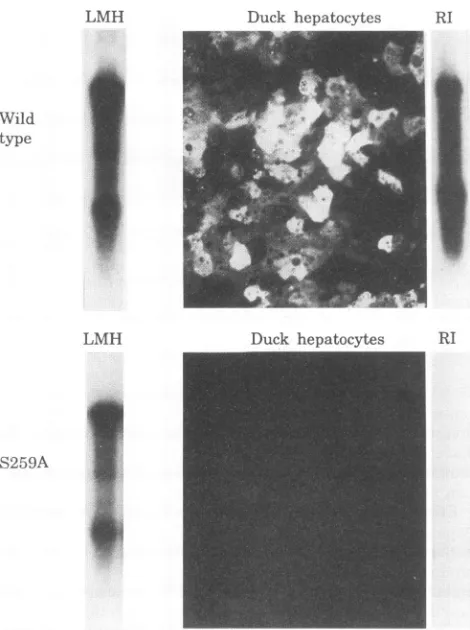
FIG. 7. Effect ofreplacementofserine 259 with alanineon infec-tivity of enveloped virus. LMH cellswere transfected with either a
wild-type viralgenome or agenomeinwhich thecapsid proteincodon 259 had beenchangedfromTCGtoGCG. Thischangedidnotalter thecodingof theoverlapping polymerasegene. Culture supernatants
were used to infect cultures of primary duck hepatocytes. Viral replicative intermediates present in the LMH cells at the time of harvest are shown in the left lanes (LMH). Immunofluorescent staining of the duck hepatocyte cell layer 14 days postinfection is
shown (duck hepatocytes). No evidence of infection of the duck
hepatocytes bymutant S259A wasseen by examination of approxi-mately1,000 fields of the size shown. Viral replicative intermediates
present in the duck hepatocyte cultures 14 days postinfection are
shown in therightlanes(RI).
nucleic acids extracted from cesium chloride-purified virus particles (datanotshown).The resultsindicatedthatthestate ofphosphorylation ofany individual sitewas not responsible for the selective assembly ofmature nucleocapsids into virus particles.
Wetestedtheinfectivityofenvelopedvirusproduced byone
of our alanine substitution mutants. We cloned the S259A mutationintoawild-type DHBVgenomecloned downstream
of the immediate-early CMV promoter (14). Transfection of LMH cells by this mutant resulted in the production of replicative intermediates at levels approximately threefold reduced from a parallel culture transfected with wild-type
genome(Fig.7, LMHlanes). Productionof enveloped virus by
this mutant wasreduced approximately fivefold (not shown).
These results were consistent with the phenotype of this
mutationassayed in thecomplementation experiments shown in Fig.3, 4, and 6.
[image:6.612.65.300.78.393.2]Supernatants of LMH cells transfected in parallel with mutant S259A or wild-type genomes were used to infect
TABLE 1. Requirementsforspecific phosphorylatedstates
Function Phosphorylation
preference Immaturecapsids
RNApackaging... Nospecific requirement
Minus-strand DNA... Serine259-phosphorylated
Plus-strand DNA... Serine245-phosphorylated
Maturecapsids
cccDNAamplification... Serine257-unphosphorylated
Virusassembly... Serine259-unphosphorylated;
serine257-unphosphorylated Viralpenetration... Serine259-phosphorylated
cultures of primary duck hepatocytes, and the cells were
assayed at 14 days postinfection for viral replicative DNA intermediates, and by immunofluorescent staining for viral capsid antigens. The hepatocyte cultures failed to show any
evidence of infectionby the mutantvirus (Fig. 7). Taking into accountthe fivefold reduction in virusyield,weestimatedfrom
the number of staining cells that the infectivity ofenveloped mutant virus was reduced by at least 2 orders ofmagnitude compared with that of wildtype.This resultwasconsistentwith the lack ofanydetectable viralDNAbySouthern blotanalysis
of themutant infected cultures (Fig. 7, RIlanes). DISCUSSION
The results ofourexperiments provideevidence that
phos-phorylationatthreespecificserines in the C terminus ofcapsid protein of DHBV influences several stepsin viral replication. Thepresumed requirementsforspecificphosphorylatedstates
are summarized in Table 1. No defects could be assigned to
alanine or aspartic acid substitution of threonine 239, and
therefore, these experiments provided no evidence for a role
forphosphorylation of thisresidue in virus replication. Rationaleandinterpretationofresults. The conclusions in Table 1 were based on the assumption that alanine could functionally replace serineat the sites ofreplacement, except for its ability to serve as a phosphoacceptor amino acid.
Similarly,we have assumed thatasparticacid could mimic the
functionofphosphoserineatthesesites. When defects associ-ated with replacementof serine byalaninewere corrected by
aspartic acid substitution, we assumed that aspartic acidwas
mimicking phosphoserine rather than serine asthewild-type functional residue. Likewise, when defects introduced by
as-particacid substitutionwerecorrectedby alanine,weassumed
that the alanine was mimicking serine rather than
phospho-serineas the functional aminoacidresidue.
Substitution of theadjacentdownstreamprolinewithglycine produced results that were more difficult to interpret but nevertheless were included in this paper. These substitutions
produced effects onvirus replication that mimicked those of
theadjacent serine-to-alanine substitution, but it isnotknown whether the proline replacements acted by inhibiting phos-phorylation of the adjacent serine. In at least one case, our
evidence indicated amorecomplicated role for proline,since evenwhentheupstreamresiduewas afunctional aspartic acid,
the downstream proline was still required for mature viral DNAsynthesis.
Themultiple effectsofphosphorylationonvirus replication.
Ourresultsprovide evidence thatserine phosphorylation has both positiveand negative effector functions in virus replica-tion. For example, aspartic acid replacement of two serines resulted in a stimulation of viral DNA synthesis relative to
alaninesubstitutionatthesesites. This result is consistent with LMH
Wild type
LMH
S259A
I
J. VIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.323.562.90.199.2]phosphoserine acting as the functional wild-type residue in activation of DNA synthesis. On the other hand, aspartic acid
replacementof at least two serines inhibited cccDNA synthesis and/or virus production relative to alanine substitution of these
residues, suggesting that nonphosphorylated serine acts as the
functional wild-type residue for these processes. The results imply that differential modification of at least three serines in thecapsid protein is required to produce all of the polypeptide species necessary for virus replication. Thus, posttranscrip-tional modification of a single capsid protein may be an
alternative to multiple capsid proteins encoded by separate genes.
In general, phosphorylation seemed to be required for
functionscarried out by immature nucleocapsids, i.e., nucleo-capsids that are not direct precursors of virus particles or
cccDNA,while phosphorylation inhibited functionscarriedout by mature nucleocapsids. The data can be interpreted to suggest that dephosphorylation of phosphorylated residues on the nucleocapsid would be required for the acquisition of mature capsid functions. However, our experiments do not
distinguish between a requirement for phosphorylation of some, asopposedto all, polypeptides in the nucleocapsid. For
example,our data indicated a requirement of phosphoserine at residue 259 for minus-strand DNA synthesis and for
unphos-phorylated serine at the same residue for virus production. Since the nucleocapsid contains many copies of the capsid
protein, bothrequirements might be met at the same time in thesamenucleocapsid.Therefore, the genetic data we present do not distinguish between the following models: (i) that the
nucleocapsid proteins must undergo sequential modification by phosphorylation and dephosphorylation in order for the
nucleocapsid to proceed through the various stages of matu-ration and virus assembly, or (ii) that the nucleocapsid is modified at selected residues before any DNA synthesis oc-curs, and thereby acquires competence for all steps in virus replication without any subsequent modifications. Our previ-ousreportshowing that the phosphorylation state of intracel-lularcapsids differs from that of mature viral capsids, however, favors thenotion that dephosphorylation does accompany the
acquisition ofmature capsid functions.
In addition to specific requirements for phosphoserine or serine in nucleocapsid maturation and assembly into virus
particles, weobtained evidence that phosphoserine at residue 259 might be required in the initiation of infection. While alanine couldreplace serine at position 259 without disrupting enveloped virus production, alanine could not substitute for
serineatthisposition during initiation of an infection. Presum-ablythedefect was due to a requirement for phosphoserine at
position 259 early during infection of hepatocytes. Phospho-serine 259 may be formed by de novo phosphorylation of serine 259during the initiation of infection or may preexist in
infectiousvirus particles.
Since alanine replacement of this residue did not inhibit cccDNAformationfrom cytoplasmic nucleocapsids, phospho-serine at residue 259 would not seem to be required for cccDNAformation from the infecting viral DNA, but for an earlier step thatintroduces the nucleocapsid of the infecting virusinto thecytoplasmof the hepatocyte. Our data suggested that phosphoserine at position 259 would be expected to inhibit virus production. Therefore, we speculate that
phos-phorylationofserine 259 may interfere with a physical associ-ationbetween thenucleocapsid and the viral envelope that is
required for virusassembly. If this were the case, phosphory-lation ofserine 259 might be actively required for dissociation of the nucleocapsid from the envelope at the site of viral
penetration.
Apart from the role ofphosphorylation,
mutations
such as S245A that generate a class II phenotype illuminate the orderly nature of viral DNA synthesis and viral assembly. Mutations that inhibited the synthesis of mature viral DNA also inhibited cccDNA regulation and virus production, two processes that depend in common on interaction of the viral nucleocapsid with the pre-S envelope protein. This result suggests that the synthesis of mature viral DNA and the acquisition by the nucleocapsid of the ability to interact with the pre-S protein may be functionally linked. We and others have repeatedly observed that only nucleocapsids that contain mature viral DNA are found in extracellular virions. Consid-ering these two observations together, we suggest that the ability of nucleocapsids to become assembled into viral enve-lopes is due to the acquisition of sites for recognition and binding of the pre-S protein concomitant with the synthesis of mature viral DNA. Such sites may constitute all or part of the packaging signal postulated by Summers and Mason (12).Mechanism of action ofphosphorylation. Our experiments raise the question of how the state of phosphorylation in a small region of the capsid protein differentially influences multiple functions of the capsid particle. We have suggested that a role of the several distinct phosphorylation states of the C terminus may be to stabilize the conformations of different functional domains that depend on the C terminus of the capsid protein. This model was indirectly suggested by the strong influence of phosphorylation at specific sites on the conformation of the C terminus of the capsid protein (20). The existence of alternative conformational and functional do-mains that depend on the particular phosphorylated state of the C terminus is consistent with the data presented here. The effects of the individual mutations that we tested would have been determined by the extent to which the conformation of the functionally specific domains depended on the particular phosphorylated state that was excluded by the mutation.
ACKNOWLEDGMENTS
We thank Tim Powell for excellent technical assistance.
This work was supported by Public Health Service grant CA-42542.
REFERENCES
1. Condreay, L., C. Aldrich, L. Coates, W. Mason, and T.-T. Wu. 1990. Efficient duck hepatitis Bvirusproduction by an avian tumor cell line. J. Virol. 64:3249-3258.
2. Eckhardt, S. G., D. R. Milich, and A. McLachlan. 1991. Hepatitis B virus core antigen has two nuclear localization sequences in the arginine-rich carboxyl terminus. J. Virol. 65:575-582.
3. Horwich, A. L., K. Furtak, J. C. Pugh, and J. Summers. 1990. Synthesis of hepadnavirus particles containing replication-defec-tive duck hepatitis B virus genomes in cultured Huh-7 cells. J. Virol. 64:642-650.
4. Kawaguchi, T.,K.Nomura,Y.Hirayama, and T. Kitagawa. 1987. Establishment and characterization of a chicken hepatocellular carcinoma cell line, LMH. Cancer Res.47:4460-4464.
5. Kunkel, T. A., J. D. Roberts, and R. A. Zakour. 1987. Rapid and efficient site specific mutagenesis without phenotypic selection. Methods Enzymol. 154:367-383.
5a.Lenhoff,R., and J. Summers. Unpublished data.
6. Mandart, E., A. Kay, and F. Galibert. 1984. Nucleotide sequence of a cloned duck hepatitis B virus genome: comparison with woodchuck and human hepatitis B virus sequences. J. Virol. 49:782-792.
7. Mason, W., C. Aldrich, J. Summers, and J. Taylor. 1982. Asym-metric replication of duck hepatitis B virus DNA in liver cells (free minus strand DNA). Proc. Natl. Acad. Sci. USA79:3997-4001. 8. Mason, W., M. Halpern, J. England, G. Seal, J. Egan, L. Coates,
C. Aldrich, and J. Summers. 1983. Experimental transmission of duck hepatitis B virus. Virology 131:375-384.
on November 9, 2019 by guest
http://jvi.asm.org/
4348 YU AND SUMMERS
9. Pugh, J., and J. Summers. 1989. Infection and uptake of duck hepatitis B virusby duck hepatocytes maintained in thepresence
ofdimethyl sulfoxide. Virology 172:564-572.
10. Pugh, J. C., A. Zweidler, and J. Summers. 1987.Characterization of themajor duck hepatitis B viruscoreparticle protein. J. Virol. 63:1371-1376.
11. Schlicht,H. J., R.Bartenschlager,andH.Schaller. 1989. The duck hepatitis B viruscoreproteincontainsahighlyphosphorylated C terminus that is essential for replication butnotfor RNA packag-ing. J. Virol.63:2995-3000.
12. Summers, J., and W. S. Mason. 1982. Replication of thegenome
of hepatitis B-like virus by reverse transcription of an RNA intermediate. Cell 29:403-415.
13. Summers, J., P. Smith, and A. L. Horwich. 1990. Hepadnaviral envelope proteins regulate amplification of covalently closed circular DNA. J.Virol. 64:2819-2824.
14. Summers, J., P. Smith, M.Huang,and M.Yu.1991. Regulatory andmorphogenetic effects of mutations in the envelope proteins ofanavianhepadnavirus. J. Virol.65:1310-1317.
15. Tuttleman, J., C. Pourcel, and J. Summers. 1986.Formation of the
pool ofcovalently closed circular viral DNA in hepadnavirus-infectedcells. Cell47:451-460.
16. Tuttleman, J. S., J.C.Pugh,andJ.W. Summers. 1986. In vitro experimentalinfection ofprimaryduckhepatocytecultures with duckhepatitisB virus. J. Virol.58:17-25.
17. Wu, T.-T.,L.Coates,C. E.Aldrich, J.Summers,and W. S. Mason. 1990. In hepatocytes infected with duck hepatitis B virus, the templatefor viral RNAsynthesis isamplifiedbyanintracellular
pathway. Virology175:255-261.
18. Yeh,C.T.,S. W.Wong,Y. K.Fung,andJ. H. Ou. 1993. Cellcycle regulationofnuclearlocalization ofhepatitisBviruscoreprotein.
Proc. Natl. Acad. Sci. USA 90:6459-6463.
19.Yu, M., and J. Summers. 1991. A domain of the hepadnaviral capsid protein specifically requiredfor DNA maturation and virus assembly.J. Virol.65:2511-2517.
20. Yu, M., and J. Summers. 1994. Phosphorylation of the duck hepatitis B virus capsid protein associated with conformational changesin the Cterminus. J. Virol.68:2965-2969.
21. Yu, M.,andJ. Summers. Unpublished observation.
J.VIROL.