0022-538X/95/$04.0010
Copyright 1995, American Society for Microbiology
Interactions of Thyroid Hormone Receptor with the Human
Immunodeficiency Virus Type 1 (HIV-1) Long Terminal
Repeat and the HIV-1 Tat Transactivator
VANDANA DESAI-YAJNIK, EMIR HADZIC, PAUL MODLINGER, SUNIL MALHOTRA,
GLEN GECHLIK,
ANDHERBERT H. SAMUELS*
Department of Medicine, New York University Medical Center, New York, New York 10016
Received 19 January 1995/Accepted 28 April 1995
Thyroid hormone (T3) receptor (T3R) regulates the human immunodeficiency virus type 1 (HIV-1) long
terminal repeat (LTR) by binding to and activating thyroid hormone response elements (TREs) embedded
within the viral NF-
k
B and Sp1 motifs. The TREs within the NF-
k
B sites are necessary for activation by T3
in the absence of Tat, while those in the Sp1 motifs function as TREs only when Tat is expressed, suggesting
that Tat and T3R interact in the cell. Transactivation of the HIV-1 LTR by T3R
a
and several receptor mutants
revealed that the 50-amino-acid N-terminal A/B region of T3R
a
, known to interact with the basal transcription
factor TFIIB, is critical for activation of both Tat-dependent and Tat-independent responsive sequences of the
LTR. A single amino acid change in the highly conserved
t
iregion in the ligand-binding domain of T3R
a
eliminates Tat-independent but not Tat-dependent activation of the HIV-1 LTR by T3. Ro 5-3335
[7-chloro-5-(2-pyrryl)-3
H
-1,4-benzodiazepin-2(
H
)-one], which inhibits Tat-mediated transactivation of HIV-1, also
in-hibits the functional interaction between Tat and T3R
a
. Binding studies with glutathione-
S
-transferase fusion
proteins and Western (immunoblot) analysis indicate that T3R
a
interacts with Tat through amino acids within
the DNA-binding domain of T3R
a
. Mutational analysis revealed that amino acid residues in the basic and
C-terminal regions of Tat are required for the binding of Tat to T3R
a
, while the N terminus of Tat is not
required. These studies provide functional and physical evidence that stimulation of the HIV-1 LTR by T3
involves an interaction between T3R
a
and Tat. Our results also suggest a model in which multiple domains of
T3R
a
interact with Tat and other factors to form transcriptionally important complexes.
Members of the steroid/thyroid hormone receptor gene
fam-ily are known to regulate transcription from the long terminal
repeats (LTRs) of a number of retroviruses (8, 60, 73). We
reported that thyroid hormone (
L-triiodothyronine; T3), via its
nucleus-associated receptor (T3R), can stimulate the human
immunodeficiency virus type 1 (HIV-1) LTR (9). T3R
regu-lates the LTR by binding to and activating thyroid hormone
response elements (TREs) embedded within the viral NF-
k
B
(
2
104 to
2
80) and Sp1 (
2
76 to
2
45) motifs. These studies
revealed that the LTR contains both Tat-independent and
Tat-dependent TREs. The TREs within the NF-
k
B sites
me-diate T3 stimulation in the absence of Tat, while those in the
Sp1 motifs function as TREs only when Tat is expressed,
sug-gesting that Tat and T3R interact in the cell. Figure 1 depicts
two pathways by which T3 and T3R may regulate the HIV-1
LTR. Pathway A involves the binding of T3R to the viral
NF-
k
B sites, which leads to an increase in the transcription of
viral genes. This increases expression of Tat, which then may
functionally interact with T3R bound to the Sp1 sites to further
enhance viral gene expression (pathway B).
HIV-1 Tat is a potent activator that is essential for viral
replication and transcription (7, 11). The actions of Tat are
enhanced by factors which bind to the NF-
k
B elements and, in
particular, to the Sp1 elements (3, 4, 67). The Tat gene encodes
an 86-amino-acid polypeptide specified by two exons (7, 11)
(Fig. 2A). Activation of HIV-1 by Tat requires the trans-acting
responsive (TAR) element found within sequences from
1
19
to
1
44 (10, 22, 24, 28, 54–56) and thus is present at the 5
9
end
of all HIV-1 transcripts (Fig. 1). Only the first 58 amino acids
of Tat are necessary for transactivation (61). Tat can be divided
into five structural domains (Fig. 2A). The N-terminal
proline-rich region (amino acids 2 to 18) contains three acidic residues,
and mutations within this region abolish transactivation (19,
36, 51). The cysteine-rich region consists of amino acids 22 to
37, and modification of any of the cysteines or the His at
position 33 dramatically reduces Tat activity (20, 23, 36, 57,
58). A core region (amino acids 38 to 48) is highly conserved
among all lentivirus Tat proteins. The arginine-rich basic
re-gion (amino acids 49 to 57) is important for nuclear
localiza-tion (57, 68) and mediates specific binding to TAR RNA (74).
Amino acids 72 to 86 (exon 2) appear to be dispensable for
efficient transactivation of the HIV-1 LTR (61).
Although the precise mechanism of Tat action has not been
fully defined, two models have been proposed to explain the
mechanism of stimulation of HIV-1 transcripts by Tat. A
feed-back model suggests that Tat is recruited to the vicinity of the
promoter by binding to nascent TAR RNA to increase the rate
of transcription initiation (3, 67), while an RNA processivity
model proposes that Tat augments transcription elongation by
suppressing random termination of RNA polymerase II (34).
The ability of Tat to transactivate the LTR without direct
interaction with viral DNA suggests that Tat, while tethered to
TAR RNA, mediates stimulation through interactions with
promoter-bound transcription factors. Evidence that Tat can
function as a more ‘‘conventional’’ transactivator comes from
studies indicating that a GAL4-Tat fusion protein activates
transcription when bound to a GAL4 response element (67).
The thyroid/retinoid receptor gene subfamily comprises T3
receptors (T3Rs), all trans-retinoic acid receptors (RARs),
* Corresponding author. Mailing address: Departments of Medicineand Pharmacology, TH-454, New York University Medical Center, 550 First Ave., New York, NY 10016. Phone: (212) 263-6279. Fax: (212) 263-7701. Electronic mail address: [email protected].
5103
on November 9, 2019 by guest
http://jvi.asm.org/
9-cis-retinoic acid receptors (RARs and RXRs), and the
1,25-dihydroxy-vitamin D
3receptor (VDR). T3Rs, RARs, and
RXRs (40, 41) are expressed from two or three genes (
a
,
b
,
and
g
). Both T3R
a
and T3R
b
can activate the HIV-1 LTR (9).
In this study, we focused on chicken T3R
a
(cT3R
a
), a
408-amino-acid protein that can be divided into six distinct
do-mains (A to F) (Fig. 2B). The N-terminal 50-amino-acid A/B
region is the least conserved region among these nuclear
hor-mone receptors. The highly conserved 68-amino-acid C
do-main is organized into two zinc finger DNA-binding structures
(amino acids 51 to 119). Domains D, E, and F constitute the
ligand-binding domain (amino acids 120 to 408) (31, 38).
Em-bedded within the ligand-binding region of T3Rs, RARs,
VDRs, and RXRs is the heptad repeat region involved in
dimerization (15) (Fig. 2B) and a conserved 20-amino-acid
region, referred to as
ti
(15), also thought to be involved in
protein-protein interactions (37, 46, 47). In this study, we show
that both Tat-dependent and Tat-independent activation of
the HIV-1 LTR by T3 requires the 50-amino-acid A/B region
of cT3R
a
, which is known to interact with the basal
transcrip-tion factor TFIIB (27). In additranscrip-tion, we show that Tat physically
interacts with the receptor. We also provide functional
evi-dence that stimulation of the HIV-1 LTR by T3 involves an
interaction between T3R and Tat and/or the cellular factor
that is regulated by the Tat antagonist Ro 5-3335. Our results
suggest a model in which different domains of T3R play a role
in interfacing Tat with members of the transcriptional
appara-tus.
MATERIALS AND METHODS
[image:2.612.66.546.77.411.2]Cloning and plasmid construction. pU3R-III-CAT(2167/121) [p(2167/ 121)-CAT], pU3R-III-CAT(2104/180) [p(2104/180)-CAT], pU3R-III-CAT (276/180) [p(276/180)-CAT], and pU3R-III-CAT(245/180) [p(245/1 80)-CAT] (9) are deletions of the pU3R-III-CAT vector containing the HIV-1 clone FIG. 1. Tat-independent and Tat-dependent regulation of the HIV-1 LTR by T3R. The positions of the two NF-kB and three Sp1 sites along with the TATA box and the TAR region in HIV-1 LTR are indicated (see text). Although T3R is depicted as occupying only the NF-kB (I) and Sp1 (I) sites, it should be noted T3R is able to bind both of the NF-kB sites and all three Sp1 sites (9).
FIG. 2. Domain organization of the Tat protein of HIV-1 (A) and of cT3Ra (B).
on November 9, 2019 by guest
http://jvi.asm.org/
[image:2.612.82.270.568.709.2]C15 39LTR. pU3R-III-CAT(2104Sp1m/180) [p(2104Sp1m/180)-CAT] con-tains mutations in each of the three Sp1 sites. This plasmid was constructed by PCR with the pHIVNSpALLRSVCAT(2177/183) vector derived from the ARV-2 virus strain (22), in which all three wild-type Sp1 sites [(276)GAGGCG TGGCCTGGGCGGGACTGGGGAGTGGCGA(243)] are mutated [(276) GATATCTGGCCTGTCTAGATCCGTGCACTGGCGT(243)]. The 59primer contained an XhoI site followed by the distal NF-kB site sequence. The 39primer included a BglII site at121. The PCR products were cleaved with XhoI and BglII and cloned into p(2104/180)-CAT previously cleaved at2104 with XhoI and at 121 with BglII. The sequence of p(2104Spm/180)-CAT was confirmed by dideoxy sequencing. pDSV-TRE-IR-CAT (72), pDMTV-GH-TRE-CAT (71), pDMTV-DR14-CAT (45), and p59[kB(I1II)]-CAT (9) have been described previously. pDSV-CAT contains the simian virus 40 (SV40) early promoter extending to2135 and thus contains the 21-bp repeat sequences which bind Sp1. The TREs were cloned into a HindIII site at2135 and were obtained from Ron Evans (72). pDMTV-CAT, a mouse mammary tumor virus LTR-chloramphen-icol acetyltransferase (CAT) reporter which lacks the glucocorticoid response element (70, 71), was also obtained from Ron Evans. This vector is not respon-sive to T3R but becomes responrespon-sive when functional TREs are cloned upstream of the promoter at288. A TRE-IR sequence (AGGTCA TGACCT) was cloned upstream of pDMTV-CAT by using HindIII cohesive ends, and the resulting construct has been termed pDMTV-TRE-IR-CAT (14). pTRE1HIV(280)-CAT
contains the TRE-IR sequence inserted into a polylinker approximately 30 bp 59 of280 in the HIV-1 LTR. It contains all three HIV-1 Sp1 sites (276 to245) but lacks the HIV-1 NF-kB sites (13). p[DR14](282/180)-CAT contains a direct repeat (AGGTCA cagg AGGTCA), with a 4-bp gap, shown in lowercase letters [DR14], at282 of the LTR. It was constructed by PCR with primers containing the DR14 sequence and providing a 59XhoI site and a 39BglII site. The PCR
products were cleaved with XhoI and BglII and cloned into p(2167/180)-CAT previously cleaved at2167 with XhoI and at121 with BglII. p[kB I1II](245/ 180)-CAT was constructed by cloning an oligonucleotide containing both HIV-1 NF-kB sites into the XhoI site at245 of p(245/180)-CAT. p[DR14](245/ 180)-CAT was constructed in a similar manner with an oligonucleotide contain-ing the DR14 sequence.
The full-length cT3RacDNA, corresponding to amino acids 1 to 408, cloned into a pEXPRESS (pEX) vector (pEX-cT3Ra), has been described previously (16). pEX vectors contain the Rous sarcoma virus (RSV) LTR linked to a phage T7 RNA polymerase promoter, followed by a KpnI site, stop codons in all three reading frames, and an SV40 polyadenylation signal, allowing expression in both
Escherichia coli and mammalian cells and synthesis of in vitro transcripts from
purified T7 RNA polymerase. pRSV-T7-cT3Rawas made by inserting an oligo-nucleotide containing a T7 polymerase binding site with 59 and 39 HindIII
cohesive ends between the RSV LTR and the cT3RacDNA in pRSV-cT3Ra (17, 18). The oligonucleotide was designed so that the 59HindIII is inactivated
but the 39HindIII is regenerated upon insertion, allowing for future cloning at
this site. pRSV-T7-cT3Ra(13–408) was constructed by first removing the 90-bp
HindIII-PflMI fragment in the N-terminal A/B region of cT3Rafrom pRSV-T7-cT3Raand inserting an oligonucleotide which begins with an ATG codon within an NcoI site followed by the codon for amino acid 13. pRSV-T7-cT3Ra(21–408) was constructed in a similar manner, using an appropriate oligonucleotide which begins with the codon for amino acid 21. pEX-cT3R(51–408), lacking the entire A/B domain, was constructed from pEX-cT3Ra(51–157) and pEX-cT3Ra. DNA corresponding to amino acids 119 to 408 was excised from pEX-cT3Rawith MscI and Asp 718 and subcloned into pEX-cT3Ra(51–157) after digestion of the vector with MscI, which cleaves at codon 118, and Asp 718, which cleaves just after codon 157. pEX-cT3Ra(51–157) was constructed by PCR of wild-type cT3Rawith appropriate primers. pEX-cT3Ra(1–118) was formed by blunt-end ligation of pEX-cT3Ra that was cleaved with MscI and Asp 718 to remove codons 119 to 408. pEX-cT3Ra(120–408) has been described previously (62). pEX-cT3Ra(K232I) was made by site-directed mutagenesis with M13 phage (Amersham). pSV2tat72, expressing a 72-amino-acid Tat protein (21), was ob-tained from the AIDS Research and Reference Reagent Program (ARRRP, Rockville, Md.). pET8c-tat was constructed from pTat (obtained from ARRRP), which contains the Tat cDNA encoding amino acids 1 to 86. The NcoI-BamHI tat fragment of ptat was then cloned into the NcoI and BamHI sites of pET8c (Novagen). pEX-(D2–8)tat and pEX-(D2–20)tat, lacking amino acids 2 to 8 or 2 to 20 of the 86-amino-acid Tat coding region, respectively, were constructed by PCR of ptat. The PCR products were digested with NcoI and KpnI and cloned into pEX.
pGST-Tat86 expresses a fusion protein between glutathione-S-transferase (GST) and wild-type Tat (1 to 86) (30, 52). pGST-Tat72 and pGST-Tat48, which express C-terminal deletion mutants of Tat (1 to 86) (30, 52), were obtained from the ARRRP. pGEX2T, which expresses only GST (66), and pGST-TFIIB (53) have been described previously. pGST-cT3Ra(1–408) was constructed from cT3Rain pTZ18R (Pharmacia) that was digested with HindIII, which cleaves 39 of the cDNA. After partial digestion with NcoI, the NcoI-HindIII fragment corresponding to the full-length cT3RacDNA was blunt-end ligated into the
BamHI site of pGEX2T (66). pGST-cT3Ra(120–408) and pGST-cT3Ra(D120– 199) were constructed by partial digestion of pGST-cT3Ra(1–408) with NcoI, followed by religation. pGST-cT3Ra(1–51), pGST-cT3Ra(1–119), and pGST-cT3Ra(1–151) express amino acids 1 to 51, 1 to 119, and 1 to 151 of cT3Ra, respectively, as fusion proteins with GST. They were constructed by PCR of
cT3Rawith a 59primer that introduced a BamHI site directly followed by an
NcoI site and a 39primer which introduced a BglII site. The PCR products were digested with BamHI and BglII and cloned into the BamHI site of pGEX2T.
Cell transfections and CAT assays.HeLa cells were cultured and transfected by electroporation (9). After electroporation, cells were cultured in Dulbecco’s modified Eagle’s medium (Gibco) supplemented with 15 mM HEPES (N-2-hydroxyethylpiperazine-N9-2-ethanesulfonic acid) plus 0.1 mg of pyruvate, 50mg of streptomycin sulfate, and 50mg of penicillin per ml (DHAP medium) con-taining 10% (vol/vol) hormone-depleted calf serum (12, 59) with and without 1 mM T3 for 48 h, before harvesting for CAT assays. Cells treated with Ro 5-3335 (32) (Hoffmann-La Roche) were incubated for 24 h with the indicated amounts of this compound (Ro 5-3335 was dissolved in 100% dimethyl sulfoxide at a stock concentration of 5 mM). CAT expression was measured by determining the extent of [14
C]chloramphenicol (50 mCi/mmol; New England Nuclear) acetyla-tion as described previously (12, 26). The protein concentraacetyla-tion was determined (39), and amounts of cell protein assayed for CAT activity were varied to maintain the percent [14
C]chloramphenicol acetylated in the linear range (,30%) during a 16-h incubation. CAT activity values were usually normalized to represent the percent [14
C]chloramphenicol acetylated by a specific amount of cell protein in 16 h at 378C. All experiments were performed in duplicate, and the results represent the means of at least three independent transfections. Within each transfection, duplicate samples varied by less than 10%.
Purification of GST fusion proteins.GST and GST fusion proteins were expressed by standard techniques (66). Overnight bacterial cultures were diluted 1:100 with Luria broth and grown for 4 h with the appropriate antibiotics at 378C before being induced with 1 mM IPTG (isopropylthiogalactopyranoside) for 20 min. Cells were pelleted and resuspended in 10 ml of phosphate-buffered saline (PBS; 137 mM NaCl, 16 mM Na2HPO4, 4 mM NaH2PO4) containing 0.5 mM
EDTA, antipain (20mg/ml), pepstatin (100mg/ml), and 1 mM phenylmethylsul-fonyl fluoride (PMSF). Cells were lysed on ice by mild sonication and centrifuged at 12,0003g for 30 min at 48C. The supernatant was mixed for 20 min at 48C in a 50-ml polypropylene tube on a Orbitron rotator with 0.5 ml of 50% glutathi-one-agarose beads (Sigma). Fusion proteins coupled to the glutathiglutathi-one-agarose beads were washed three times in PBS and stored in the same buffer at 48C as a 50% (vol/vol) slurry. The concentration and size of GST and GST fusion proteins were determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) with a known quantity of molecular weight standards (data not shown).
Protein labeling and cT3Ra-Tat binding assays.L-[35S]cysteine-labeled Tat
and cT3Raproteins were prepared by using TNT reticulocyte lysates (Promega). SDS–15% urea–sodium phosphate–polyacrylamide gels, which can resolve low-molecular-weight proteins, were used in experiments involving35S-labeled Tat
proteins. Approximately 2.53104cpm of35S-labeled protein was incubated with
50 to 100 ng of GST or GST fusion protein immobilized on glutathione-agarose beads in 300ml of buffer A (50 mM KCl, 25 mM HEPES [pH 7.9], 6% glycerol, 5 mM EDTA, 5 mM MgCl2, 1 mM dithiothreitol [DTT], 0.05% Triton X-100) for
45 min at 48C on an Orbitron rotator. Beads were collected by centrifugation at 48C for 5 min at;5003g and washed three times with 1 ml of buffer A. The
bound proteins were eluted with SDS and analyzed by SDS gel electrophoresis followed by autoradiography. Where indicated, 1mM T3 was included in the binding reaction mix. In some studies, a 200-fold molar excess of the indicated unlabeled TRE relative to the amount of cT3Rawas included in the binding reaction mix.35
S-labeled wild-type or mutant cT3Raor Tat proteins were ana-lyzed by electrophoresis and autoradiography to ensure that equal amounts of input radioactivity of all of the labeled proteins were used in the GST binding assays.
Western (immunoblot) analysis.One microgram of purified cT3Ra(16) was incubated with either GST, GST-Tat86, or GST-TFIIB (50 to 100 ng) bound to glutathione-agarose beads. Bound protein was eluted with SDS loading buffer and electrophoresed in an SDS–10% polyacrylamide gel. Proteins were electro-blotted onto a nitrocellulose filter at 48C in Towbin’s buffer (25 mM Tris [pH 7.5], 192 mM glycine, 20% methanol) at 250 mA constant current. The filter was blocked for 8 h at 48C in TBS buffer (1 mM EDTA [pH 8.0], 20 mM Tris [pH 7.5], 100 mM NaCl) containing 5% nonfat milk with constant agitation. The filter was then incubated in the same buffer with rabbit anti-cT3Raantibody 28 (62) (1:1,000 dilution) overnight at 48C with constant agitation. After several washes with TBS buffer, the filter was incubated for 2 h at 258C in TBS buffer containing 5% nonfat milk and 0.1mCi of125I-protein A per ml. The filter was then washed
several times in TBS buffer and autoradiographed.
RESULTS
Ro 5-3335 modulates Tat-dependent cT3R
a
-mediated
stim-ulation of the HIV-1 LTR.
T3Rs regulate the HIV-1 LTR by
binding to and activating TREs within the viral NF-
k
B and Sp1
sites (9). Mutations within the NF-
k
B motifs, which eliminated
binding of cT3R
a
, also abolished stimulation by T3, indicating
that sequences in the Sp1 region do not independently mediate
activation by T3. The Sp1 region, however, was converted to a
functionally strong TRE by expression of Tat. This suggests
on November 9, 2019 by guest
http://jvi.asm.org/
that Tat might interact with and modify the activity of cT3R
a
bound in proximity to the TATA box (Fig. 1). Alternatively,
ligand-occupied cT3R
a
could enhance transcriptional
activa-tion by Tat. Either model suggests that the two proteins
inter-act functionally directly or indirectly in vivo. Further evidence
for this interaction comes from studies with Ro 5-3335
[7-chloro-5-(2-pyrryl)-3H-1,4-benzodiazepin-2(H)-one], which
inhibits transcriptional activation of HIV-1, HIV-2, and
azido-thymidine (AZT)-resistant clinical isolates by Tat (32). HeLa
cells were cotransfected with p(
2
76/
1
80)-CAT, containing the
three Sp1 binding sites, the TATA box, and the TAR element,
as well as vectors expressing cT3R
a
and Tat (Fig. 3). Cells
were then incubated for 24 h with the indicated amounts of Ro
5-3335 with and without 1
m
M T3. Incubation was for only 24
h since longer times (40 to 48 h) resulted in less activation at
3 and 10
m
M Ro 5-3335, possibly due to a mild toxic effect on
the cells. Because of the 24-h incubation time, the fold
stimu-lation by T3 was reduced by about 50% compared with that
after 40 to 48 h of incubation. As previously described, we
found that 1
m
M Ro 5-3335 completely blocked Tat
stimula-tion of the HIV-1 LTR (32) (data not shown). The funcstimula-tional
interaction between Tat and cT3R
a
was completely blocked by
1
m
M Ro 5-3335 (Fig. 3), supporting the idea that cT3R
a
and
Tat interact to facilitate the Tat-dependent activation of the
HIV-1 LTR by cT3R
a
. Studies on the activation of p
D
SV-TRE-IR-CAT and p
D
MTV-DR
1
4-CAT by cT3R
a
indicated
that the inhibitory effects of this compound on Tat were not
due to a toxic effect on the cells or to a nonspecific effect on the
HIV-1 LTR (Table 1, experiment 2).
In experiments without Tat, T3 stimulated p(
2
167/
1
21)-CAT expression sixfold, and Ro 5-3335 alone did not activate
the HIV-1 LTR above basal levels (Fig. 4). Surprisingly, the
combination of Ro 5-3335 and T3 resulted in up to 40-fold
stimulation compared with that of T3 alone (6-fold). Other
pro-moters containing TREs did not show this effect (Table 1,
exper-iment 2). To our knowledge, this is the first example of a
com-pound that enhances transcriptional activation by T3 in a
promoter-selective fashion. Other benzodiazepam derivatives
(chlordiazepoxide HCl, 4
9
-chlorodiazepam, flunitrazepam,
des-methyldiazepam, flurazepam 2HCl, diazepam, and PK-11195) did
not enhance the T3-dependent activation of the LTR (data not
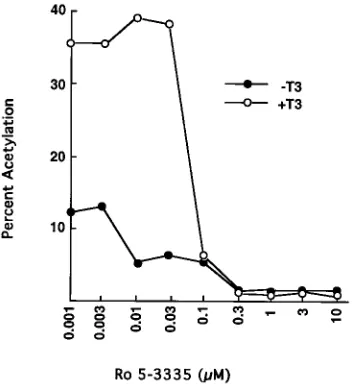
shown). Concentrations of Ro 5-3335 of between 0.3 and 10
m
M
were optimal (Fig. 5). The stimulation by T3 alone was sevenfold
(not illustrated). This was similar to the fold stimulation observed
when 0.001
m
M Ro 5-3335 was incubated with T3. Thus, the
concentrations of Ro 5-3335 which mediate synergistic
stimula-tion with T3 are similar to those that inhibit Tat-dependent T3
activation of the HIV-1 LTR, suggesting that the same cellular
factor may be involved in both of these events.
The region of the LTR important for synergistic stimulation
was determined by using two 5
9
deletion mutants of the HIV-1
[image:4.612.90.266.72.264.2]FIG. 3. Ro 5-3335, a Tat antagonist, inhibits the Tat-dependent activation of the HIV-1 LTR by cT3Ra. HeLa cells were cotransfected with p(276/180)-CAT (10mg) and expression vectors for cT3Ra(5mg) and Tat (1 ng), followed by incubation for 24 h with various concentrations of Ro 5-3335 with and without 1 mM T3. Procedures for transfection and assay of CAT activity are described in Materials and Methods. CAT assays were performed with 50mg of cell protein. Results represent the means of at least three independent transfections.
TABLE 1. Ro 5-3335- and T3-dependent synergistic regulation of the HIV-1 LTR is promoter specific and requires sequences within the HIV-1 Sp1 sitesa
Expt no. Transfection
CAT activityb Fold stimulation
Control (C) T3 Ro1T3 T3/C (T31Ro)/C
1 p(2104/180)-CAT 1.5 9 30 6.0 20.0
p(276/180)-CAT 0.7 0.4 0.3 0.5 0.4
2 p(2167/121)-CAT 2.9 16 62 5.5 21.3
p59[kB(I1II)]-CAT 0.3 4 2.9 13.3 9.6
pDSV-TRE-IR-CAT 4.8 73 76 15.2 15.8
pDMTV-DR14-CAT 4.3 84 56 19.5 13.0
3 p(245/180)-CAT 1.2 0.6 0.5 0.5 0.4
p[kB I1II](245/180)-CAT 0.5 3.6 4.7 7.2 9.4
p[DR14](245/180)-CAT 2.7 39 34 14.4 12.5
4 p[DR14](282/180)-CAT 128 280 892 2.1 6.9
pTRE1HIV(280)-CAT 7.9 20 51 2.5 6.4
5 p(2104/180)-CAT 22 80 212 3.7 9.6
p(2104Sp1m/180)-CAT 2.2 27 17 12.2 7.7
a
HeLa cells were cotransfected with the indicated CAT reporter (10mg) and cT3Raexpression vector (5mg). Cells were incubated for 24 h with 1mM T3 with or without 1mM Ro 5-3335 (Ro). Procedures are described in Materials and Methods. Results represent the means of at least three independent transfections.
b
Percent acetylated per 50mg of protein per 16 h.
on November 9, 2019 by guest
http://jvi.asm.org/
LTR (Fig. 1). As previously found, deletion of the two NF-
k
B
sites [p(
2
76/
1
80)-CAT] resulted in loss of T3 stimulation (9)
(Table 1, experiment 1). The same deletion resulted in loss of
the synergistic regulation, suggesting that the TREs within the
NF-
k
B sites (
2
104/
2
80) and not the Sp1 sites (
2
76/
2
45) were
important for the effect. p59[
k
B(I
1
II)]-CAT, containing the
HIV-1 NF-
k
B sites upstream of the rat angiotensinogen
min-imal promoter (9), was responsive to T3, as reported previously
(9), but Ro 5-3335 did not enhance the T3 stimulation (Table
1, experiment 2). Two other TRE-containing reporter genes,
p
D
SV-TRE-IR-CAT and p
D
MTV-DR
1
4-CAT, were also
un-responsive to Ro 5-3335 (Table 1, experiment 2).
Since the NF-
k
B sites out of context of the HIV-1 LTR did
not mediate the synergistic effect, we examined the role of
sequences within the Sp1 sites. The HIV-1 NF-
k
B (I
1
II) sites
or the DR
1
4 sequence was cloned at position
2
45 of p(
2
45/
1
80)-CAT, which lacks all three Sp1 sites (Table 1, experiment
3). Although p(
2
45/
1
80)-CAT was not T3 responsive, both
p[
k
B I
1
II](
2
45/
1
80)-CAT and p[DR
1
4](
2
45/
1
80)-CAT
showed significant stimulation by T3. We also examined
pTRE
1HIV(
2
80)-CAT and p[DR
1
4](
2
82/
1
80)-CAT, which
contain the TRE-IR or DR
1
4 sequences, respectively,
up-stream of the three HIV-1 Sp1 sites. The TRE-IR sequence in
pTRE
1HIV(
2
80)-CAT is approximately 30 bp 5
9
of
2
80 in the
LTR (13) and is not directly adjacent to the Sp1 sites (
2
76/
2
45). The DR
1
4 sequence in p[DR
1
4](
2
82/
1
80)-CAT is at
2
82 of the HIV-1 LTR. Both reporters exhibited synergistic
stimulation with T3 and Ro 5-3335 (Table 1, experiment 4). In
contrast, p
D
SV-TRE-IR-CAT contains several Sp1 sites in the
SV40 early promoter but did not show the synergistic effect
(Table 1, experiment 2). Thus, the synergistic effect appears to
be independent of the position or sequence of the TRE but is
dependent on sequences within the Sp1 sites of the HIV-1
LTR. Other evidence to support this notion comes from
stud-ies comparing the activity of p(
2
104Sp1m/
1
80)-CAT, which
contains mutations in each of three Sp1 sites, with the
wild-type CAT reporter (Table 1, experiment 5). The basal activity
of the p(
2
104Sp1m/
1
80)-CAT reporter was reduced in
accor-dance with previously described results (22). As a result of
the differences in basal expression, the fold stimulation of
the Sp1-mutated LTR-CAT reporter was greater than that of
the wild-type HIV-1 LTR-CAT reporter. In contrast to the
p(
2
104/
1
80)-CAT reporter, with which Ro 5-3335 enhanced
the extent of T3 stimulation, Ro 5-3335 did not further
en-hance the T3 effect with the mutant reporter. In fact, Ro
5-3335 caused a slight decrease in the extent of stimulation of
the mutant reporter. This study further supports the notion
that the sequences in the HIV-1 Sp1 sites are important in
mediating the synergistic effect.
Tat-independent and Tat-dependent T3-mediated activation
of the HIV-1 LTR by wild-type and mutant cT3R
a
.
Transacti-vation of the HIV-1 LTR by cT3R
a
and several mutants was
determined in the absence of Tat by using p(
2
167/
1
21)-CAT
and in the presence of Tat by using p(
2
76/
1
80)-CAT (Table
2). The N-terminal deletion mutants shown in Table 2 all bind
T3 and DNA elements as homodimers or as heterodimers with
an RXR similar to wild-type cT3R
a
(27). In the absence of Tat
(Table 2, experiment 1), cT3R
a
(13–408) and cT3R
a
(21–408)
activated the HIV-1 LTR reporter similarly to wild-type
cT3R
a
(1–408). cT3R
a
(51–408), which lacks the entire
50-ami-no-acid A/B region, did not mediate T3 stimulation. As
previ-ously reported, T3 inhibited the basal activity of p(
2
76/
1
80)-CAT in the presence of cT3R
a
by threefold without Tat (9)
(Table 2, experiment 2). Expression of Tat reversed this
inhi-bition to significant stimulation. Tat-dependent T3 stimulation
of the HIV-1 LTR was also found with cT3R
a
(13–408) and
cT3R
a
(21–408) but not cT3R
a
(51–408), indicating that amino
acids 21 to 50 are critical for activation of both Tat-dependent
(Sp1) and Tat-independent (NF-
k
B) responsive sequences of
the LTR. cT3R
a
(K232I), which contains a single Lys-to-Ile
change at amino acid 232 in the highly conserved
ti
domain
(Fig. 2B), poorly stimulated gene expression via the NF-
k
B
TREs in p(
2
167/
1
21)-CAT (Table 2, experiment 1). Previous
studies showed that cT3R
a
(K232I) does not mediate
T3-de-pendent activity from p
D
MTV-TRE-IR-CAT and p
D
MTV-TRE-GH-CAT but does bind T3 and DNA response elements
FIG. 4. Ro 5-3335 markedly enhances the T3-dependent activation of the HIV-1 LTR by cT3Rain the absence of Tat. HeLa cells were cotransfected with p(2167/121)-CAT (10mg) and an expression vector for cT3Ra(5mg). Cells received either 0.3 or 1mM Ro 5-3335 with or without 1mM T3 for 24 h. CAT assays were performed with 25mg of cell protein. Results represent the means of at least three independent transfections and are expressed as fold stimulation (Treatment/control).
FIG. 5. Dose-response study for Ro 5-3335 and T3-dependent synergistic regulation of the HIV-1 LTR. HeLa cells were cotransfected with p(2167/1 21)-CAT (10mg) and expression vector for cT3Ra(5mg), followed by incubation for 24 h with various concentrations of Ro 5-3335 with and without 1mM T3. The fold stimulation by T3 alone was sevenfold (not illustrated). This was similar to the fold stimulation observed when 0.001mM Ro 5-3335 was incubated with T3. CAT assays were performed with 30mg of cell protein. Results represent the means of at least three independent transfections. C, control.
on November 9, 2019 by guest
http://jvi.asm.org/
as a homodimer as well as a heterodimer with an RXR similar
to wild-type cT3R
a
(29). Surprisingly, cT3R
a
(K232I) behaved
like wild-type cT3R
a
(1–408) in mediating Tat-dependent T3
stimulation of the HIV-1 LTR via sequences in the Sp1 motif
in p(
2
76/
1
80)-CAT (Table 2, experiment 2).
Protein-protein interactions of Tat with cT3R
a
.
These and
earlier studies (9) suggest a functional interaction between
cT3R
a
and Tat. Binding assays with GST fusion proteins were
used to identify a physical interaction between these two
pro-teins (Fig. 6). GST protein alone bound poorly to
35S-labeled
control lysate, labeled Tat, and labeled cT3R
a
(lanes 1 to 3).
cT3R
a
(27), human T3R
a
1 (hT3R
a
1) (13), and hT3R
b
1 (1)
have been shown to interact with TFIIB, a basal transcription
factor critical for the formation of the preinitiation complex.
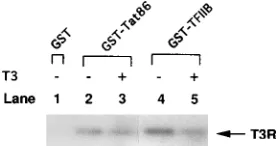
GST-TFIIB did not bind to any factors in the control lysate
(Fig. 6, lane 4), but did bind to cT3R
a
, as expected (lane 5).
Inclusion of T3 in the binding reaction mix reduced the
amount of cT3R
a
bound to GST-TFIIB (Fig. 6, lane 6) (27).
While control lysate did not bind to GST-cT3R
a
(Fig. 6, lane
7),
35S-labeled Tat bound to GST-cT3R
a
(lane 8), and T3 did
not affect this interaction (lane 9). Ro 5-3335 also did not affect
the interaction between GST-cT3R
a
and Tat (data not
shown).
The physical interaction between cT3R
a
and Tat was
con-firmed by Western immunoblot analysis with bacterially
ex-pressed and purified cT3R
a
and GST-Tat. This was done to
exclude the possibility that a reticulocyte factor present in the
lysate-expressed receptor was responsible for this interaction.
The bound proteins were separated by SDS-PAGE,
trans-ferred to nitrocellulose, and immunoblotted with rabbit
anti-cT3R
a
antibody 28 (62). Purified cT3R
a
did not bind GST
alone (Fig. 7, lane 1) but did bind GST-Tat86 (lanes 2 and 3)
and GST-TFIIB (lanes 4 and 5). T3 decreased the affinity of
cT3R
a
for GST-TFIIB (Fig. 7, lane 5) but not for GST-Tat86
(lane 3).
Domains of cT3R
a
essential for the Tat-cT3R
a
interaction.
A series of
35S-labeled cT3R
a
mutants and GST-Tat72 were
used to delineate the regions of cT3R
a
essential for the
inter-action with Tat (Fig. 8). GST-Tat has been shown to have the
same biological activity as wild-type Tat in transient
transfec-tion studies as well as in cell-free transcriptransfec-tion studies (49, 69).
Both wild-type cT3R
a
(1–408) and cT3R
a
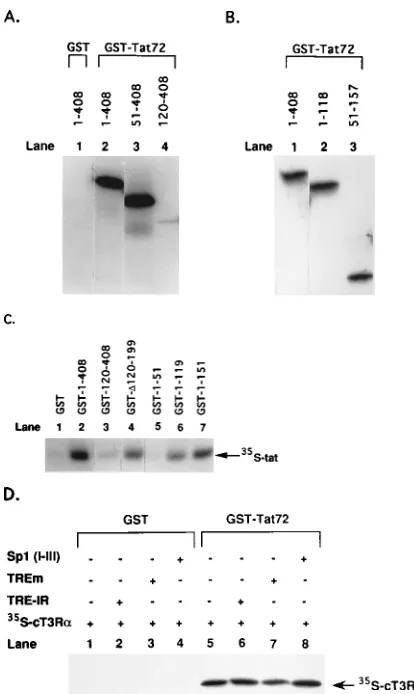
(51–408) bound with
similar affinity to GST-Tat72 (Fig. 8A, lanes 2 and 3).
How-ever, cT3R
a
(120–408), containing only the ligand-binding
do-main, bound poorly to GST-Tat72 (lane 4). These results
sug-gested that Tat interacts primarily with the A/B/C region of
cT3R
a
. We next studied cT3R
a
mutants containing only the
A/B/C domains [cT3R
a
(1–118)] or all of the C domain plus
part of the D domain [cT3R
a
(51–157)]. Both deletion mutants
of cT3R
a
bound GST-Tat72 as efficiently as the wild-type
receptor, suggesting that amino acids in the DNA-binding
do-main (51 to 118) are crucial for the Tat-cT3R
a
interaction
(Fig. 8B, lanes 2 and 3).
To confirm these findings, we studied the binding of
35[image:6.612.366.506.72.145.2]
S-labeled Tat to wild-type GST-cT3R
a
(1–408) and several
GST-cT3R
a
mutants. GST-cT3R
a
(120–408), containing only the
ligand-binding domain of cT3R
a
, did not bind to Tat (Fig.
8C, lane 3). However, an internal deletion mutant,
GST-cT3R
a
(
D
120–199), in which amino acids 120 to 199 of cT3R
a
were deleted, bound to Tat with an affinity similar to that
obtained with GST-cT3R
a
(1–119) (Fig. 8C, lane 6). Of the
FIG. 6. Specific interaction between immobilized cT3Ra and HIV-1 Tat. Wild-type cT3Rawas cloned into pGEX2T to express a GST-cT3Rafusion protein in E. coli. The fusion protein was purified by immobilization on gluta-thione-agarose beads. GST (negative control) and GST-TFIIB (positive control) were also expressed and purified. cDNAs for cT3Raand Tat(1–86) were tran-scribed and translated in vitro with T7 RNA polymerase in rabbit reticulocyte lysates in the presence ofL-[35
S]cysteine. Control lysate containing no plasmid DNA was also radiolabeled (35
[image:6.612.61.298.92.263.2]S-Cont. Lys.). The labeled proteins with or with-out 1mM T3 were incubated at 48C for 45 min with 100 ng of GST or GST fusion proteins immobilized on glutathione-agarose beads. After several washes, bound proteins were eluted and electrophoresed in a 15% urea–SDS–sodium phos-phate–polyacrylamide gel to resolve the Tat protein and autoradiographed. Pro-cedures for GST fusion protein purification and GST binding assays are de-scribed in Materials and Methods.
FIG. 7. Thyroid hormone decreases the affinity of purified cT3Rafor TFIIB but not for HIV-1 Tat. Bacterially expressed and purified cT3Rawas incubated with either GST, GST-TFIIB, or GST-Tat86 (50 to 100 ng) immobilized on glutathionine-agarose beads with or without 1mM T3. The bound proteins were separated by SDS-PAGE, transferred to nitrocellulose, and immunoblotted with rabbit anti-cT3Raantibody 28 (62). Detection of cT3Rawas achieved by125
[image:6.612.61.298.474.610.2]I-protein A reaction followed by autoradiography.
TABLE 2. Domains of cT3Racrucial for Tat-independent and Tat-dependent activation of the HIV-1 LTRa
Expt
no. Transfection
Cat activityb Fold
stimulation, T3/C Control (C) T3
1 p(2167/121)-CAT alone 20 21 1.0
11–408 22 126 5.7
113–408 15 100 6.6
121–408 29 176 6.0
151–408 16 21 1.3
1K232I 1.2 2.4 2.0
2 p(276/180)-CAT alone 0.9 0.7 0.7
1Tat 9.8 10 1.0
11–408 4.8 1.8 0.3
11–4081Tat 13 81 6.2
113–4081Tat 33 242 7.3
121–4081Tat 27 159 5.8
151–4081Tat 31 37 1.1
1K232I1Tat 20 103 5.1
aHeLa cells were transfected with the appropriate HIV-1 LTR-CAT vector
alone (10mg), or with expression vectors for Tat (1 ng) and/or wild-type or mutant cT3Ra(5mg), as indicated. Procedures for transfection and assay of CAT activity are described in Materials and Methods. Results represent the means of at least three independent transfections.
bPercent acetylated per 20mg of protein per 16 h.
on November 9, 2019 by guest
http://jvi.asm.org/
three GST-cT3R
a
N-terminal mutants studied, only
GST-cT3R
a
(1–51) did not bind to Tat (Fig. 8C, lane 5), confirming
that amino acids 51 to 118/119 of cT3R
a
are important for the
cT3R
a
-Tat interaction. Since we observed that Tat interacts
with the DNA-binding region of cT3R
a
, we examined whether
this interaction could occur under conditions in which cT3R
a
is completely bound to its DNA response element. Thus, we
carried out binding assays with GST-Tat72 and
35S-labeled
cT3R
a
with and without a 200-fold molar excess of unlabeled
synthetic TREs relative to the amount of cT3R
a
. The TREs
studied were the high-affinity TRE-IR, a mutant form of the
TRE-IR (TREm) which does not bind to cT3R
a
(14), and an
oligonucleotide containing the three Sp1 sites (I, II, III) of the
HIV-1 LTR, which binds to cT3R
a
and is involved in the
Tat-dependent T3-mediated activation of the HIV-1 LTR (9)
(Fig. 8D).
35S-labeled cT3R
a
did not bind to GST alone (Fig.
8D, lane 1) or to GST incubated with the various response
elements (lanes 2 to 4). The selective binding of
35S-labeled
cT3R
a
to GST-Tat72 was not inhibited by the inclusion of any
of the TREs in the binding reaction mix (Fig. 8D, lanes 6 to 8).
Domains of Tat important for the Tat-cT3R
a
interaction.
Several C-terminal deletion mutants of Tat in the form of GST
fusion proteins were used to determine the regions of Tat
necessary for binding to cT3R
a
(Fig. 9A).
35S-labeled cT3R
a
bound equally well to Tat86 (Fig. 9A, lane 2) and
GST-Tat72 (lane 3) but poorly to GST-Tat48 (lane 4). This suggests
that the arginine-rich basic region (amino acids 49 to 57) and
possibly amino acids 58 to 72 of Tat are important for the
interaction with cT3R
a
. N-terminal deletion mutants of Tat (1
to 86) which lack amino acids 2 to 8 (Fig. 9B, lane 3) or 2 to 20
(Fig. 9B, lane 4) bound cT3R
a
as efficiently as wild-type Tat.
DISCUSSION
[image:7.612.71.278.68.415.2]Tat is a strong positive regulator of HIV-1 replication and
gene expression. Several cellular factors that may interact with
Tat-TAR have been identified (i.e., Sp1 [33], Tat binding
pro-tein-1 [TBP-1] [44], MSS1 [65], TBP-7 [48], p68 [42, 43],
TRBP-1 [25], the TRP2 family [64], and TRP-185 [64]). A
number of these proteins, however, have not been well
char-acterized, and their role in the Tat response has not yet been
defined. We previously reported that the T3Rs, which are
widely expressed in various cells and tissues, can mediate
T3-dependent activation of the LTR alone via TREs within the
NF-
k
B sites and from sequences in the Sp1 sites when Tat is
expressed (9). In this study, we provide further functional as
well as physical evidence for this interaction. These and
pre-vious studies (9, 67) lend support to the notion that at least one
action of Tat is to function as a transcriptional activator by
interacting with other transcription factors (directly or
indi-rectly via other proteins) to promote transcriptional initiation.
Ro 5-3335 acts to inhibit the action of Tat and to enhance
the stimulation of the HIV-1 LTR by cT3R
a
.
The
benzodiaz-epine Ro 5-3335 inhibits Tat-dependent activation of the
HIV-1 LTR (32) and was used to study the functional
inter-action of cT3R
a
with Tat. At 1
m
M, Ro 5-3335 completely
inhibited Tat-dependent stimulation of the HIV-1 LTR by T3
and synergistically activated the HIV-1 LTR with T3 in the
absence of Tat. The direct target of Ro 5-3335 has been
sug-gested to be a cellular factor, and the compound may act either
by influencing the expression of that factor or by interfering
with the binding of the factor to Tat or to TAR RNA (75). Our
finding that similar concentrations of Ro 5-3335 (Fig. 3 and 5)
inhibit Tat activity and enhance T3-mediated stimulation of
the LTR in the absence of Tat implies that the same cellular
FIG. 8. C domain of cT3Raessential for Tat-cT3Rainteraction. (A) Binding of35
S-labeled wild-type cT3Ra(1–408), cT3Ra(51–408), and cT3Ra(120–408) to GST and GST-Tat72. (B) Binding of35
S-labeled cT3Ra(1–408), cT3Ra(1–118), and cT3Ra(51–157) to GST-Tat72. (C) Binding of 35
S-labeled Tat(1–86) to GST-cT3Ra(1–408) or GST-cT3Ra mutants (designated 120–408,D120–199, 1–51, 1–119, and 1–151). (D) Binding of35
S-labeled cT3Ra(1–408) to GST-Tat72 with and without a 200-fold molar excess of unlabeled DNA response elements relative to the amount to cT3Ra. The sequence of the TRE-IR is AGGTCA TGACCT, and the sequence of the mutated TRE-IR (TREm) is ACGTCA TGACGT. The sequence of the Sp1 (I, II, III) element, with the three Sp1 motifs underlined, is (280)CAGGGAGGCGTGGCCTGGGCGGGACT GGGGAGTGGC(245). In vitro-translated and35S-labeled proteins were
incu-bated with GST or GST fusion proteins immobilized on glutathione-agarose beads. After washing, the bound proteins were eluted, analyzed by SDS-PAGE, and visualized by autoradiography.
FIG. 9. Amino acid residues in the basic and C-terminal regions of Tat are required for binding to cT3Ra, while the N terminus is not required. (A) Binding of35
S-labeled cT3Rato GST-Tat86 and its C-terminal deletions GST-Tat72 and GST-Tat48. (B) Binding of35
S-labeled Tat(1–86) and the N-terminal Tat mu-tants (D2–8)tat and (D2–20)tat to GST-cT3Ra. wt, wild type.
on November 9, 2019 by guest
http://jvi.asm.org/
factor (Ro factor) may be involved in these responses. Ro
5-3335 and T3 activated p(
2
167/
1
21)-CAT synergistically,
in-dicating that the TAR sequence is not required for the activity
of the Ro factor. Inhibition of Tat activity by Ro 5-3335
ap-pears to be specific to HIV-1 or heterologous genes driven by
the HIV-1 LTR, since other retroviral promoters which are not
Tat responsive (i.e., RSV) are not affected by this compound
(32). The synergistic effect of Ro 5-3335 and T3 also appears to
be specific to the HIV-1 LTR and requires sequences within
the Sp1 motifs of the LTR (Table 1). The importance of the
HIV-1 Sp1 sites for Tat function has been well documented (3,
4, 67). Studies with a GAL4-Tat fusion protein indicate that
GAL4-Tat acts as a transactivator only with GAL4–HIV-1
LTR–CAT reporters containing all three HIV-1 Sp1 sites (67).
A model to explain both the Ro 5-3335-dependent inhibition
of Tat and the Ro 5-3335- and T3-dependent synergistic
reg-ulation of the LTR is described below. The HIV-1 LTR
con-tains recognition sequences for many known cellular
transcrip-tion factors, which may overlap those for other
promoter-specific factors, permitting a single sequence to be utilized by
more than one factor. For example, T3R can bind sequences
within the NF-
k
B and Sp1 motifs in the LTR (9). Our results
suggest that the Ro factor is bound to sequences within the Sp1
sites (Table 1, experiment 5). This factor, however, does not
appear to be Sp1, since the synergistic effect of T3 and Ro
5-3335 was not observed with the SV40 promoter (e.g.,
D
SV-TRE-IR-CAT; Table 1). We suggest that the Ro factor binds
to sequences within the Sp1 sites and interacts with the
tran-scriptional apparatus and Tat to facilitate the Tat-mediated
activation of the LTR. Ro 5-3335 acts to either degrade or
block the Ro factor, so that it is unable to interact with Tat
and/or the basal transcription apparatus, resulting in inhibition
of Tat activity. The Ro factor also limits the maximal activation
potential of T3R, possibly by competing for a limiting
tran-scription factor required for T3 stimulation. Inactivation of the
Ro factor by Ro 5-3335 permits maximal activation of the LTR
by T3. Thus, in cells expressing T3R, Tat, and the Ro factor,
Ro 5-3335 would inhibit Tat-dependent activation but not
T3-mediated activation by T3R via the NF-
k
B sites. Consistent
with this model is the observation that Ro 5-3335 does not
block the direct interaction of cT3R
a
with Tat (data not
shown).
Regions of cT3R
a
necessary for dependent and
Tat-independent activation of the HIV-1 LTR by thyroid hormone.
We studied the regions of cT3R
a
essential for transactivation
of the LTR in the absence and presence of Tat by using various
cT3R
a
mutants (Table 2). We identified a 30-amino-acid
se-quence in the A/B region of cT3R
a
(amino acids 21 to 50) that
is critical for T3-dependent activation of a variety of genes and
TREs, including the Tat-dependent and Tat-independent
se-quences of the HIV-1 LTR (Table 2). Mutations in this region
eliminate T3-dependent activity even though the protein binds
T3 and DNA similarly to the wild-type receptor (27). We have
also recently found that deletion of these 30 amino acids in the
A/B region markedly reduces the binding of cT3R
a
to TFIIB
and the extent of T3 stimulation of the rat growth hormone
and malic enzyme TREs (27). This suggests that cT3R
a
(51–
408) does not activate the HIV-1 LTR because it lacks amino
acids 21 to 50, which are required for the interaction of cT3R
a
with TFIIB. Interaction of cT3R
a
with TFIIB may recruit one
or more new additional factors to the complex, leading to
transcriptional activation. Tat was reported to not bind TFIIB
but to directly bind to TATA-binding protein (TBP), a
com-ponent of the multiprotein TFIID complex (35). This
interac-tion, which occurred through amino acids 36 to 50 of Tat, was
proposed to be involved in Tat action (35).
Our studies also suggest that the highly conserved
20-amino-acid
ti
region of cT3R
a
(Fig. 2B) plays a novel role in the
activation of the HIV-1 LTR in the presence of Tat. The
ti
region has been suggested to be involved in protein-protein
interactions of the receptor (37, 46, 47). A single Lys-to-Ile
change at amino acid 232 [cT3R
a
(K232I)] results in a protein
that binds ligand and DNA (29) and TFIIB (27) but poorly
activates transcription from a variety of synthetic and native
TREs (the optimized TRE-IR and DR
1
4 and the native rat
growth hormone and malic enzyme TREs) when linked to a
variety of heterologous promoters. We also found that
cT3R
a
(K232I) did not stimulate the HIV-1 LTR via TREs in
the NF-
k
B sites (Table 2). However, cT3R
a
(K232I) showed
the same level of Tat-dependent T3-mediated stimulation of
the HIV-1 LTR as wild-type cT3R
a
. Thus, cT3R
a
may also
mediate transcriptional activation via Tat by using activation
domains distinct from that utilized with other factors.
C-terminal basic region of Tat interacts with cT3R
a
.
Dele-tion mutant analysis indicates that amino acids 49 to 72, which
include the basic and C-terminal regions of Tat, are required
for binding to cT3R
a
, while the N terminus (amino acids 2 to
20) and cysteine-rich region appear to be dispensable (Fig. 9).
In contrast, the N terminus and the cysteine-rich region of Tat
are required for transactivation (19, 20, 23, 36, 51, 57, 58) and
have been proposed to interact with cellular factors (51). The
basic region involved in cT3R
a
binding also appears to be
involved in nuclear localization of Tat (57, 68) and in
mediat-ing specific bindmediat-ing of Tat to TAR RNA (74). Other studies
have shown that amino acids 30 to 62 of Tat are primarily
responsible for Sp1 binding (33), while amino acids 36 to 50 are
required for interaction of Tat with TBP (35). Furthermore,
since only amino acids 49 to 57 of Tat are necessary for TAR
RNA binding (5) and nuclear localization (36, 57, 68),
se-quences flanking this region may promote proteprotein
in-teractions. N-terminal Tat mutants (
D
2–6,
D
3–19,
D
2–35, and
D
9–18) have been reported to be functionally inactive (36), and
the mutants used in this study (
D
2–8 and
D
2–20) also did not
activate transcription from the HIV-1 LTR and did not
en-hance T3 stimulation (data not shown). Deletion of amino
acids 1 to 48 or 1 to 58 was not studied, since removal of these
sequences results in an inactive Tat protein (61).
Model to explain the regulation of the HIV-1 LTR by cT3R
a
,
Tat, and Ro 5-3335.
The finding that Tat and cT3R
a
interact
via the receptor DNA-binding domain suggests that this region
of the receptor may also play a role in protein-protein
inter-actions. This is also supported by the finding that c-jun
func-tionally interacts with T3R to yield an activity greater than that
of either factor alone (63), and this occurs through direct
protein-protein interactions via the DNA-binding domain of
T3R (76). Although Tat and cT3R
a
interact directly in vitro in
the absence or presence of a DNA response element,
Tat-mediated T3-dependent transcriptional activation may require
additional auxiliary proteins which interact with the C terminus
of the receptor. Our results suggest the following model for the
interaction of cT3R
a
and Tat in regulating the HIV-1 LTR.
DNA-associated T3R interacts with Tat bound to nascent
tran-scripts of TAR RNA. This interaction may be direct, as shown
in this study, or via another protein, such as the putative Ro
factor. The finding that Ro 5-3335 can influence both Tat
activity and T3 stimulation suggests that the Ro factor may
bridge or stabilize interactions of T3R and Tat. Additional
receptor domains may link the receptor and Tat to other
tran-scription factors. Thus, the N terminus of cT3R
a
, which binds
to TFIIB (27), anchors the receptor and Tat and any associated
proteins to the transcriptional apparatus. The ligand-binding
domain of T3R, which contains a T3-dependent
on November 9, 2019 by guest
http://jvi.asm.org/
tion domain (2, 6, 50), presumably acts to recruit or interact
with other proteins involved in transcriptional activation. The
finding that the cT3R
a
(K232I) mutant mediates
Tat-depen-dent but not Tat-indepenTat-depen-dent T3 stimulation of the HIV-1
LTR suggests that different factors interact with different
gions of the ligand-binding domain to mediate the two
re-sponses. These findings further support a role for Tat as a
transcriptional activator of HIV-1 genes and provide a working
model to explain the mechanism by which Tat interacts with
promoter-bound transcription factors to regulate the HIV-1
LTR. T3Rs are expressed in a wide variety of tissues, including
T and B lymphocytes (9), monocytes-macrophages (9), and
other target tissues for HIV-1 (9), suggesting that T3 may
regulate HIV-1 in these cells or in vivo. Studies are under way
to determine whether T3R plays a role in the life cycle of
HIV-1 or whether it influences expression of the integrated
provirus.
ACKNOWLEDGMENTS
We thank the following investigators for providing us with reagents: Joseph Fondell and Robert Roeder [pTRE1HIV(280)-CAT], Colleen
Nelson [pDMTV-DR14-CAT], Micheal Green [pGST-TFIIB], Rich-ard Gaynor [pHIVNSpALLRSVCAT(2177/183)], and Ming-Chu Hsu [Ro 5-3335, Hoffmann-La Roche]. We also thank Bruce M. Raaka and John Casanova for their assistance in the development of the manuscript and Jian-Shen Qi for encouragement and many useful discussions.
V.D.-Y. is an Aaron Diamond Foundation Fellow, and this work was supported in part by a grant (HRI817-5332F) from the Aaron Dia-mond Foundation. This work was also supported by research grants from the NIH (DK16636) (to H.H.S.) and an NIH short term training grant for students in health professional schools (5T35DK07421) (to P.M. and S.M.). E.H. is a graduate student in the Cell and Molecular Biology Program at the New York University Medical Center (DK16636). G.G. participated in these studies during a research rota-tion as a fourth-year medical student at the New York University School of Medicine.
REFERENCES
1. Baniahmad, A., I. Ha, D. Reinberg, S. Tsai, M.-J. Tsai, and B. O’Malley. 1993. Interactions of human thyroid hormone receptorbwith the transcrip-tion factor TFIIB may mediate target gene derepression and activatranscrip-tion by thyroid hormone. Proc. Natl. Acad. Sci. USA 90:8832–8836.
2. Baniahmad, A., S. Y. Tsai, B. W. O’Malley, and M.-J. Tsai. 1992. Kindred S thyroid hormone receptor is an active and constitutive silencer and a repres-sor for thyroid hormone and retinoic acid responses. Proc. Natl. Acad. Sci. USA 89:10633–10637.
3. Berkhout, B., A. Gatignol, A. B. Rabson, and K.-T. Jeang. 1990. TAR-independent activation of the HIV-1 LTR: evidence that tat requires specific regions of the promoter. Cell 62:757–767.
4. Berkhout, B., and K.-T. Jeang. 1992. Functional roles for the TATA pro-moter and enhancers in basal and tat-induced expression of the human immunodeficiency virus type 1 long terminal repeat. J. Virol. 66:139–149. 5. Calnan, B. J., S. Biancalana, D. Hudson, and A. D. Frankel. 1991. Analysis
of arginine-rich peptides form the HIV tat protein reveals unusual features of RNA-protein recognition. Genes Dev. 5:201–210.
6. Casanova, J., E. Helmer, S. Selmi-Ruby, J.-S. Qi, M. Au-Fliegner, V.
Desai-Yajnik, N. Koudinova, F. Yarm, B. M. Raaka, and H. H. Samuels.1994. Functional evidence for ligand-dependent dissociation of thyroid hormone and retinoic acid receptors from an inhibitory cellular factor. Mol. Cell. Biol.
14:5756–5765.
7. Dayton, A. I., J. G. Sodroski, C. A. Rosen, W. C. Goh, and W. A. Haseltine. 1986. The trans-activator gene of the human T-cell lymphotrophic virus type III is required for replication. Cell 44:941–947.
8. DeFranco, D., and K. R. Yamamoto. 1986. Two different factors act sepa-rately or together to specify functionally distinct activities at a single tran-scriptional enhancer. Mol. Cell. Biol. 6:993–1001.
9. Desai-Yajnik, V., and H. H. Samuels. 1993. The NF-kB and Sp1 DNA motifs of the human immunodeficiency virus type 1 long terminal repeat function as novel thyroid hormone response elements. Mol. Cell. Biol. 13:5057–5069. 10. Feng, S., and E. C. Holland. 1988. HIV-1 trans-activation requires the loop
sequence within tat. Nature (London) 334:165–167.
11. Fisher, A. G., M. B. Feinberg, S. F. Josephs, M. E. Harper, L. M. Marselle,
G. Reyes, M. A. Gonda, A. Aldovini, C. Debouk, R. C. Gallo, and F.
Wang-Staal.1986. The transactivator gene of HTLV-III is essential for virus rep-lication. Nature (London) 320:367–371.
12. Flug, F., R. P. Copp, J. Casanova, Z. D. Horowitz, L. Janocko, M. Plotnick,
and H. Samuels.1987. Cis-acting elements of the rat growth hormone gene which mediate basal and regulated expression by thyroid hormone. J. Biol. Chem. 262:6373–6382.
13. Fondell, J. D., A. L. Roy, and R. G. Roeder. 1993. Unliganded thyroid hormone receptor inhibits formation of a functional preinitiation complex: implications for active repression. Genes Dev. 7:1400–1410.
14. Forman, B. M., J. Casanova, B. M. Raaka, J. Ghysdael, and H. H. Samuels. 1992. Half-site spacing and orientation determines whether thyroid hormone and retinoic acid receptors and related factors bind to DNA response ele-ments as monomers, homodimers, or heterodimers. Mol. Endocrinol. 6:429– 442.
15. Forman, B. M., and H. H. Samuels. 1990. Dimerization among nuclear hormone receptors. New Biol. 2:587–594.
16. Forman, B. M., and H. H. Samuels. 1991. pEXPRESS: a family of expression vectors containing a single transcription unit active in prokaryotes, eu-karyotes and in vitro. Gene 105:9–15.
17. Forman, B. M., C.-R. Yang, M. Au, J. Casanova, J. Ghysdael, and H. H.
Samuels.1989. A domain containing leucine zipper like motifs mediate novel in vivo interactions between the thyroid hormone and retinoic acid receptors. Mol. Endocrinol. 3:1610–1626.
18. Forman, B. M., C.-R. Yang, F. Stanley, J. Casanova, and H. H. Samuels. 1988. c-erbA protooncogenes mediate thyroid hormone-dependent and in-dependent regulation of the rat growth hormone and prolactin genes. Mol. Endocrinol. 2:902–911.
19. Frankel, A. D., S. Biancalana, and D. Hudson. 1989. Activity of synthetic peptides from the tat protein of human immunodeficiency virus type I. Proc. Natl. Acad. Sci. USA 86:7397–7401.
20. Frankel, A. D., D. S. Bredt, and C. A. Pabo. 1988. tat protein from human immunodeficiency virus forms a metal-linked dimer. Science 240:70–73. 21. Frankel, A. D., and C. O. Pabo. 1988. Cellular uptake of the tat protein from
human immunodeficiency virus. Cell 55:1189–1193.
22. Garcia, J. A., K. W. Foon, R. Mitsuyasu, and R. B. Gaynor. 1987. Interac-tions of cellular proteins involved in the transcriptional regulation of the human immunodeficiency virus. EMBO J. 6:3761–3770.
23. Garcia, J. A., D. Harrich, L. Pearson, R. Mitsuyasu, and R. B. Gaynor. 1988. Functional domains required for tat-induced transcriptional activation of the HIV-1 long terminal repeat. EMBO J. 7:3143–3147.
24. Garcia, J. A., D. Harrich, E. Soutanakis, F. Wu, R. Mitsuyasu, and R. B.
Gaynor.1989. Human immunodeficiency virus type 1: LTR TATA and TAR region sequences required for transcriptional regulation. EMBO J. 8:765– 778.
25. Gatignol, A., A. Kumar, A. Rabson, and K. T. Jeang. 1989. Identification of cellular proteins that bind to the human immunodeficiency virus type I trans-activation responsive TAR element RNA. Proc. Natl. Acad. Sci. USA
86:7828–7832.
26. Gorman, C. M., L. F. Moffat, and B. H. Howard. 1982. Recombinant ge-nomes which express chloramphenicol acetyltransferase in mammalian cells. Mol. Cell. Biol. 2:1044–1051.
27. Hadzic, E., V. Desai-Yajnik, E. Helmer, S. Guo, S. Wu, N. Koudinova, J.
Casanova, B. M. Raaka, and H. H. Samuels.A 10-amino-acid sequence in the N-terminal A/B domain of chicken thyroid hormone receptorais es-sential for transcriptional activation and interaction with the general tran-scription factor TFIIB. Mol. Cell. Biol., in press.
28. Hauber, J., and B. Cullen. 1988. Mutational analysis of the trans-activation-responsive region of the human immunodeficiency virus type 1 long terminal repeat. J. Virol. 62:673–679.
29. Helmer, E. B., B. M. Raaka, and H. H. Samuels. Hormone-dependent and -independent transcriptional activation by thyroid hormone receptors is me-diated by different mechanisms. Submitted for publication.
30. Herrmann, C. H., and A. P. Rice. 1993. Specific interactions of the human immunodeficiency virus tat proteins with a cellular protein kinase. Virology
197:601–608.
31. Horowitz, Z. D., C.-R. Yang, B. M. Forman, J. Casanova, and H. H. Samuels. 1989. Characterization of the domain structure of chick c-erbA by deletion mutation: in vitro translation and cell transfection studies. Mol. Endocrinol.
3:148–156.
32. Hsu, M.-C., A. D. Schutt, M. Holly, L. W. Slice, M. I. Sherman, D. D.
Richman, M. J. Potash, and D. J. Volsky.1991. Inhibition of HIV replication in acute and chronic infections in vitro by a tat antagonist. Science 254:1799– 1802.
33. Jeang, K.-T., R. Chun, N. H. Lin, A. Gatignol, C. G. Glabe, and H. Fan. 1993. In vitro and in vivo binding of human immunodeficiency virus type 1 tat protein and Sp1 transcription factor. J. Virol. 67:6224–6233.
34. Kao, S.-Y., A. F. Calman, P. A. Luciw, and B. M. Peterlin. 1987. Anti-termination of transcription within the long terminal repeat of HIV-1 by tat gene product. Nature (London) 330:489–493.
35. Kashanchi, F., G. Piras, M. F. Radonovich, J. F. Duvall, A. Fattaey, C.-M.
Chiang, R. G. Roeder, and J. N. Brady.1994. Direct interaction of human TFIID with the HIV-1 transactivator tat. Nature (London) 367:295–299.