Multiple Functions of Cellular FLIP Are Essential for
Replication of Hepatitis B Virus
Ah Ram Lee,
aKeo-Heun Lim,
aEun-Sook Park,
aDoo Hyun Kim,
aYong Kwang Park,
aSoree Park,
aDong-Sik Kim,
bGu-Choul Shin,
aHong Seok Kang,
aJuhee Won,
aHeewoo Sim,
aYea Na Ha,
aByeongjune Jae,
aSeong Il Choi,
cKyun-Hwan Kim
a,d,eaDepartment of Pharmacology, Center for Cancer Research and Diagnostic Medicine, IBST, School of Medicine,
Konkuk University, Seoul, Republic of Korea
bDivision of HBP Surgery and Liver Transplantation, Department of Surgery, Korea University College of
Medicine, Seoul, Republic of Korea
cDepartment of Biotechnology, College of Life Science and Biotechnology, Yonsei University, Seoul, Republic
of Korea
dKU Open Innovation Center, Konkuk University, Seoul, Republic of Korea
eResearch Institute of Medical Sciences, Konkuk University, Seoul, Republic of Korea
ABSTRACT
Hepatitis B virus (HBV) infection is a leading cause of liver diseases;
however, the host factors which facilitate the replication and persistence of HBV are
largely unidentified. Cellular FLICE inhibitory protein (c-FLIP) is a typical
antiapop-totic protein. In many cases of liver diseases, the expression level of c-FLIP is altered,
which affects the fate of hepatocytes. We previously found that c-FLIP and its
cleaved form interact with HBV X protein (HBx), which is essential for HBV
replica-tion, and regulate diverse cellular signals. In this study, we investigated the role of
endogenous c-FLIP in HBV replication and its underlying mechanisms. The
knock-down of endogenous c-FLIP revealed that this protein regulates HBV replication
through two different mechanisms. (i) c-FLIP interacts with HBx and protects it from
ubiquitin-dependent degradation. The N-terminal DED1 domain of c-FLIP is required
for HBx stabilization. (ii) c-FLIP regulates the expression or stability of hepatocyte
nu-clear factors (HNFs), which have critical roles in HBV transcription and maintenance
of hepatocytes. c-FLIP regulates the stability of HNFs through physical interactions.
We verified our findings in three HBV infection systems: HepG2-NTCP cells,
differenti-ated HepaRG cells, and primary human hepatocytes. In conclusion, our results
iden-tify c-FLIP as an essential factor in HBV replication. c-FLIP regulates viral replication
through its multiple effects on viral and host proteins that have critical roles in HBV
replication.
IMPORTANCE
Although the chronic hepatitis B virus (HBV) infection still poses a
major health concern, the host factors which are required for the replication of HBV
are largely uncharacterized. Our studies identify cellular FLICE inhibitory protein
(c-FLIP) as an essential factor in HBV replication. We found the dual roles of c-FLIP in
regulation of HBV replication: c-FLIP interacts with HBx and enhances its stability
and regulates the expression or stability of hepatocyte nuclear factors which are
es-sential for transcription of HBV genome. Our findings may provide a new target for
intervention in persistent HBV infection.
KEYWORDS
hepatitis B virus, HBx, c-FLIP, hepatocyte nuclear factor, transcriptional
control
D
espite the development of efficient prophylactic vaccines, chronic hepatitis B virus
(HBV) infection is still a major health concern, affecting more than 350 million
people worldwide (1). HBV infection significantly increases the probability of
develop-Received2 March 2018Accepted24 May 2018
Accepted manuscript posted online6 June 2018
CitationLee AR, Lim K-H, Park E-S, Kim DH, Park YK, Park S, Kim D-S, Shin G-C, Kang HS, Won J, Sim H, Ha YN, Jae B, Choi SI, Kim K-H. 2018. Multiple functions of cellular FLIP are essential for replication of hepatitis B virus.
J Virol 92:e00339-18.https://doi.org/10.1128/
JVI.00339-18.
EditorJ.-H. James Ou, University of Southern California
Copyright© 2018 American Society for
Microbiology.All Rights Reserved.
Address correspondence to Kyun-Hwan Kim, [email protected].
crossm
August 2018 Volume 92 Issue 16 e00339-18 Journal of Virology jvi.asm.org 1
on November 6, 2019 by guest
http://jvi.asm.org/
ing liver diseases such as inflammation, cirrhosis, and hepatocellular carcinoma (HCC).
HBV X protein (HBx), a multifunctional regulatory protein, plays a pivotal role in viral
liver pathogenesis (2) and especially in HBV replication (3, 4). In this regard,
under-standing how HBx exerts its function and identifying the host binding partners which
are necessary for HBx function are important for preventing viral replication and
HBV-induced liver diseases. HBx interacts with various host proteins and modulates
several cellular events, including gene transcription, signal transduction, and epigenetic
modifications (5). HBx contributes to the establishment of viral infection (6, 7) and
regulates the transcription of viral covalently closed circular DNA (cccDNA) through
interaction with chromatin-modifying enzymes (8–10). In addition, our recent study
revealed that HBx evades the innate antiviral response by interacting with host proteins
and may lead to development of chronic infection (11). Recently, HBx was shown to
interact with DDB1-containing E3 ubiquitin ligase to promote the degradation of the
structural maintenance of chromosomes (Smc) 5/6 complex and to facilitate the
transcription of extrachromosomal DNA templates such as cccDNA (12, 13). However,
the host factors required for the stability or activity of HBx are largely unidentified.
Transcription of HBV cccDNA is regulated by several liver-enriched transcription
factors such as hepatocyte nuclear factors (HNFs), C/EBP, and Sp1, which bind to HBV
enhancers and promoter regions (14). HNF1
␣
and HNF4
␣
are master regulators of
hepatocyte differentiation and maintenance (15) and essential for cccDNA transcription
(14, 16), whereas HNF3

is generally known to inhibit HBV transcription (14, 17). The
expression of these HNFs is regulated by the mitogen-activated protein kinase signaling
pathway (18, 19). The host proteins which enhance the expression or stability of HNFs
are also largely unidentified.
The cellular FLICE inhibitory protein (c-FLIP) is a master antiapoptotic protein against
death receptor-mediated apoptosis (20). c-FLIP is targeted by several viruses and is
involved in viral infection and pathogenesis (21–24). We previously showed that HBx
interacts with c-FLIP and alters the susceptibility to death-inducing signals (25). In
addition, we recently found that p22-FLIP, a cleavage product of c-FLIP, forms a ternary
complex with HBx and NEMO, which activates NF-
B signaling (26) and mediates the
TNF-
␣
-induced suppression of HBV (18). These findings prompted us to investigate the
role of c-FLIP in HBV replication and its underlying mechanisms.
In this study, we demonstrate that c-FLIP regulates HBV replication through multiple
different mechanisms: c-FLIP interacts with HBx and increases its stability and also
regulates the expression or stability of HNFs. We suggest that c-FLIP is an essential host
factor for the replication of HBV.
RESULTS
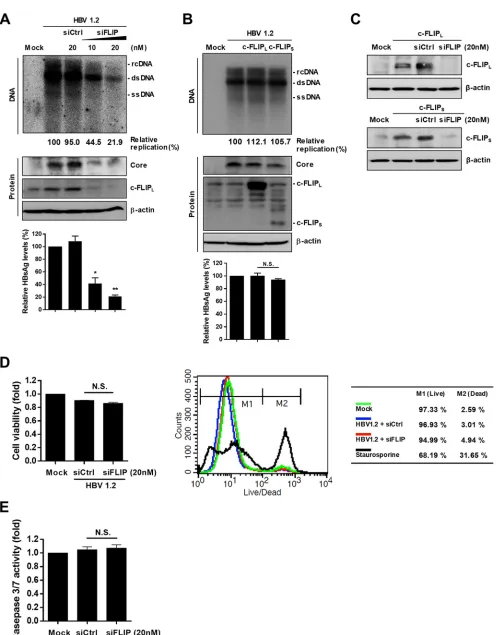
c-FLIP is necessary for robust HBV replication.
Since we previously showed that
c-FLIP binds HBx, an essential viral protein for HBV replication (25), we examined
whether the interaction between endogenous c-FLIP and HBx affects the replication of
HBV. To test this, we first silenced the expression of endogenous c-FLIP by small
interfering RNA (siRNA) and examined the replication of HBV in HepG2 cells. The
silencing of c-FLIP expression by siRNA (siFLIP) dramatically downregulated HBV
repli-cation, as well as the expression of viral core and surface antigens (Fig. 1A). Since there
are two major c-FLIP isoforms, long FLIP (c-FLIP
L) and short FLIP (c-FLIP
S), we tested
whether the overexpression of c-FLIP
Lor c-FLIP
Swould increase viral replication.
Unexpectedly, overexpression of neither isoform significantly increased the levels of
HBV replication or expression of core and surface antigens (Fig. 1B). siFLIP silenced both
the c-FLIP
Land c-FLIP
Sisoforms (Fig. 1C). Although c-FLIP is an antiapoptotic protein,
its knockdown did not reduce cell viability, as analyzed by three different assays: XTT
(Fig. 1D, left panel), Live/Dead flow cytometry (Fig. 1D, right panel), and caspase 3/7
activity (Fig. 1E). Taken together, these results indicate that c-FLIP is necessary for
robust HBV replication and that the endogenous level of c-FLIP is sufficient to support
replication in HepG2 cells.
on November 6, 2019 by guest
http://jvi.asm.org/
FIG 1Endogenous c-FLIP is required for robust replication of HBV in hepatocytes. (A) Effect of c-FLIP silencing on HBV replication and viral protein expression. HepG2 cells were cotransfected with the indicated plasmids and siRNAs. At 72 h posttransfection, the cells were lysed and subjected to Southern and Western
(Continued on next page)
August 2018 Volume 92 Issue 16 e00339-18 jvi.asm.org 3
on November 6, 2019 by guest
http://jvi.asm.org/
[image:3.585.42.542.61.696.2]c-FLIP regulates the expression level of HBx.
Since viral replication was strongly
suppressed by the knockdown of c-FLIP and c-FLIP interacts with HBx, which is required
for viral replication in HepG2 cells, we sought to determine whether c-FLIP alters the
level of HBx. The level of HBV genome-driven HBx expression was remarkably reduced
when c-FLIP was silenced by siRNA (Fig. 2A). The mRNA level of ectopically expressed
HBx was unaffected by c-FLIP knockdown (Fig. 2B, left panel). However, the level of the
HBx protein was dramatically reduced by c-FLIP knockdown (Fig. 2B, right panel),
indicating that c-FLIP may regulate the expression or stability of HBx. Since HBx is
known as an aggregation-prone protein (27–30), we tested whether c-FLIP silencing
would enhance the formation of insoluble aggregates of HBx. However, the level of HBx
was decreased in both the soluble supernatant and insoluble pellet fractions (Fig. 2B,
right panel). These results suggest that the reduction in viral replication by c-FLIP
knockdown (Fig. 1A) is due to the reduced level of HBx.
Next, we tested whether the level of HBx reduced by c-FLIP silencing can be
recovered by c-FLIP
Lor c-FLIP
Ssupplementation. Both the levels of HBV genome-driven
and ectopic expression of HBx were considerably recovered by c-FLIP
Lor c-FLIP
Ssupplementation (Fig. 2C). When c-FLIP
Lor c-FLIP
Swas overexpressed, the
genome-driven HBx expression was barely increased (Fig. 2D, left), consistent with the result of
HBV replication shown in Fig. 1B. When HBx was overexpressed, the coexpression of
c-FLIP
Lonly slightly increased the HBx level in HepG2 cells (Fig. 2D, right) but strongly
increased it in Huh7 cells (Fig. 2E). To test whether the siFLIP-mediated reduction in HBx
level is responsible for reduced replication, we tested the effect of HBx
supplementa-tion on replicasupplementa-tion. HBx overexpression in c-FLIP knockdown HepG2 cells significantly
restored viral replication (Fig. 2F). Taken together, these results suggest that c-FLIP is
essential for maintaining the steady-state level of HBx.
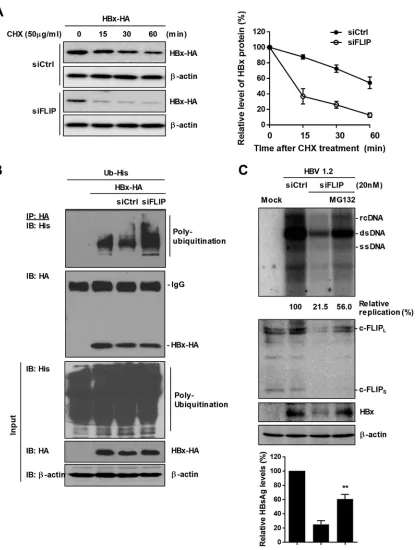
Knockdown of c-FLIP promotes the ubiquitin-dependent degradation of HBx.
To investigate the mechanism of the decrease in HBx stability by c-FLIP silencing, we
analyzed HBx stability by a cycloheximide (CHX) chase assay (Fig. 3A). When the
synthesis of new proteins was inhibited by CHX, the stability of HBx was greatly
decreased by c-FLIP knockdown (Fig. 3A), whereas the level of HBx mRNA was not
changed (Fig. 2B). These data suggest that c-FLIP knockdown facilitates the
degrada-tion of the HBx protein. We performed an
in vivo
ubiquitination assay and found that
c-FLIP silencing promotes HBx polyubiquitination (Fig. 3B). Accordingly, treatment with
MG-132, a proteasome inhibitor, considerably recovered the levels of HBx protein,
replication, and HBsAg production that were reduced by c-FLIP knockdown (Fig. 3C).
These results demonstrate that c-FLIP knockdown accelerates the degradation of the
HBx protein through the ubiquitin-dependent proteasomal pathway.
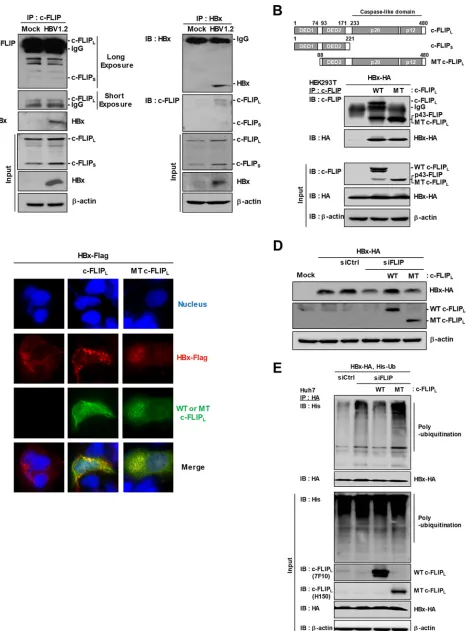
DED1 domain of c-FLIP is required for HBx stability.
The data presented above
suggest that c-FLIP protects HBx from ubiquitination through physical interaction. As
we have previously shown that HBx interacts with c-FLIP (25), we verified the
interac-tion in the experimental setting outlined above. Endogenous c-FLIP interacted with
HBV genome– driven HBx and
vice versa
(Fig. 4A).
Both c-FLIP
Land c-FLIP
Scontain two N-terminal death effector domains, DED1 and
DED2, which prevent caspase activation. Unlike c-FLIP
S, c-FLIP
Lcontains a caspase-like
domain (p20 and p12), as depicted in Fig. 4B (upper panel). Since HBx interacts with
both c-FLIP
Sand c-FLIP
L, we determined which of the two DEDs binds HBx and is
FIG 1Legend (Continued)
blot analyses. The levels of secreted HBsAg in culture supernatants were quantified by ELISA. Data are expressed as means⫾the standard deviations (SD). The statistical significance of the differences was assessed by the Studentttest:*,P⬍0.05;**,P⬍0.01. (B) Effect of ectopic overexpression of c-FLIP on the levels of HBV replication. The experimental procedures were as in panel A. Data are means⫾the SD. N.S., not significant. (C) Validation of siRNA-mediated c-FLIP knockdown. HepG2 cells were cotransfected with the indicated plasmids and siRNAs. At 48 h posttransfection, cells were harvested and subjected to Western blot analysis. (D) Effect of c-FLIP silencing on cell viability. HepG2 cells were cotransfected with the indicated plasmids and siRNAs. At 48 h posttransfection, cell viability was determined by XTT assay and fluorescence-activated cell sorting (FACS) analysis. For FACS analysis, treatment with staurosporine (2M) for 24 h before harvesting was used as a positive control for cell death. Data are means⫾the SD. N.S., not significant. (E) Analysis of the effect of c-FLIP knockdown on caspase 3/7 activity. Cells were prepared as in panel D. N.S., not significant. At least three independent experiments were performed.
on November 6, 2019 by guest
http://jvi.asm.org/
FIG 2Effects of c-FLIP levels on HBx levels. (A to E) HepG2 cells were cotransfected with the indicated plasmids and siRNAs. At 48 h posttransfection, cells were lysed, and supernatants and cell pellet fractions were used for immunoblotting and RT-PCR. (A) Effect of c-FLIP silencing on HBx levels. (B) Effect of c-FLIP silencing on mRNA and protein levels of HA-tagged HBx, which were determined by RT-PCR (left panel) and Western blot analysis (right panel), respectively. (C) Restoration of reduced HBx expression by overexpression of c-FLIP. (D) Effect of c-FLIP overexpression on HBx levels. (E) Effect of c-FLIP overexpression on HBx levels in Huh7 cells. Cells were cotransfected with the indicated plasmids and harvested at the indicated time points. Cell lysates were subjected to Western blot analysis. (F) Restoration of HBV replication by supplementation of HBx. HepG2 cells were cotransfected with the indicated plasmids and siRNAs. At 72 h
(Continued on next page)
August 2018 Volume 92 Issue 16 e00339-18 jvi.asm.org 5
on November 6, 2019 by guest
http://jvi.asm.org/
[image:5.585.47.542.64.669.2]responsible for its stability. We constructed a c-FLIP
Lmutant that lacked DED1 (Fig. 4B,
upper panel) and performed an immunoprecipitation assay. Both the wild-type (WT)
and the mutant c-FLIP
Lforms bound to HBx (Fig. 4B, lower panel). Immunofluorescence
microscopy analysis showed that, although the cellular distribution of c-FLIP was
slightly changed by the absence of DED1, both WT and mutant c-FLIP
Lcolocalized with
HBx (Fig. 4C). These results suggest that, at least under these experimental conditions,
DED2 is sufficient for binding to HBx. However, the c-FLIP
Lmutant did not restore the
HBx level reduced by c-FLIP knockdown, whereas WT c-FLIP
Lrescued it (Fig. 4D). The
in
vivo
ubiquitination assay showed that, unlike the WT form, the c-FLIP
Lmutant did not
inhibit but rather slightly increased HBx polyubiquitination (Fig. 4E). These results
demonstrate that, although DED2 of c-FLIP is involved in its interaction with HBx, DED1
is required for HBx stabilization. Consistent with these findings, the domains or regions
neighboring the substrate-binding domains of chaperones such as Hsp70 and Hsp90
have been reported to be also important for substrate stabilization (31, 32).
c-FLIP also controls HBV replication through an HBx-independent pathway.
The above data demonstrated that c-FLIP knockdown inhibited HBV replication by
reducing the level of HBx, an essential factor for HBV replication (3, 4). Next, we
examined whether c-FLIP knockdown affects the level of HBV transcription. The
knock-down of c-FLIP greatly decreased the levels of HBV RNAs (Fig. 5A), whereas c-FLIP
overexpression had no considerable effect (Fig. 5B). The much higher reduction in HBV
transcription by c-FLIP knockdown than expected prompted us to test whether the
decrease in viral replication was solely dependent on the reduced level of HBx.
Surprisingly, c-FLIP knockdown strongly reduced the replication and transcription of
both WT (HBV1.2) and HBx-null [HBV1.2(X
⫺
)] HBV (Fig. 5C) in HepG2 cells, although the
effect was stronger for WT HBV (Fig. 5D). The expression of HBsAg was also decreased
in both cases (Fig. 5C, lower panel). These unexpected findings suggest that c-FLIP
regulates HBV replication via HBx-dependent and HBx-independent pathways.
HBx is well known to affect HBV replication in HepG2 cells but not in Huh7 cells.
Under our experimental conditions, the replication level of HBV1.2(X
⫺
) was slightly
reduced in Huh7 cells (87.4% of that of HBV1.2) (Fig. 5E). To further confirm the
HBx-independent pathway, we analyzed HBV replication in Huh7 cells after c-FLIP
knockdown (Fig. 5F). The levels of viral replication and antigen expression were also
decreased in Huh7 cells by silencing of c-FLIP, supporting the involvement of
HBx-independent regulation. Similar to the lack of its effect on the viability of HepG2 cells,
c-FLIP knockdown did not reduce Huh7 cell viability, as determined by three different
assays (Fig. 5G and H). Collectively, these findings suggest that endogenous c-FLIP
regulates HBV replication through two different mechanisms.
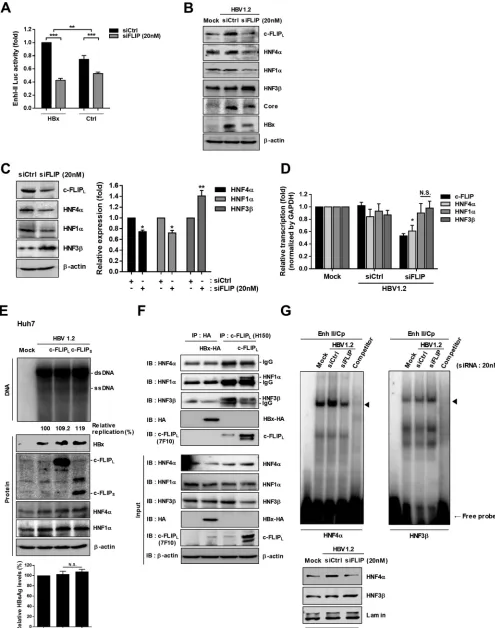
Knockdown of c-FLIP inhibits HBV transcription via dysregulation of HNFs.
To
elucidate how c-FLIP knockdown reduces HBV transcription, we examined the effect of
c-FLIP on HBV enhancer/promoter activities. Since transcription of the HBV genome is
mainly regulated by two essential enhancers, I and II (33, 34), we constructed a reporter
plasmid containing both enhancers and analyzed the effect of c-FLIP silencing on their
activity in the presence or absence of HBx. Consistent with the replication data (Fig. 5C
and D), a luciferase assay also revealed that c-FLIP silencing reduces the enhancer
activity through two independent pathways (Fig. 6A). These results indicate that c-FLIP
knockdown suppresses HBV replication through downregulation of enhancer I and II
activities.
Among several HNFs, the activity of HBV enhancers is mainly regulated by the
proviral factors HNF1
␣
and HNF4
␣
and the antiviral factor HNF3

(14, 35, 36).
Accord-ingly, we examined whether c-FLIP silencing alters the expression levels of these HNFs.
The expression levels of HNF1
␣
and HNF4
␣
were downregulated by c-FLIP knockdown,
FIG 2Legend (Continued)
posttransfection, cell lysates were subjected to Southern and Western blot analyses. The level of HBsAg in culture supernatants was determined by enzyme-linked immunosorbent assay (ELISA). Data are means⫾the SD. Statistical significance of the differences was assessed by the Studentttest:*,P⬍0.05. At least three independent experiments were performed.
on November 6, 2019 by guest
http://jvi.asm.org/
FIG 3Knockdown of c-FLIP accelerates the proteasome-mediated degradation of HBx. HepG2 cells were cotransfected with the indicated plasmids and siRNAs. (A) CHX chase assay of HBx. CHX was added for indicated time before harvesting. HA-tagged HBx was detected by Western blotting (left panel) and its levels were quantified (right panel). Data are means⫾the SD. (B) Polyubiquitination assay of HA-tagged HBx. At 42 h posttransfection, cells were treated with MG132 (20M) for 6 h and subjected to immunoprecipi-tation with HA antibody. The levels of polyubiquitinated HBx were determined by Western blotting with His antibody. (C) Recovery of HBx stability by proteasome inhibition. At 42 h posttransfection, cells were treated with MG132 (20M) for 6 h before harvesting, and cell lysates were subjected to Western blotting. For Southern blot analysis, at 24 h posttransfection, the cells were treated with MG132 (20M) for 48 h before harvesting. The levels of secreted HBsAg were evaluated by ELISA. Data are means⫾the SD. Statistical significance of the differences was assessed by the Studentttest:**, P⬍0.01. At least three independent experiments were performed.
August 2018 Volume 92 Issue 16 e00339-18 jvi.asm.org 7
on November 6, 2019 by guest
http://jvi.asm.org/
[image:7.585.48.465.67.617.2]FIG 4Death-effector domain 1 (DED1) of c-FLIP is required for maintaining HBx stability. (A) Interaction between endogenous c-FLIP and HBV genome– driven HBx. HepG2 cells were transfected with the HBV1.2 plasmid; at 48 h posttransfection, cell lysates were immunoprecipitated with c-FLIP (left) or HBx
(Continued on next page)
on November 6, 2019 by guest
http://jvi.asm.org/
[image:8.585.60.527.69.700.2]while that of HNF3

was upregulated (Fig. 6B). Similar results were obtained even in the
absence of HBV (Fig. 6C). To check the regulation was transcriptional event, we
performed real-time PCR. Interestingly, the transcript level of HNF4
␣
was significantly
reduced, whereas those of HNF1
␣
and HNF3

were not affected by c-FLIP knockdown
(Fig. 6D), suggesting HNF1
␣
and HNF3

are regulated at a posttranscriptional level.
Overexpression of c-FLIP in HBx-insensitive Huh7 cells also increased the expression level
of HNF4
␣
, accompanied by a slight increase in HBV replication (by 10 to 20%) (Fig. 6E).
Therefore, we tested whether HBx or c-FLIP interacts with HNFs. We found that
HNF1
␣
and HNF3

, but not HNF4
␣
, interacted with c-FLIP
L(Fig. 6F). Interestingly, the
level of endogenous c-FLIP was sufficient to fully interact with HNF3

. These results
suggest that c-FLIP regulates the stability of HNF1
␣
and HNF3

through physical
interaction. The overexpression of c-FLIP decreased the level of HNF3

(Fig. 6F, lower
panel), whereas c-FLIP knockdown increased it (Fig. 6B). No interaction was observed
between HBx and HNFs (Fig. 6F). The electrophoretic mobility shift assay (EMSA) results
demonstrated that c-FLIP knockdown reduced HNF4
␣
binding to the enhancer region
but enhanced HNF3

binding (Fig. 6G). Taken together, these results indicate that
c-FLIP regulates the levels of HNFs through different mechanisms: it regulates the
transcription of HNF4
␣
and the stability of the HNF1
␣
and HNF3

proteins through
physical protein-protein interactions.
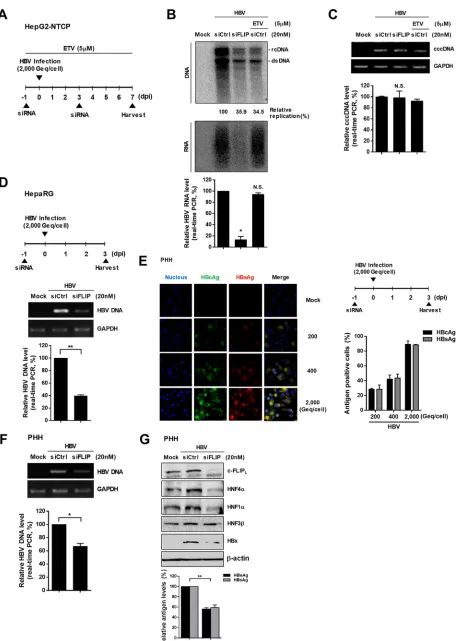
Knockdown of c-FLIP inhibits viral replication in different HBV infection
sys-tems.
To check whether our findings are physiologically relevant, we performed similar
experiments in different HBV infection systems (HepG2-NTCP cells, differentiated
HepaRG cells, and primary human hepatocytes [PHHs]). Similar to the transfection
system in HepG2 cells (Fig. 1A), c-FLIP knockdown reduced HBV replication in
HepG2-NTCP and differentiated HepaRG cells (Fig. 7B and D). The cells were treated with a high
dose of entecavir (ETV), a potent inhibitor of HBV polymerase, to discriminate the level
of HBV replication that originated from the inoculated viruses. c-FLIP knockdown
reduced the HBV DNA levels to a similar extent as did ETV and also strongly reduced
the level of viral RNA, suggesting that c-FLIP knockdown effectively inhibits HBV
transcription in this infection system. However, the cccDNA level was not changed by
c-FLIP knockdown at 7 days postinfection (dpi) (Fig. 7C).
We further validated our finding using freshly isolated PHHs. The optimal inoculum
dose for HBV infection in PHHs was also determined by immunofluorescence, and the
2,000-genome equivalents (GEq)/cell dose, which resulted in a high expression level of
viral antigens (approximately 90% infection efficiency), was used for further studies (Fig.
7E). The knockdown of c-FLIP reduced both HBV replication (Fig. 7F) and expression of
HBsAg and HBeAg (Fig. 7G). Similar to the results for HepG2 cells, c-FLIP knockdown
downregulated HNF4
␣
and HNF1
␣
, whereas the change in the of HNF3

level seemed
marginal (Fig. 7G). Of note, the expression level of HBV genome-driven HBx was
considerably decreased by c-FLIP knockdown, suggesting that both the HBx-dependent
and HBx-independent mechanisms exist in PHHs. Cell viability was unaffected by viral
infection or siRNA transfection in all tested infection systems (data not shown).
Collec-tively, our data suggest that endogenous c-FLIP is essential for viral replication during
the natural course of HBV infection.
DISCUSSION
In the present study, we demonstrated a novel role of c-FLIP in hepatocytes: it
interacts with the HBx protein, an essential factor for HBV replication, and increases HBx
FIG 4Legend (Continued)
(right) antibody. Proteins were detected by immunoblotting with the indicated antibodies. (B) Top: schematic presentation of c-FLIPL, c-FLIPS, and mutated
c-FLIPL. Bottom: HEK293T cells were cotransfected with the indicated plasmids, and lysates were subjected to immunoprecipitation with c-FLIP antibody.
Proteins were detected by immunoblotting with the indicated antibodies. (C) Colocalization of Flag-tagged HBx with c-FLIPLor mutated c-FLIPL. HepG2
cells growing on a coverslip were transfected with the indicated plasmids. At 48 h posttransfection, cells were subjected to immunofluorescence analysis. (D) Recovery of HBx stability by c-FLIP supplementation. HepG2 cells were cotransfected with the indicated plasmids and siRNA; at 48 h posttransfection, cell lysates were subjected to immunoblotting with the indicated antibodies. (E) Effect of mutated c-FLIPLon polyubiquitination of HA-tagged HBx. The
experimental procedures were as in Fig. 3B. At least three independent experiments were performed.
August 2018 Volume 92 Issue 16 e00339-18 jvi.asm.org 9
on November 6, 2019 by guest
http://jvi.asm.org/
FIG 5c-FLIP inhibits HBV replication via an HBx-independent pathway. (A to F) HepG2 or Huh7 cells were cotransfected with the indicated plasmids and siRNAs and harvested 72 h posttransfection. (A) Inhibition of HBV transcription by c-FLIP knockdown. Cell lysates were subjected to Northern blot analysis. (B) Effect
(Continued on next page)
on November 6, 2019 by guest
http://jvi.asm.org/
[image:10.585.48.537.64.702.2]stability. The knockdown of c-FLIP strongly downregulated HBV replication by
accel-erating HBx degradation. Because c-FLIP is required for HBx stability, it acts as an
essential factor in HBV replication. In addition, c-FLIP controls the expression or stability
of HNFs, which are critical for the transcription of the HBV genome and maintenance
of hepatocytes. Our findings were validated in HepG2-NTCP cells, differentiated
HepaRG cells, and PHHs, suggesting that endogenous c-FLIP has an important role in
the natural course of HBV infection. Collectively, our data demonstrate that c-FLIP
regulates the replication of HBV by two different mechanisms: (i) its chaperone-like
function for client proteins via physical interaction and (ii) transcriptional control.
Regulatory proteins alone such as HBx and HNFs are often unstable and prone to
aggregation. Thus, it is of fundamental importance to understand how their stability
can be modulated depending on biological necessity. Regulatory proteins interact with
a variety of other proteins, including chaperones. In this regard, we previously
pro-posed the concept of binding partner-mediated protein folding or stabilization as a
novel chaperoning function of binding partners (27, 37, 38); cellular macromolecules,
including chaperones, commonly have inherent chaperoning function for proteins
physically associated with them due to their large excluded volume and surface charge.
So far, chaperones have been widely believed to control proteostasis (protein
homeo-stasis), including folding assistance, prevention of aggregation, control of degradation,
and the modulation (increase or decrease) of stability of regulatory proteins (39, 40). For
example, Hsp90 controls stability of many regulatory proteins (40). Mechanistically, the
modulation of the stability of HBx and HNFs by c-FLIP (Fig. 2A and 6) is very similar to
the modulation of the stability of regulatory proteins by Hsp90. We think that c-FLIP is
an example of a binding partner that acts as a noncanonical molecular chaperone for
unstable HBx and HNF1
␣
(Fig. 6B).
Chaperones may facilitate the degradation of their binding partners; this
chaperone-assisted degradation regulates several disease-associated cellular processes (41, 42). We
previously found that hepatocystin/80K-H interacts with HBx, thereby enhancing HBx
degradation and eventually inhibiting HBV replication (43). In this study, we found that
c-FLIP interacts with HNF3

and enhances its degradation (Fig. 6). In this respect, c-FLIP
acts as a molecular chaperone for HNF3

that assists its degradation.
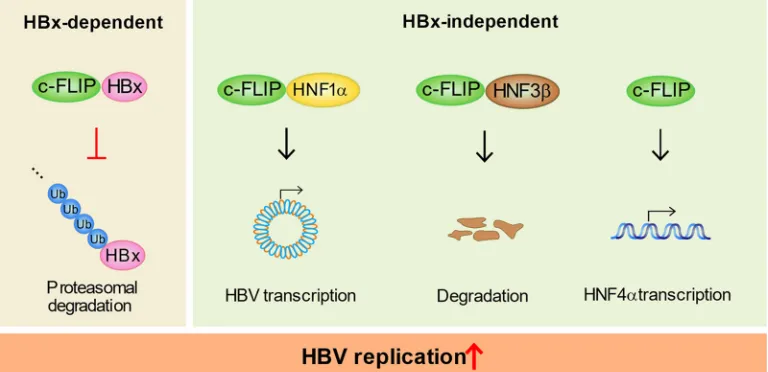
Collectively, our data suggest c-FLIP as a novel chaperone that can enhance the
degradation of target proteins or protect them from degradation through physical
interactions. This novel chaperoning function of c-FLIP controls HBV replication through
two different mechanisms (Fig. 8). This example would provide new insights into the
control of stability of other regulatory proteins in cells.
The HBx-dependent mechanism related to c-FLIP-mediated control of HBV
replica-tion is supported by the following observareplica-tions. (i) As shown in Fig. 2 to 4, c-FLIP
interacts with HBx and its knockdown enhanced the degradation of CMV-driven HBx
protein but did not affect the level of CMV-driven HBx mRNA (the CMV promoter is
independent of HNFs). (ii) The relative level of replication inhibition by siFLIP was
significantly lower for HBV1.2(X
⫺
) (50% inhibition) than for HBV1.2 (80% inhibition)
(Fig. 5D). We think that the inhibition of HBV1.2(X
⫺
) replication was mainly through the
HNF-dependent pathway (because there was no HBx expression), whereas that of
HBV1.2 replication was due to the combined effects of HBx- and HNF-dependent
FIG 5Legend (Continued)
of c-FLIP overexpression on HBV transcription. The levels of HBV RNAs were analyzed by Northern blotting. pg/pre C RNA, pregenomic and precore HBV RNA; pre-S/S RNA, HBV RNAs for envelope proteins. (C) Effect of c-FLIP silencing on replication and transcription of WT (HBV1.2) or X-null HBV[HBV1.2(X⫺)]. Cells were subjected to Southern, Northern, and Western blot analyses. The presence of HBsAg in culture supernatants was analyzed by ELISA. Data are means⫾ the SD. Statistical significance of the differences was assessed by the Studentttest:**,P⬍0.01. (D) Inhibitory effect of c-FLIP silencing on replication was quantified using Multi-Gauge V3.0 and plotted. Data are means⫾the SD.**,P⬍0.01;***,P⬍0.001. (E and F) Effect of HBx (E) and c-FLIP silencing (F) on HBV replication in Huh7 cells. (E) Cells were transfected with the plasmids for WT (HBV1.2) or X-null HBV [HBV1.2(X⫺)], and the replication and protein levels were determined by Southern and Western blotting, respectively. (F) Huh7 cells were cotransfected with the indicated plasmids and siRNAs; at 72 h posttransfection, the levels of replication and secreted HBsAg were analyzed by Southern blotting and ELISA, respectively. Data are means⫾the SD.*,P⬍
0.05;**,P⬍0.01. (G and H) Effect of c-FLIP silencing on in Huh7 cell viability as determined by XTT, FACS, and caspase 3/7 activity analyses. The experimental procedures were as in Fig. 1. N.S., not significant. At least three independent experiments were performed.
August 2018 Volume 92 Issue 16 e00339-18 jvi.asm.org 11
on November 6, 2019 by guest
http://jvi.asm.org/
FIG 6c-FLIP regulates the expression of hepatocyte nuclear factors. (A) Effect of the knockdown of endogenous c-FLIP on the activities of viral enhancers I and II in the presence or absence of HBx. HepG2 cells were cotransfected with the pEnh I/II-luc,-gal, HBx-HA, pGL3-basic, and siRNAs as indicated. At 48 h
(Continued on next page)
on November 6, 2019 by guest
http://jvi.asm.org/
[image:12.585.42.537.72.700.2]pathways. Therefore, the net enhanced inhibition of HBV1.2 replication in comparison
with that of HBV1.2(X
⫺
) can be attributed to the HBx-dependent mechanism. (iii)
Similarly, the relative inhibition of enhancer activity by siFLIP was significantly higher
when HBx was present (Fig. 6A).
When hepatocytes are infected with HBV, the intrahepatic immune cells are
acti-vated and secrete proinflammatory cytokines and chemokines such as interferons,
interleukins, and tumor necrosis factor alpha (TNF-
␣
) (44, 45). These factors are crucial
for HBV clearance, especially at the early phase of HBV infection. TNF-
␣
was shown to
induce the expression of c-FLIP through the NF-
B signaling pathway (46, 47), and we
also showed that TNF-
␣
enhances the generation of p22-FLIP, an N-terminal cleavage
product of c-FLIP, which strongly suppresses HBV (18). Because c-FLIP binds to HBx and
increases its stability, HBV may have evolved to escape the TNF-
␣
-induced anti-HBV
pressure by hijacking c-FLIP. The HBx interaction with c-FLIP may inhibit the generation
of p22-FLIP, an effect that would be beneficial for HBV propagation. In line with this
suggestion, HBx is required for initiation and maintenance of viral replication during the
natural course of HBV infection (48).
However, in the chronic phase of HBV infection, the reactivity of HBV-specific T cells
is often weak or absent (this decrease in reactivity is called T-cell exhaustion), which
leads to poor immune responses (49, 50) and evasion of immune surveillance (51).
Accordingly, the proinflammatory cytokines, including TNF-
␣
, are poorly induced (44,
52, 53). Therefore, the basally expressed c-FLIP will be intact because the conversion of
c-FLIP to p22-FLIP would be blocked. The unprocessed c-FLIP may stabilize HBx and
facilitate HBV replication, contributing to persistent HBV infection. It seems likely c-FLIP
is a double-edged sword in regulating HBV replication, depending on the level of
TNF-
␣
. HBx may turn the antiviral nature of c-FLIP to its advantage and convert it into
a proviral factor.
Being a regulator of apoptosis, c-FLIP is tightly regulated (54). Many studies have
revealed that c-FLIP is frequently overexpressed in various types of cancers, including
colorectal, melanoma, malignant mesothelial, cervical, and gastric cancers (22, 55–58).
Furthermore, the expression level of c-FLIP is closely related to cancer pathogenesis (22,
59–61). Importantly, c-FLIP is highly expressed in HCC patients (62, 63) and plays a
critical role in HCC development (64). It is possible that the elevated level of c-FLIP in
patients with HBV-related HCC contributes to HBx stabilization; together with the
antiapo-ptotic function of c-FLIP, this stabilization may enhance the oncogenic effect of HBx.
The class I histone deacetylase inhibitors valproic acid and suberoylanilide
hy-droxamic acid downregulate c-FLIP expression and increase TRAIL-mediated apoptosis
(59, 65). Therefore, it would be interesting to test the effect of these drugs on the
suppression of HBV and HBV-related HCC. In this respect, it may be relevant that
specific silencing or downregulation of c-FLIP not only apparently attenuated
HBV-induced HCC but also increased apoptosis in HCC tissues (66, 67).
In conclusion, our study demonstrates a novel function of c-FLIP as an essential
factor in HBV replication.
FIG 6Legend (Continued)
posttransfection, cells were subjected to luciferase activity analysis. Data are means⫾the SD. Statistical significance of the differences was assessed by the Studentttest:**,P⬍0.01;***,P⬍0.001. (B to D) Effect of c-FLIP knockdown on the expression of hepatocyte nuclear factors (HNFs). HepG2 cells were cotransfected with the indicated plasmids and siRNAs; at 48 h posttransfection, the expression levels of each protein or mRNA in the presence of HBV (B) or absence of HBV (C) were analyzed by immunoblotting (B and C) and real-time PCR (D). Expression levels were quantified using Multigauge V3.0 and plotted (C, right panel). The relative transcription levels of target genes were normalized to that of the GAPDH gene. Data are means⫾the SD. N.S., not significant;
*,P⬍0.05;**,P⬍0.01. (E) Effect of c-FLIP overexpression on HBV replication and HNF expression in Huh7 cells. The experimental procedures were as in Fig. 1B. Data are means⫾the SD. N.S., not significant. (F) Interaction of endogenous HNFs with HA-tagged HBx or c-FLIP. HepG2 cells were transfected with the indicated plasmids; at 48 h posttransfection, cell lysates were subjected to immunoprecipitation with HA or c-FLIPLantibody. Each protein was detected with
the indicated antibody. (G) EMSA. HepG2 cells were cotransfected with the indicated plasmids and siRNAs; at 48 h posttransfection, nuclear extracts were prepared and used in EMSA. The oligonucleotides corresponding to the binding sites of HNF4␣or HNF3were used as binding probes (left panel). The levels of HNF proteins in nuclear extracts were assessed by Western blotting (right panel). The nonlabeled Enh II probe was used as a cold competitor. At least three independent experiments were performed.
August 2018 Volume 92 Issue 16 e00339-18 jvi.asm.org 13
on November 6, 2019 by guest
http://jvi.asm.org/
FIG 7Effect of c-FLIP knockdown on viral replication in different HBV infection systems. (A) Experimental scheme of HBV infection of HepG2-NTCP cells. Cells were infected with HBV inoculum 1 day after c-FLIP knockdown and were harvested 7 dpi. Cells were treated with 5M entecavir (ETV)
(Continued on next page)
on November 6, 2019 by guest
http://jvi.asm.org/
[image:14.585.44.500.65.706.2]MATERIALS AND METHODS
Cell culture and transfection.The human hepatoma cell lines (HepG2 and Huh7) and the human embryonic kidney cell line (HEK293T) were obtained from the American Type Culture Collection. All cell lines were maintained in Dulbecco modified Eagle medium (Welgene, Gyeongsan, South Korea) supple-mented with 10% fetal bovine serum (FBS; Capricorn, Ebsdorfergrund, Germany) and 1% penicillin-streptomycin (Gibco, Grand Island, NY), at 37°C in a 5% CO2humidified incubator. The HepAD38 cell line,
which stably produces HBV particles, was cultured in the presence or absence of tetracycline (0.3g/ml). The HepG2-NTCP cell line, which constitutively expresses NTCP, was cultured in the presence of G418 (200g/ml). Differentiated HepaRG cells (Biopredic International, Saint-Gregoire, France) were main-tained according to the supplier’s protocol. PHHs were cultured in Williams’ medium E (Gibco) containing cell maintenance supplements (CM4000; Gibco) and 1% penicillin-streptomycin (PMM). All transfections were performed at 70 to 80% confluence using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions.
Plasmids and reagents.The replication-competent HBV constructs, WT HBV 1.2mer and X-null HBV 1.2mer (X⫺) (genotype D), were kindly provided by W. S. Ryu (Yonsei University, Seoul, South Korea). The plasmids expressing HBx-HA (subtype ayw), HBx-Flag, c-FLIPL, c-FLIPs, and His-Ub were described in our
previous studies (25, 26, 43). A fragment encoding mutated c-FLIPLlacking the death-effector domain 1
(DED1) was amplified by PCR using the c-FLIPLconstruct as a template and then subcloned into the
pcDNA3.1(⫹) vector (Invitrogen). The reporter plasmid for HBV enhancer I and II (18) was cloned using the pGL3-basic vector (Promega, Madison, WI). Primary antibodies for immunoblotting against the following proteins or tags were used: c-FLIPL/S(7F10; Enzo Life Sciences, Farmingdale, NY), c-FLIPL(H-150;
Santa Cruz Biotechnology, Dallas, TX), HNF4␣(⌯-171; Santa Cruz), HNF1␣(⌯-140; Santa Cruz), HNF3 (RY-7; Santa Cruz), HBx (BioVendor, Heidelberg, Germany), Core (B0586; Dako, Carpinteria, CA), HA (H6908; Sigma-Aldrich, Saint Louis, MO), His tag (D291-3; MBL International, Woburn, MA), and-actin (A5441; Sigma). Horseradish peroxidase-conjugated secondary antibody for immunoblotting was pur-chased from Sigma, and Alexa Fluor-conjugated secondary antibody for immunofluorescence analysis was obtained from Invitrogen. The proteasome inhibitor MG132 (catalog no. 474790) was purchased
FIG 7Legend (Continued)
as shown on the scheme. (B and C) Effect of c-FLIP knockdown on HBV replication and cccDNA level in HepG2-NTCP cells. HBV DNA levels were determined by Southern blotting. HBV RNA levels were analyzed by Northern blotting and real-time PCR. The levels of cccDNA were quantified by real-time PCR and normalized to that of the GAPDH gene. Amplification of cccDNA of the correct size was confirmed by electrophoresis (upper panel). Data are means⫾the SD. Statistical significance of the differences was assessed by the Studentttest:*,P⬍0.05; N.S., not significant. (D) Effect of c-FLIP knockdown on HBV replication in differentiated HepaRG cells. Cells were infected with HBV inoculum 1 day after c-FLIP knockdown and were harvested at 3 dpi. The levels of HBV DNA were analyzed by semiquantitative PCR (upper panel) and real-time PCR (bottom panel). GAPDH was used as a normalization control. Data are means⫾the SD. Statistical significance of the differences was assessed by the Studentttest:**,P⬍0.01. (E) Titration of HBV to optimize the inoculum dose for infection of PHHs. Infection efficiency was determined by immunofluorescence analysis using antibodies against viral core and surface proteins at 3 dpi. Antigen positive cells were counted. Data are means⫾ the SD of at least three different regions. (F and G) Effect of c-FLIP knockdown on replication (F) and HNF expression (G) in PHHs. HBV DNA levels were analyzed by semiquantitative PCR (upper panel) and real-time PCR (bottom panel). GAPDH was used as a normalization control. The levels of HNFs and HBx were determined by Western blotting, and those of HBeAg and HBsAg were determined by ELISA. Data are means⫾the SD.
*,P⬍0.05;**,P⬍0.01. At least three independent experiments were performed.
FIG 8A hypothetical model of c-FLIP as an essential factor in HBV replication. The novel chaperone-like function of c-FLIP controls HBV replication through two different mechanisms. c-FLIP regulates viral replication through its interaction-mediated chaperone-like function to HBx and HNFs, which have critical roles in HBV replication. c-FLIP also regulates the transcription of HNF4␣, a master regulator of cccDNA transcription and hepatocyte differentiation.
August 2018 Volume 92 Issue 16 e00339-18 jvi.asm.org 15
on November 6, 2019 by guest
http://jvi.asm.org/
[image:15.585.42.426.73.259.2]from Calbiochem (Darmstadt, Germany). The inhibitor of protein biosynthesis CHX and the inducer of apoptosis staurosporine (S6942) were obtained from Sigma.
HBV infection.PHH cells were isolated from virus-free human liver tissue specimens by using a slightly modified two-step collagenase perfusion technique as described in our earlier studies (18, 68). HepAD38 cells were used to collect HBV particles in the absence of tetracycline. Culture supernatants were collected, centrifuged, and passed through a 0.45-m-pore size filter. HBV particles were precipi-tated using 6% PEG8000 (Sigma) at 4°C overnight and resuspended with phosphate-buffered saline (PBS) containing 25% FBS. HBV DNA was quantified by dot blot analysis. Approximately 106HepG2-NTCP,
differentiated HepaRG, or PHH cells were seeded onto a 6-well plate coated with collagen I (Gibco). Cells were infected with 2,000 HBV GEq/cell containing 4% PEG8000 and 2.5% dimethyl sulfoxide (DMSO; Sigma) at 37°C for 18 to 20 h. The cells were then washed three times with PBS, maintained in PMM containing 2% DMSO, and harvested at 3 or 7 days postinfection as shown in each figure panel.
Western blotting.At 48 h posttransfection, the cells were harvested and lysed with RIPA buffer containing a protease inhibitor cocktail (Roche, Mannheim, Germany). The lysates were boiled for 5 min in sodium dodecyl sulfate (SDS) sample buffer. Proteins were separated by SDS-PAGE and transferred onto polyvinylidene difluoride membrane (Millipore, Billerica, MA). The membrane was blocked with 5% nonfat milk in Tris-buffered saline containing 0.1% Tween 20 (TBS-T) and incubated with primary antibodies at 4°C overnight. After a wash with TBS-T, the membrane was incubated with secondary antibody at room temperature for 1 h. The signal was detected by enhanced chemiluminescence (AbClon, Seoul, South Korea) using an LAS-4000 digital imaging system (Fujifilm, Tokyo, Japan) and analyzed using Multi-Gauge software V3.0 (Fujifilm).
Immunoprecipitation.At 48 h posttransfection, the cells were harvested and lysed with radioim-munoprecipitation assay (RIPA) buffer containing a protease inhibitor cocktail. The lysates were diluted 1:5 with RIPA buffer, followed by precleaning with protein G-agarose (Roche) at 4°C for 2 h. The clarified lysates were incubated with primary antibody in an orbital shaker at 4°C overnight. The immunocom-plexes were precipitated with protein G-agarose at 4°C for 4 h, washed three times with cold PBS, boiled for 5 min in SDS sample buffer, and analyzed by Western blotting.
In vivo ubiquitination assay.A plasmid encoding His-tagged ubiquitin was cotransfected with plasmids or siRNA indicated in the figures. At 43 h posttransfection, MG132 (20M) was added for 5 h. The cells were lysed with SDS lysis buffer and boiled for 10 min. An aliquot (5% of lysate volume) was used for Western blotting as input control, and the rest was used for immunoprecipitation: it was diluted 1:5 with TBS containing a protease inhibitor cocktail, followed by incubation with anti-HA antibody at 4°C overnight. The levels of polyubiquitination were analyzed by Western blotting with anti-His tag antibody. Southern blotting.Core-associated HBV replication was determined by Southern blotting as de-scribed in our previous report (69). Briefly, at 72 h posttransfection, cell pellets were lysed with HEPES buffer containing 1% NP-40. To remove transfected plasmid DNA, cell lysates were treated with DNase I (Sigma) and mung bean nuclease (TaKaRa, Kusatsu, Shiga, Japan) at 37°C for 20 min. Core particles were precipitated with polyethylene glycol solution by 2 h on ice, and capsids were digested with proteinase K (20 mg/ml; Roche) at 37°C for 2 to 3 h in the presence of 0.5% SDS. HBV DNA was extracted with a mixture of phenol-chloroform-isoamyl alcohol (25:24:1; Sigma) and precipitated with ethanol and 3 M sodium acetate. Purified DNA was separated on a 1% agarose gel and transferred onto a Hybond-N⫹
nylon membrane (GE Healthcare, Buckinghamshire, UK). HBV DNA was detected with highly pure randomly primed probes labeled with [␣-32P]dCTP (Perkin-Elmer, Waltham, MA) and quantified using a
phosphorimager (Fujifilm).
Northern blotting.HBV RNA was detected by Northern blotting as described previously (18, 68). Total RNA was extracted using TRIzol (Invitrogen) according to the manufacturer’s protocol. Total RNA (20g) was separated on a 1% formaldehyde-agarose gel and transferred onto a Hybond-N⫹nylon
membrane. To detect HBV RNA, the membrane was hybridized with the probe used for Southern blotting.
Electrophoretic mobility shift assay. The binding of nuclear proteins to the DNA probe was examined as described previously (18). Briefly, at 48 h posttransfection, nuclear fractions were obtained using a nuclear and cytoplasmic extraction kit (Thermo Fisher Scientific, Waltham, MA). We designed double-stranded oligonucleotides harboring the binding sites of HNF4␣and HNF3from the HBV Enhancer II region (HNF4␣, 5=-GGAGGAGATTAGGTTAAAGGTCTTT-3=, nucleotides [nt] 1742 to 1768; HNF3, 5=-CTTCAAAGACTGTTTGTTTAAAGAC-3=, nt 1706 to 1732). The oligonucleotides were end labeled with [␥-32P]dCTP using T4-polynucleotide kinase. The nuclear extract (2g) and the probes (2 pmol) were
incubated in reaction buffer (20 mM Tris-HCl [pH 7.5], 60 mM KCl, 25 mM MgCl2, 1 mM dithiothreitol, 1%
NP-40, 7% glycerol) for 30 min at room temperature. The DNA-protein complexes were separated on a 6% polyacrylamide gel and analyzed using a Phosphorimager (Fujifilm).
RNA interference and RT-PCR.Customized siRNA against the 5=end of c-FLIP mRNA, which encodes the DED1 region, was purchased from ST Pharm (Seoul, South Korea): sense, 5=-UGAAGAAGCACUUGA UACATT(dTdT)-3=; and antisense, 5=-UGUAUCAAGUGCUUCUUCATT(dTdT)-3=. Cells were transfected with annealed siRNA using Lipofectamine 2000. Total RNA (2g) was used to synthesize cDNA with Moloney murine leukemia virus (M-MLV) reverse transcriptase (iNtRON Biotechnology, Seongnam, South Korea). Reverse transcription-PCR (RT-PCR) was performed using cDNA and specific primers for GAPDH and HBx as previously described (18, 68). For c-FLIPL, the following primers were used: forward, 5=-GAGCACCGA
GACTACGACAG-3=; and reverse, 5=-GTGAAGATCCAGGAGTGGGC-3=. PCR products were confirmed by electrophoresis on a 1.2% agarose gel.
Real-time PCR.Real-time PCR was performed using SYBR green PCR master mix (Applied Biosystems, Foster City, CA) and the primers used for RT-PCR (for GAPDH and c-FLIP). Primers for HNF4␣, HNF1␣, and
on November 6, 2019 by guest
http://jvi.asm.org/
HNF3were described previously (18, 68). cDNA was amplified in an ABI Prism 7500 system (Applied Biosystems) under the following conditions: denaturation at 94°C for 5 min, followed by 40 cycles of 94°C for 30 s and 55°C for 1 min, and a final extension at 55°C for 5 min. Transcript levels were quantified by the comparative ΔΔCTmethod relative to a control sample (70).
For the analysis of HBV DNA in HBV-infected cells, total genomic and viral DNA was extracted using a QIAamp DNA minikit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. Briefly, cells were lysed with buffer containing proteinase K to release genomic and HBV DNA. Semiquantitative PCR was performed using 20 ng of extracted DNA as follows: denaturation at 94°C for 5 min, followed by 25 cycles of 94°C for 30 s, 60°C for 30 s, and 72°C for 45 s, and then a final extension for 5 min at 72°C. Serial dilutions of a plasmid containing the HBV monomer digested with EcoRI were used as quantification standard. PCR products were analyzed by electrophoresis on a 1.2% agarose gel.
Real-time PCR was performed with 100 ng of extracted DNA using FAM probes and a LightCycler 480 Probes Master (Roche) in a LightCycler 480 PCR Instrument (Roche) under the following conditions: denaturation at 95°C for 5 min, followed by 45 cycles of 95°C for 10 s, 58°C for 10 s, and 72°C for 15 s, followed by cooling to 40°C over 30 s. The primers targeting HBV DNA were as follows: forward (nt 256 to 274), 5=-CTCGTGGTGGACTTCTCTC-3=; and reverse (nt 404 to 421), 5=-CTGCAGGATGAAGAGGAA-3=. Relative quantification was determined by the LightCycler 480 Software 1.5 (Roche).
To analyze HBV RNA in HepG2-NTCP cells, total RNA (2g) was used to synthesize cDNA with M-MLV reverse transcriptase (iNtRON Biotechnology). cDNA was amplified using SYBR green PCR master mix (Applied Biosystems), and primers were used to detect HBV DNA and GAPDH in an ABI Prism 7500 system (Applied Biosystems) under the following conditions: denaturation at 94°C for 5 min, followed by 40 cycles of 94°C for 30 s and 60°C for 1 min, and then a final extension at 55°C for 5 min. Transcript levels were quantified by the comparative ΔΔCTmethod relative to a control sample.
The level of HBV cccDNA in HepG2-NTCP cells was determined by real-time PCR as described previously (71), with minor modifications. To ensure the specificity of cccDNA detection, aliquots of extracted DNA were treated with 5 U of T5 exonuclease (M0363S; New England BioLabs, Evry, France) at 37°C for 30 min, and the reaction was stopped by adding EDTA to a final concentration of 11 mM. Real-time PCR was performed in a LightCycler 480 PCR Instrument (Roche) in a 20-l reaction volume containing 20 ng of T5-treated DNA, 3 mM MgCl2, 0.5M concentrations of each forward and reverse
primer, 0.2 M 3=-fluorescein (FL)-labeled probe, and 0.4 M 5=-Red640 (R640)-labeled probe. The forward and reverse primers were 5=-CTCCCCGTCTGTGCCTTCT-3=(nt 1548 to 1566) and 5=-GCCCCAAA GCCACCCAAG-3=(nt 1903 to 1886), respectively. FRET hybridization probes were 5=-GTTCACGGTGGTCT CCATGCAACGT-FL-3= and 5=-R640-AGGTGAAGCGAAGTGCACACGGACC-p-3=. Amplification of cccDNA and GAPDH was performed as follows: denaturation at 95°C for 10 min, followed by 45 cycles of 95°C for 10 s, 62°C for 10 s, and 72°C for 20 s, and then cooling to 40°C over 30 s. cccDNA levels were quantified by the comparative ΔΔCTmethod relative to that of the GAPDH gene, which was amplified using the
primers described above. PCR products were analyzed by electrophoresis on a 1.2% agarose gel. Luciferase assay.Approximately 2.5⫻105HepG2 cells per well were seeded on 12-well plates. At
48 h posttransfection, the cells were lysed, and the luciferase activity in the lysates was determined using the luciferase assay system (Promega) according to the manufacturer’s instructions. The signals were measured in a luminometer.
XTT assay and Live/Dead flow cytometry.Approximately 2.5⫻105HepG2 or Huh7 cells per well
were seeded on 12-well plates. For XTT assay, at 48 h posttransfection, cells were incubated with XTT and PMS reagents (Welgene) at 37°C for 1 h. The optical density (OD) values were measured at 450 and 690 nm using a spectrophotometer.
To determine the populations of live and dead cells, cells were detached with trypsin at 48 h posttransfection. After a washing step with PBS, the cells were stained with fluorescent dye for 30 min at 4°C using a Live/Dead fixable dead cell stain kit (L34970; Thermo Fisher Scientific) according to the manufacturer’s instructions. The cells were fixed with 4% paraformaldehyde and analyzed by using a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA).
Caspase 3/7 activity assay.Approximately 2.5⫻105HepG2 or Huh7 cells per well were seeded on
12-well plates. At 48 h posttransfection, the cells were incubated with Caspase-Glo reagent at room temperature for 1 h. The caspase 3/7 activity was evaluated using the Caspase-Glo 3/7 Assay (Promega) according to the manufacturer’s protocol. The signals were measured with a luminometer.
Measurement of HBeAg and HBsAg by ELISA.At 72 h posttransfection, culture supernatants were collected and diluted 1: 20 for HBeAg and 1:50 for HBsAg. The levels of secreted HBeAg and HBsAg were quantified using a Diagnostic Kit for Hepatitis B e/s Antigen (Wantai Bio-Pharm, Beijing, China) according to the manufacturer’s instructions. The OD values were measured at 450 nm using a spectrophotometer. Immunofluorescence microscopy analysis.Cells seeded on coverslips were fixed with 4% parafor-maldehyde and permeabilized with 0.2% Triton X-100. Coverslips were blocked with 3% bovine serum albumin (BSA) and incubated with primary antibody diluted 1:300 with PBS containing 3% BSA at 4°C overnight. After washing with PBS, Alexa Fluor-conjugated secondary antibody (Invitrogen) and DAPI (4=,6=-diamidino-2-phenylindole; Sigma) were used to detect immune complexes and nuclei, respectively.
ACKNOWLEDGMENTS
This study was supported by Konkuk University in 2015.
Author contributions were as follows: study conception and design (A.R.L., K.-H.L.,
Y.K.P., and K.-H.K.), acquisition of data (A.R.L., K.-H.L., and E.-S.P.), analysis and
interpre-tation of data (A.R.L., K.-H.L., E.-S.P., D.H.K., Y.K.P., S.I.C., and K.-H.K.), material support
August 2018 Volume 92 Issue 16 e00339-18 jvi.asm.org 17
on November 6, 2019 by guest
http://jvi.asm.org/
(the isolation, differentiation, or maintenance of HepaRG cells and primary human
hepatocytes) (D.-S.K., G.-C.S., S.P., H.S.K., J.W., H.S., Y.N.H., and B.J.), draft of the
manu-script (A.R.L. and K.-H.K.), and study supervision (K.-H.K.).
REFERENCES
1. Trepo C, Chan HL, Lok A. 2014. Hepatitis B virus infection. Lancet 384:2053–2063.https://doi.org/10.1016/S0140-6736(14)60220-8. 2. Ali A, Abdel-Hafiz H, Suhail M, Al-Mars A, Zakaria MK, Fatima K, Ahmad
S, Azhar E, Chaudhary A, Qadri I. 2014. Hepatitis B virus, HBx mutants and their role in hepatocellular carcinoma. World J Gastroenterol 20: 10238 –10248.https://doi.org/10.3748/wjg.v20.i30.10238.
3. Tang H, Delgermaa L, Huang F, Oishi N, Liu L, He F, Zhao L, Murakami S. 2005. The transcriptional transactivation function of HBx protein is important for its augmentation role in hepatitis B virus replication. J Virol 79:5548 –5556.https://doi.org/10.1128/JVI.79.9.5548-5556.2005. 4. Bouchard MJ, Wang LH, Schneider RJ. 2001. Calcium signaling by HBx
protein in hepatitis B virus DNA replication. Science 294:2376 –2378.
https://doi.org/10.1126/science.294.5550.2376.
5. Ng SA, Lee C. 2011. Hepatitis B virus X gene and hepatocarcinogenesis. J Gastroenterol 46:974 –990.https://doi.org/10.1007/s00535-011-0415-9. 6. Chen HS, Kaneko S, Girones R, Anderson RW, Hornbuckle WE, Tennant BC, Cote PJ, Gerin JL, Purcell RH, Miller RH. 1993. The woodchuck hepatitis virus X gene is important for establishment of virus infection in woodchucks. J Virol 67:1218 –1226.
7. Zoulim F, Saputelli J, Seeger C. 1994. Woodchuck hepatitis virus X protein is required for viral infection in vivo. J Virol 68:2026 –2030. 8. Pollicino T, Belloni L, Raffa G, Pediconi N, Squadrito G, Raimondo G,
Levrero M. 2006. Hepatitis B virus replication is regulated by the acety-lation status of hepatitis B virus cccDNA-bound H3 and H4 histones. Gastroenterology 130:823– 837.https://doi.org/10.1053/j.gastro.2006.01 .001.
9. Belloni L, Pollicino T, De Nicola F, Guerrieri F, Raffa G, Fanciulli M, Raimondo G, Levrero M. 2009. Nuclear HBx binds the HBV minichromo-some and modifies the epigenetic regulation of cccDNA function. Proc Natl Acad Sci U S A 106:19975–19979. https://doi.org/10.1073/pnas .0908365106.
10. Alarcon V, Hernandez S, Rubio L, Alvarez F, Flores Y, Varas-Godoy M, De Ferrari GV, Kann M, Villanueva RA, Loyola A. 2016. The enzymes LSD1 and Set1A cooperate with the viral protein HBx to establish an active hepatitis B viral chromatin state. Sci Rep 6:25901. https://doi.org/10 .1038/srep25901.
11. Shin GC, Kang HS, Lee AR, Kim KH. 2016. Hepatitis B virus-triggered autophagy targets TNFRSF10B/death receptor 5 for degradation to limit TNFSF10/TRAIL response. Autophagy 12:2451–2466.https://doi.org/10 .1080/15548627.2016.1239002.
12. Murphy CM, Xu Y, Li F, Nio K, Reszka-Blanco N, Li X, Wu Y, Yu Y, Xiong Y, Su L. 2016. Hepatitis B virus X protein promotes degradation of SMC5/6 to enhance HBV replication. Cell Rep 16:2846 –2854.https://doi .org/10.1016/j.celrep.2016.08.026.
13. Decorsiere A, Mueller H, van Breugel PC, Abdul F, Gerossier L, Beran RK, Livingston CM, Niu C, Fletcher SP, Hantz O, Strubin M. 2016. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature 531:386 –389.https://doi.org/10.1038/nature17170.
14. Kim DH, Kang HS, Kim KH. 2016. Roles of hepatocyte nuclear factors in hepatitis B virus infection. World J Gastroenterol 22:7017–7029.https:// doi.org/10.3748/wjg.v22.i31.7017.
15. Watt AJ, Garrison WD, Duncan SA. 2003. HNF4: a central regulator of hepatocyte differentiation and function. Hepatology 37:1249 –1253.
https://doi.org/10.1053/jhep.2003.50273.
16. Quasdorff M, Hosel M, Odenthal M, Zedler U, Bohne F, Gripon P, Dienes HP, Drebber U, Stippel D, Goeser T, Protzer U. 2008. A concerted action of HNF4␣and HNF1␣links hepatitis B virus replication to hepatocyte differentiation. Cell Microbiol 10:1478 –1490. https://doi.org/10.1111/j .1462-5822.2008.01141.x.
17. Banks KE, Anderson AL, Tang H, Hughes DE, Costa RH, McLachlan A. 2002. Hepatocyte nuclear factor 3inhibits hepatitis B virus replication in vivo. J Virol 76:12974 –12980.https://doi.org/10.1128/JVI.76.24.12974 -12980.2002.
18. Park YK, Park ES, Kim DH, Ahn SH, Park SH, Lee AR, Park S, Kang HS, Lee JH, Kim JM, Lee SK, Lim KH, Isorce N, Tong S, Zoulim F, Kim KH. 2016. Cleaved c-FLIP mediates the antiviral effect of TNF-␣against hepatitis B
virus by dysregulating hepatocyte nuclear factors. J Hepatol 64:268 –277.
https://doi.org/10.1016/j.jhep.2015.09.012.
19. Zhao Z, Hong W, Zeng Z, Wu Y, Hu K, Tian X, Li W, Cao Z. 2012. Mucroporin-M1 inhibits hepatitis B virus replication by activating the mitogen-activated protein kinase (MAPK) pathway and down-regulating HNF4␣in vitro and in vivo. J Biol Chem 287:30181–30190.https://doi .org/10.1074/jbc.M112.370312.
20. Krueger A, Baumann S, Krammer PH, Kirchhoff S. 2001. FLICE-inhibitory proteins: regulators of death receptor-mediated apoptosis. Mol Cell Biol 21:8247– 8254.https://doi.org/10.1128/MCB.21.24.8247-8254.2001. 21. Wang W, Zhou J, Shi J, Zhang Y, Liu S, Liu Y, Zheng D. 2014. Human T-cell
leukemia virus type 1 Tax-deregulated autophagy pathway and c-FLIP expression contribute to resistance against death receptor-mediated apoptosis. J Virol 88:2786 –2798.https://doi.org/10.1128/JVI.03025-13. 22. Wang W, Wang S, Song X, Sima N, Xu X, Luo A, Chen G, Deng D, Xu Q,
Meng L, Lu Y, Ma D. 2007. The relationship between c-FLIP expression and human papillomavirus E2 gene disruption in cervical carcinogene-sis. Gynecol Oncol 105:571–577.https://doi.org/10.1016/j.ygyno.2007.01 .051.
23. Stefanidou M, Ramos I, Mas Casullo V, Trepanier JB, Rosenbaum S, Fernandez-Sesma A, Herold BC. 2013. Herpes simplex virus 2 (HSV-2) prevents dendritic cell maturation, induces apoptosis, and triggers re-lease of proinflammatory cytokines: potential links to HSV-HIV synergy. J Virol 87:1443–1453.https://doi.org/10.1128/JVI.01302-12.
24. Bleumink M, Kohler R, Giaisi M, Proksch P, Krammer PH, Li-Weber M. 2011. Rocaglamide breaks TRAIL resistance in HTLV-1-associated adult T-cell leukemia/lymphoma by translational suppression of c-FLIP expres-sion. Cell Death Differ 18:362–370.https://doi.org/10.1038/cdd.2010.99. 25. Kim KH, Seong BL. 2003. Proapoptotic function of HBV X protein is mediated by interaction with c-FLIP and enhancement of death-inducing signal. EMBO J 22:2104 –2116.https://doi.org/10.1093/emboj/ cdg210.
26. Lim KH, Choi HS, Park YK, Park ES, Shin GC, Kim DH, Ahn SH, Kim KH. 2013. HBx-induced NF-B signaling in liver cells is potentially mediated by the ternary complex of HBx with p22-FLIP and NEMO. PLoS One 8:e57331.https://doi.org/10.1371/journal.pone.0057331.
27. Lim KH, Kim KH, Choi SI, Park ES, Park SH, Ryu K, Park YK, Kwon SY, Yang SI, Lee HC, Sung IK, Seong BL. 2011. RPS3a overexpressed in HBV-associated hepatocellular carcinoma enhances the HBx-induced NF-B signaling via its novel chaperoning function. PLoS One 6:e22258.https:// doi.org/10.1371/journal.pone.0022258.
28. Henkler F, Hoare J, Waseem N, Goldin RD, McGarvey MJ, Koshy R, King IA. 2001. Intracellular localization of the hepatitis B virus HBx protein. J Gen Virol 82:871– 882.https://doi.org/10.1099/0022-1317-82-4-871. 29. Song CZ, Bai ZL, Song CC, Wang QW. 2003. Aggregate formation of
hepatitis B virus X protein affects cell cycle and apoptosis. World J Gastroenterol 9:1521–1524.https://doi.org/10.3748/wjg.v9.i7.1521. 30. Urban S, Hildt E, Eckerskorn C, Sirma H, Kekule A, Hofschneider PH. 1997.
Isolation and molecular characterization of hepatitis B virus X-protein from a baculovirus expression system. Hepatology 26:1045–1053.
https://doi.org/10.1002/hep.510260437.
31. Ryu K, Kim CW, Kim BH, Han KS, Kim KH, Choi SI, Seong BL. 2008. Assessment of substrate-stabilizing factors for DnaK on the folding of aggregation-prone proteins. Biochem Biophys Res Commun 373:74 –79.
https://doi.org/10.1016/j.bbrc.2008.05.186.
32. Wayne N, Bolon DN. 2010. Charge-rich regions modulate the anti-aggregation activity of Hsp90. J Mol Biol 401:931–939.https://doi.org/ 10.1016/j.jmb.2010.06.066.
33. Yuh CH, Ting LP. 1990. The genome of hepatitis B virus contains a second enhancer: cooperation of two elements within this enhancer is required for its function. J Virol 64:4281– 4287.
34. Su H, Yee JK. 1992. Regulation of hepatitis B virus gene expression by its two enhancers. Proc Natl Acad Sci U S A 89:2708 –2712.https://doi.org/ 10.1073/pnas.89.7.2708.
35. Li J, Ning G, Duncan SA. 2000. Mammalian hepatocyte differentiation requires the transcription factor HNF-4␣. Genes Dev 14:464 – 474.
on November 6, 2019 by guest
http://jvi.asm.org/
36. Parviz F, Matullo C, Garrison WD, Savatski L, Adamson JW, Ning G, Kaestner KH, Rossi JM, Zaret KS, Duncan SA. 2003. Hepatocyte nuclear factor 4␣controls the development of a hepatic epithelium and liver morphogenesis. Nat Genet 34:292–296.https://doi.org/10.1038/ng1175. 37. Choi SI, Kwon S, Son A, Jeong H, Kim KH, Seong BL. 2013. Protein folding in vivo revisited. Curr Protein Pept Sci 14:721–733.https://doi.org/10 .2174/138920371408131227170544.
38. Choi SI, Han KS, Kim CW, Ryu KS, Kim BH, Kim KH, Kim SI, Kang TH, Shin HC, Lim KH, Kim HK, Hyun JM, Seong BL. 2008. Protein solubility and folding enhancement by interaction with RNA. PLoS One 3:e2677.
https://doi.org/10.1371/journal.pone.0002677.
39. Hartl FU, Bracher A, Hayer-Hartl M. 2011. Molecular chaperones in protein folding and proteostasis. Nature 475:324 –332.https://doi.org/ 10.1038/nature10317.
40. Citri A, Harari D, Shohat G, Ramakrishnan P, Gan J, Lavi S, Eisenstein M, Kimchi A, Wallach D, Pietrokovski S, Yarden Y. 2006. Hsp90 recognizes a common surface on client kinases. J Biol Chem 281:14361–14369.
https://doi.org/10.1074/jbc.M512613200.
41. Arndt V, Rogon C, Hohfeld J. 2007. To be, or not to be–molecular chaperones in protein degradation. Cell Mol Life Sci 64:2525–2541.
https://doi.org/10.1007/s00018-007-7188-6.
42. Bailey CK, Andriola IF, Kampinga HH, Merry DE. 2002. Molecular chap-erones enhance the degradation of expanded polyglutamine repeat androgen receptor in a cellular model of spinal and bulbar muscular atrophy. Hum Mol Genet 11:515–523.https://doi.org/10.1093/hmg/11.5 .515.
43. Shin GC, Ahn SH, Choi HS, Lim KH, Choi do, Kim YKP, Kim KH. 2013. Hepatocystin/80K-H inhibits replication of hepatitis B virus through interaction with HBx protein in hepatoma cell. Biochim Biophys Acta 1832:1569 –1581.https://doi.org/10.1016/j.bbadis.2013.04.026. 44. Ait-Goughoulte M, Lucifora J, Zoulim F, Durantel D. 2010. Innate antiviral
immune responses to hepatitis B virus. Viruses 2:1394 –1410.https://doi .org/10.3390/v2071394.
45. Cavanaugh VJ, Guidotti LG, Chisari FV. 1997. Interleukin-12 inhibits hepatitis B virus replication in transgenic mice. J Virol 71:3236 –3243. 46. Xiao CW, Yan X, Li Y, Reddy SA, Tsang BK. 2003. Resistance of human
ovarian cancer cells to tumor necrosis factor alpha is a consequence of nuclear factorB-mediated induction of Fas-associated death domain-like interleukin-1beta-converting enzyme-domain-like inhibitory protein. Endo-crinology 144:623– 630.https://doi.org/10.1210/en.2001-211024. 47. Micheau O, Lens S, Gaide O, Alevizopoulos K, Tschopp J. 2001. NF-B
signals induce the expression of c-FLIP. Mol Cell Biol 21:5299 –5305.
https://doi.org/10.1128/MCB.21.16.5299-5305.2001.
48. Lucifora J, Arzberger S, Durantel D, Belloni L, Strubin M, Levrero M, Zoulim F, Hantz O, Protzer U. 2011. Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J Hepatol 55: 996 –1003.https://doi.org/10.1016/j.jhep.2011.02.015.
49. Ye B, Liu X, Li X, Kong H, Tian L, Chen Y. 2015. T-cell exhaustion in chronic hepatitis B infection: current knowledge and clinical significance. Cell Death Dis 6:e1694.https://doi.org/10.1038/cddis.2015.42. 50. Chisari FV, Isogawa M, Wieland SF. 2010. Pathogenesis of hepatitis B
virus infection. Pathol Biol (Paris) 58:258 –266.https://doi.org/10.1016/j .patbio.2009.11.001.
51. Li X, Wang Y, Chen Y. 2014. Cellular immune response in patients with chronic hepatitis B virus infection. Microb Pathog 74:59 – 62.https://doi .org/10.1016/j.micpath.2014.07.010.
52. Bauer T, Sprinzl M, Protzer U. 2011. Immune control of hepatitis B virus. Dig Dis 29:423– 433.https://doi.org/10.1159/000329809.
53. Chen SH, Wu HL, Kao JH, Hwang LH. 2012. Persistent hepatitis B viral replication in a FVB/N mouse model: impact of host and viral factors. PLoS One 7:e36984.https://doi.org/10.1371/journal.pone.0036984. 54. Bagnoli M, Canevari S, Mezzanzanica D. 2010. Cellular FLICE-inhibitory
protein (c-FLIP) signalling: a key regulator of receptor-mediated apop-tosis in physiologic context and in cancer. Int J Biochem Cell Biol 42:210 –213.https://doi.org/10.1016/j.biocel.2009.11.015.
55. Longley DB, Wilson TR, McEwan M, Allen WL, McDermott U, Galligan L, Johnston PG. 2006. c-FLIP inhibits chemotherapy-induced colorectal cancer cell death. Oncogene 25:838 – 848.https://doi.org/10.1038/sj.onc .1209122.
56. Xiao C, Yang BF, Song JH, Schulman H, Li L, Hao C. 2005. Inhibition of
CaMKII-mediated c-FLIP expression sensitizes malignant melanoma cells to TRAIL-induced apoptosis. Exp Cell Res 304:244 –255.https://doi.org/ 10.1016/j.yexcr.2004.11.002.
57. Rippo MR, Moretti S, Vescovi S, Tomasetti M, Orecchia S, Amici G, Catalano A, Procopio A. 2004. FLIP overexpression inhibits death receptor-induced apoptosis in malignant mesothelial cells. Oncogene 23:7753–7760.https://doi.org/10.1038/sj.onc.1208051.
58. Zhou XD, Yu JP, Liu J, Luo HS, Chen HX, Yu HG. 2004. Overexpression of cellular FLICE-inhibitory protein (FLIP) in gastric adenocarcinoma. Clin Sci (Lond) 106:397– 405.https://doi.org/10.1042/CS20030238. 59. Zheng Z, Cheng S, Wu W, Wang L, Zhao Y, Shen Y, Janin A, Zhao WL.
2014. c-FLIP is involved in tumor progression of peripheral T-cell lym-phoma and targeted by histone deacetylase inhibitors. J Hematol Oncol 7:88.https://doi.org/10.1186/s13045-014-0088-y.
60. Ili CG, Brebi P, Tapia O, Sandoval A, Lopez J, Garcia P, Leal P, Sidransky D, Guerrero-Preston R, Roa JC. 2013. Cellular FLICE-like inhibitory protein long form (c-FLIPL) overexpression is related to cervical cancer progression. Int J Gynecol Pathol 32:316 –322.https://doi.org/ 10.1097/PGP.0b013e31825d8064.
61. Dutton A, Young LS, Murray PG. 2006. The role of cellular FLICE inhibi-tory protein (c-FLIP) in the pathogenesis and treatment of cancer. Expert Opin Ther Targets 10:27–35.https://doi.org/10.1517/14728222.10.1.27. 62. Du X, Bao G, He X, Zhao H, Yu F, Qiao Q, Lu J, Ma Q. 2009. Expression and
biological significance of c-FLIP in human hepatocellular carcinomas. J Exp Clin Cancer Res 28:24.https://doi.org/10.1186/1756-9966-28-24. 63. Jiang R, Xia Y, Li J, Deng L, Zhao L, Shi J, Wang X, Sun B. 2010. High
expression levels of IKK␣and IKK are necessary for the malignant properties of liver cancer. Int J Cancer 126:1263–1274.https://doi.org/ 10.1002/ijc.