0022-538X/88/030673-07$02.00/0
Copyright C)1988, AmericanSociety forMicrobiology
Mutational Analysis
of the
trans-Activation-Responsive Region
of
the
Human
Immunodeficiency Virus
Type
I
Long
Terminal
Repeat
JOACHIM HAUBERtAND BRYAN R. CULLENt*
Departmentof MolecularGenetics, RocheResearch Center,
Hoffmann-La
Roche Inc., Nutley, NewJersey 07110Received30September 1987/Accepted4December1987
We usedsite-directed mutagenesis to delineate sequences within the humanimmunodeficiency virustypeI
(HIV-I) long terminal repeat (LTR) required for trans-activation by the viral tat gene product. We demonstratedthat sequences 3' to LTR position +44 are
dispensable
fortrans-activation but that almostallofthe mutations tested located between positions -17 and +44 greatly reduced trans-activation at both the
transcriptional and posttranscriptional levels. However, displacement of the HIV-1 LTR trans-activation-responsiveregion (TAR)3' by insertion ofupto 32 basepairsbetween the LTR TATA box and capsite had
little effect on trans-activation. An analysis of the DNase I hypersensitivity profile of the HIV-I LTR in
transfectedcultures suggested the presence of at least two DNase
I-hypersensitive
sites, includingone whichextends into the viralTARelement; however, neither of these sitesappeared to besignificantlyaffectedbytat
coexpression. These results allow moreprecise
delineation
of thesequences important for TAR function andsuggest that the TAR may be recognizedbyahost-specific DNA-binding protein rather than by the tat protein
directly.
The pathogenic human retrovirus human immunodefi-ciency virus type I (HIV-I) encodes not only the three structural genes (gag, pol, and env) common to all known replication-competent retroviruses but also at least four other, apparently nonstructural, gene products (4). Two of these, termedartltrs andtat,havebeenshownto actintrans to increasethe level of synthesis ofHIV-I-specific proteins (1, 5, 6, 8,16, 22,25, 30, 35, 39).Inthecaseoftat,evidence hasbeen presented forabimodalmechanism of action which involves both anincrease in the steady-state level of HIV-I-specificmRNAandanincrease inthe translational utiliza-tion of that mRNA (5, 39).
More
recently, it has been demonstrated that tat increases HIV-I mRNA levels by enhancingthe rateoftranscription rather thanby stabilizing HIV-I-specific mRNAs (12).This
result is consistent with the predominantly nuclear subcellular localization of tat protein(12). Themeansbywhich thesecond, posttranscrip-tional action of tat on HIV-I gene expression is effected remains unknown. However, evidence has been presented suggesting that HIV-I mRNAs are poorlyutilized
by the cellular translationalmachinery
in theabsence oftatcoex-pression (5, 8).
Inthis study, weattempted to address the mechanismof action of tat via mutational analysis of the HIV-I long terminalrepeat (LTR) sequence originally identified as the core trans-activation-responsive (TAR) region (31). This
sequence, whichextendsfrom -17to +80within theHIV-I
LTR, exhibits a number of striking inverted and direct
repeats (Fig. 1), and it has indeed been suggested that the
LTR sequence from + 1 to +59 can form a stable mRNA
stem-loop
structure important for the posttranscriptionalcomponent ofHIV-Itrans-activation (22, 24).Alternatively,
directrepeats arefrequentlyobserved within transcriptional regulatory sequences such as enhancers or promoters (19),
*Correspondingauthor.
tPresent address: Howard Hughes Medical Institute,
Depart-ment of Medicine, Duke University Medical Center, Box 3025, Durham, NC 27710.
and therefore might also be involved in the transcriptional
action of tat. By using constructions in which the HIV-I
LTR was fused to areporter gene, the human interleukin-2
(IL-2) gene (5), we measured the effects of differenttargeted
LTR mutations on both the basal and tat trans-activated
levels of IL-2 protein production in transfected cells. For
severalmutations, we also quantitated the steady-state level
of the reporter gene mRNA and deduced the relative
trans-lational efficiencies of the mRNAs encoded by different
mutants. These results allow moreprecisedetermination of
the location of TAR and more accurate assessment of the roles of particular repeat sequences. Finally, we
demon-strated the existence of at least two sites of DNase I
hypersensitivity within the HIV-I LTR. The location of
these sites, including one site which extends into the TAR
region, was, however, not markedly affected by
coexpres-sion of the tatgene product.
MATERIALS AND METHODS
Construction of molecular clones. All molecular clones
usedin this work werederived from the vectorpXF3/ori(5),
which contains the simian virus 40 origin of replication
insertedinto the pBR322derivative pXF3. The
cytomegalo-virusimmediate-earlypromoter-based IL-2expression
vec-tor pBC12/CMV/IL-2 and the tat expression vector pBC12/CMV/t2have been previously described, as has the
internal control plasmid pBC12AI (5). All HIV-I LTR
mu-tants were derived from pBC12/HIV/IL-2, which contains
the human IL-2 gene undercontrol of a 728-base-pair (bp)
HIV-I DNA fragment (5). This HIV-I sequence is derived
from the replication-competent HXB-3 strain ofHIV-I (27)
and includes theentire LTR U3 region, as well as 84 bp of
the LTR R region (Fig. 1). pBC12/HIV-Xho was derived
frompBC12/HIV-IL-2byoligonucleotide-directed
mutagen-esis by using the procedure described by Morinaga et al.
(21). The oligonucleotide used, an 18-mer, introduced a contiguous 2-bp mutation at positions +10 and +11 (TG-*GA) and created a unique XhoI site (Fig. 1). pBC12/HIV/IL-2 and pBC12/HIV-Xho were then used to
673
on November 10, 2019 by guest
http://jvi.asm.org/
A
-40 C T CACAT C C TCCAT A T A A6CACC TGCTTTTTC C C T C TAC
TATA Pvu11 U3-J
+I 666TCTCTCTB6TTAGACCAGATCTGAGCCTGBGAUCTCT .40
AF agi11 Sac
Xho
+41 CT 66 C TA ACT A 6 6 AA C C CAC TCCT TA A6C C T CAATAA AGC0 4
31B
+81 CT TC9ta a9t
HindIII
B
Ic
+1 IC%I%
/-cap-G G U U C U C UC CU UACC CA6ACA6C C
. . I I I |II
CCCAABBBAUCAAUCBBUCUCUCB C
+59 4
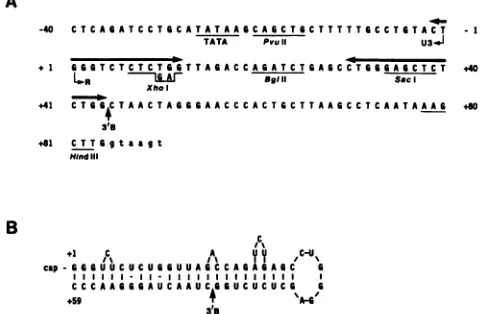
FIG. 1. Sequence of the HIV-I LTR TAR region included in pBC12/HIV/IL-2. (A)Apartial sequenceof the LTRof the HXB3
strain of HIV-Iused in this report is shown(capital letters). This sequenceisidenticaltothe HXB3sequencepreviously published by
Ratneretal. (28),except atposition +52, wherewe observedaG
rather thananA. AG has been observed in this positionin some
other HIV-I isolates (32, 38). Also shown are the locations of
relevant restriction sites, including the XhoI site introduced by oligonucleotide-directed mutagenesis at position +9. The TATA
box and the 3' border of thecoreTARregion mappedinthis work
(3'B)areindicated. The location ofanimperfectdirectrepeatwhich isdiscussed in thetextis also shown(<-*). (B)Apossible secondary
structureinvolvingthe first59ntofthe HIV-I mRNAis shown. This
structurehasanestimated freeenergyof -37 kcal/mol(22, 24)and hasbeen shown to occurin a synthetic SP6 transcript byinvitro enzymatic analysis (22). The residual stem present in the
pD+45/+77mutant, which is deletedto thelocation indicated by 3'B, has a free energy of -17 kcal/mol as determined by the
procedureof Tinoco et al.(36).
generate series of internalHIV-I LTR deletion(pD)mutants
by cleavageatvarious restrictionenzymesites,followedby
bluntendingwith Klenow DNApolymeraseI andreligation.
Mutants generated by this procedure include pD-18/+7
(PvuII to XhoI), pD-18/+20 (PvuII to BglII), pD+9/+20
(XhoItoBglII),pD+9/+38(XhoItoSacl), pD+9/+77(XhoI
to HindIII), pD+25/+38 (BglII to
Sacl),
and pD+35/+38(Sacl
sitedeletion). Ineachcase,the numbersin theplasmiddesignationdescribe theinclusiveextentof the deletion(Fig.
1).Asecondsetof HIV-I LTRdeletionmutantswasderived
by processive BAL 31 digestion ofpBC12/HIV/IL-2 after
cleavagewithHindlll.After BAL31digestion (International
Biotechnologies, Inc.;slowform),the DNAwasbluntended
with Klenow DNA polymerase I and ligated to a Hindlll
linker(5'-GAAGCTTC-3').TheDNAwasthencleaved with HindIIIand with MstII, which cuts at a unique site in the HIV-I DNA at position -526. The resultant fragments of <608bpwereisolated fromapreparative2%agarosegeland recloned into the larger parental pBC12/HIV/IL-2 vector HindIII-MstII fragment. The resultant clones were
charac-terized
as to deletion size and tested for trans-activationphenotype. Onlythetwo smallest deletions withanegative
phenotype (pD+42/+77andpD+44/+77)and the twolargest
deletions with a positive phenotype (pD+45/+77 and
pD+46/+77) are described. The final four LTR mutants,
termed
pl
mutants, were derivedby insertion of nucleotidesequences at differentlocations within the LTR. p14/+7(a
4-nucleotide [nt] insertion at position +7) was derived by
filling in the XhoI site in pBC12/HIV-Xho. Similarly,
p14/+20 was derived by filling in the BglII site in
pBC12/HIV/LTR. pI8/-18 and p132/-18 were derived from
pBC12/HIV/IL-2 viaatwo-step process.Initially, the unique
HIV-I LTR HindlIl site was filled in to generate p14/+78.
Thisclone, which hasanNheI siteatposition +82 in place
of the HindlIl site, exhibits the same trans-activation
re-sponse as the wild-type clone pBC12/HIV/IL-2 (data not
shown). Subsequently,one orfour of the 8-nt HindlIl linkers
wereintroduced into the unique LTR PvuII site atposition
-18 toyield, respectively,pI8/-18 and pI32/-18. All of the
mutants weresequenced by the dideoxynucleotide sequenc-ing procedure (33) after being subcloned into the vector M13mp9 (20).
Cell culture and DNA transfection. COS cells were
main-tainedaspreviously described (5) and were transfected with
2.5
Rg
ofplasmidDNA per 100-mm(diameter) culture withDEAE-dextran and chloroquine (5). Cell culture medium
and transfected-cell RNA were harvested at 72 h after
transfection. Secreted IL-2 levels were measuredby using
theIL-2-dependent murineT-celllineCTLL(5, 29).
RNAanalysis. TotalRNA waspreparedfrom transfected
cultures(5), and RNAsamples,asindicated in the text,were
then usedtoquantitatethe levelofRNA expressionby using
S1nucleaseprotection analysis (5).The S1probesused have
been previously described (5). The 750-nt 3'-end-labeled probe used (see Fig. 2) rescues a 175-nt fragment derived
fromthe RNAencoded by the IL-2expression vectors, as
wellas a156-ntfragment derived fromthe RNAencoded by
the internal referencevectorpBC12AI. Wehave previously
shown that the level ofRNAencodedbyRoussarcomavirus LTR-based pBC12AI is unaffected by tatcoexpression (5).
Theprobes used (see Fig. 3)wereuniquely5'end labeled eitheratposition +82 atthe wild-type HIV-I LTRHindlIl site or at +84attheintroducedNheI site ofpI32/-18. The probesextendtothe HIV-I LTRU3region Aval site (-159 inpBC12/HIV/IL-2). Transcripts initiating withinthe HIV-I LTRrescue aprobe fragmentwhoselengthisdeterminedby thedistance of the transcript initiation sitefromthe site of the label(5).The RNAderived from HIV-I-infectedH9cells (26; see Fig. 3)was agift ofM.Dukovich andW. Greene.
DNaseIhypersensitivity analysis. Nuclei from transfected
cultureswerepreparedat72h
posttransfection
before diges-tion with various amounts of DNase I (Worthington Diag-nostics; DPFF) (9, 10).TreatmentofthenuclearDNAwith therestrictionenzymeBamHI, agarosegel electrophoresis, Southernblotting,
and filterhybridization
with thenick-translatedprobe werecarriedout asdescribedby Frittonet
al. (9, 10). A 261-bp XbaI-to-BamHI human IL-2 gene fragment derived from
pBC12/HIV/IL2
wasusedfor prepa-ration ofthe probeusedforhybridization.RESULTS
Effect ofTARmutationson reporter geneprotein synthesis.
Theexpression vector
pBC12/HIV/IL-2
containsareportergene,the human IL-2 gene, under controlofa728-bpHIV-I
DNAfragmentwhich includes the entire456-bpHIV-I LTR U3
region,
aswellas84bpof the LTR Rregion
(5;Fig.
1).Thecore TAR sequence has previously been shown to be
contained within an HIV-I LTR sequence
extending
from approximately -17 to +80(31; where the site oftranscrip-tion initiatranscrip-tion is +1). Ourinitialapproachtothe determina-tion ofthefunctional
significance
of differentregions
ofTAR wasprocessive5'deletion fromtheHIV-I LTRHindlllsiteat +80, with BAL 31 exonuclease, to determine the siteof
the 3' border of the TAR sequence. It has previouslybeen
shown that deletionofsequences 3' to
position
+37 resultson November 10, 2019 by guest
http://jvi.asm.org/
[image:2.612.62.307.77.234.2]in loss of tat responsiveness by the HIV-I LTR (5, 39).
Numerousdeletionmutants werecharacterized, and thetwo
smallest deletions that lacked a significant tat response
(pD+42/+77 andpD+44/+77) and the twolargest deletions
which demonstrated tat responsiveness (pD+45/+77 and
pD+46/+77) were sequenced and characterized (Table 1). The results demonstrated that all four BAL 31 deletion
mutants exhibited approximately the same level of IL-2
protein expression in the absence oftat. However, pD+
42/+77andpD+44/+77 yieldedonly a small,abouttwofold trans-activation of IL-2 expression in the presence oftat.
The twomutantspD+45/+77 andpD+46/+77, which differ
by only 1 or 2 nt, respectively, from the negative deletion
mutant pD+44/+77, in contrast, yielded very significant
(-24-fold) trans-activationin response to tat.This isslightly lower than that observed for the wild-type clone pBC12/HIV/IL-2,thussuggestingsomerole forsequences 3' to position +44. Nevertheless, these results allowed place-ment of the 3' border of the core TAR element at nt +44 withinthe HIV-I LTR Rregion(Fig. 1).
Several other BAL 31 deletion mutants were also exam-ined and yielded the expected trans-activation phenotype; i.e., all of the deletions tested extending 5' to +45 were inactive, whereas all of the deletions terminating3' to +44
were active (data not shown). One extensive deletion,
pD+9/+77, whichremovesessentiallythe entire HIV-I LTR Rregion,including thesequencesproposed toformastable mRNA stem-loop, was examined in detail (Table 1). As expected, this deletion was not responsive to tat. Interest-ingly, this mutation still demonstrated thesamebasallevelof IL-2 expression seen with the wild-type construction pBC12/HIV/IL-2.
TheHIV-ILTRcontains anumberof direct and inverted
repeats. An almost perfect 14-bp direct repeat occurs
[image:3.612.57.297.456.650.2]be-tweenpositions-2and+12andagainbetween +30 and +44
TABLE 1. Levelsofexpression fromtheHIV-ILTR mutants in thepresence orabsence oftata
IL-2production
Clonetransfected (U/ml) Avg
trans-activation'
-tat +tat
BC12/HIV/IL-2 96 6,148 64
pD+46/+77 192 6,148 24
pD+45/+77 128 3,072 23
pD+44/+77 128 192 2.1
pD+42/+77 9 192 1.8
pD+9/+77 128 192 1.4
pBC12/HIV-Xho 96 6,148 64
pD-18/+7 128 192 2.0
pD-18/+20 96 192 1.5
pD+9/+20 192 768 4
pD+9/+38 192 192 1.0
pD+25/+38 96 96 1.0
pD+35/+38 128 192 1.4
pI4/+7 192 6,148 32
p14/+20 192 1,536 8
p18/-18 128 4,096 28
p132/-18 256 3,072 14
a Culturesweretransfected with equimolaramountsofeachHIV-I con-struction,together with eitherthetatexpressionvectorpBC12/CMV/t2orthe
negativecontrol vectorpXF3/ori. SecretedIL-2levelsweredeterminedat72 hposttransfection, and results obtained inarepresentative experiment are
shown. Mock-transfected cultures yielded no detectable IL-2activity (<2
U/mI).
bThe averagelevel of trans-activation ofHIV-I LTR-drivenIL-2
expres-sion due to tatcoexpression is shown(averageoftwo tosix independent transfections).
(Fig. 1). This particularrepeat is of interest because ofthe above observation that the 3' border of the second repeat element precisely coincides with the 3' border of the TAR
core sequence. To address the significance of these and
other reportedrepeatelements, we constructed a number of additional mutants between HIV-I LTR positions -17 and
+38 within pBC12/HIV/IL-2. Initially, weused
oligonucle-otide-directed mutagenesis(21)togenerate a2-bp changeat positions+10 and +11within repeatelement 1(Fig. 1).This
LTR mutant, termed pBC12/HIV-Xho, demonstrated a
trans-activation phenotype indistinguishable from that of
the parental clone (Table 1). Subsequently, we used
pBC12/HIV-Xho and pBC12/HIV/IL-2 to generate a series
ofdeletion mutations between the different restriction
en-zyme sites present within the TAR region (Fig. 1). All of
these deletion mutants yielded anessentially negative
trans-activation phenotype (i.e., c twofold), except for pD+9/
+20, which demonstrated a low butsignificant
trans-activa-tion of about fourfold (Table 1). Of particular interest is the
small-deletion mutant pD+35/+38, which lost 4 bp from
within the 3' repeatelement and which was trans-activation
negative. Allof thedeletion mutants retained essentially the
same basal level of expression as the parental clone
pBC12/HIV/IL-2.
A final set ofmutants was constructed which contained
smallinsertions within the TAR sequence. Insertion of 4 bp
atposition +7 (pI4/+7) had little effect on the
trans-activa-tion phenotype; however, insertion of4bp atposition +20
(pI4/+20) resulted in a significantly reduced level of
trans-activation (about eightfold). Two mutants, pI8/-18 and
p132/-18, contained different size insertions 5' to the cap
siteatposition-18adjacentto the TATAbox (Fig. 1).Both
of these mutants demonstrated levels of trans-activation
slightly but significantly lower than that of the wild-type
construction. In the case of pI32/-18, in particular, this
reductionappeared to result in part from amodestly(two- to
threefold) higher level of basal IL-2 expression.
Effect of TAR mutations on reporter gene mRNA levels. In
the previous section, weexaminedthe levelofreporter gene
expression at the protein levelfor anumber ofHIV-I LTR
mutants in the presence or absence of tat. It has been
suggested that tat-mediated enhancement ofHIV-I-specific
geneexpression is due to both anincreasein HIV-I mRNA
levels and an increase in theutilization ofthat mRNAbythe
cellular translationalmachinery (5, 39). To examinealsothe
phenotype of the HIV-I LTR mutants at these levels, we
performed an internally controlled S1 nuclease protection
assay (5) to quantitate the level of HIV-I-specific IL-2
mRNA in cultures transfected with several of the HIV-I
mutantsdescribed above(Fig. 2). Inaddition, we examined
the parental vector pBC12/HIV/IL-2 and a control,
tat-nonresponsive construction (pBC12/CMV/IL-2) which
con-tains the IL-2 gene driven by the active human cytomegalo-virus immediate-early promoter (5). The results (Fig. 2;
Table 2) demonstrated that pBC12/HIV/IL-2, pD+46/+77,
andpD+45/+77all respondedto tatwith a markedincrease
in IL-2 mRNA steady-state levels. In contrast, pD+42/+77
yielded only a slight increase in IL-2 mRNA, whereas pD+9/+77 was almost nonresponsive to tat. The
cytomeg-alovirusimmediate-earlypromoter was, as expected,highly
transcriptionally active in both the presence and absence of tat.
The S1 protection analysis presented in Fig. 2 used a 3'-end-labeled probe specific for the 3' noncoding region of
the IL-2 reporter gene mRNA. Thisprobe has theadvantage
that it also allows determination ofthe level ofan internal
on November 10, 2019 by guest
http://jvi.asm.org/
10 11 12 13 14 15
4a..*_
*I* .: i
______*:
t- + I + I L-- .-9.
.0 .0 ..
0 0
,% it Jr Af
-wSs-k
,a _ __
.. .0,_
-l....
[image:4.612.61.301.69.315.2]- O
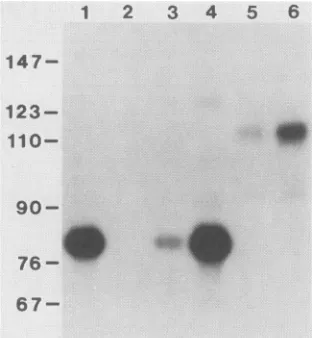
FIG. 2. Analysis of the effect of TAR mutations on the
trans-activation of HIV-I mRNA expression.COS cellsweretransfected
with the differentvectors indicatedin the absence (-)or presence
(+) of the tat expression vectorpBC12/CMV/t2. Total RNA was
harvested 72 h aftertransfection, and 4-pug sampleswereused in the
quantitative Si nuclease protectionassayvisualized here (except for
lanes 14and 15;seebelow). Levels of IL-2 mRNA (175-ntprotected
probe fragment) and reference mRNA(Ref; 156-nt protected probe fragment) in the culture are proportional to the level of rescued probe (5; Table 2). All of the cultures except the negative control (lane 1) werealso transfected with equalamountsof the reference plasmid pBC12AI. Lanes: 1, cellstransfected with pBC12/CMV/t2 alone(negative control); 14, 2 ,ug ofpositive control IL-2mRNA; 15, 20 jig of positive control IL-2 mRNA.
control insulin mRNA encoded by the tat-nonresponsive Rous sarcomavirusLTR-based reference plasmidpBC12AI
(5, 12). We also examined (Fig. 3)the RNA levelsencoded
by PBC12/HIV/IL-2 and p132/-18 in the presence or
ab-senceoftatby using Si probes5' endlabeledatthelocation of the HIV-I LTRHindlllsite(Fig. 1).Theseprobesareable
tomaptranscription initiation (cap) sites used by the HIV-I
transcripts (5). These data confirmed the large increase in
HIV-I-specific mRNA induced by tat cotransfection in
pBC12/HIV/IL-2-transfected cells (Fig. 2) and also
demon-strated that the cap site usedin the pBC12/HIV/IL-2-trans-fectedcellswasthesame asthat in HIV-I-infected H9 cells.
In contrast, the site of transcription initiation used in
pl32/-18-transfected cells, which contain a 32-bpinsertion
mutation between theHIV-I LTRTATA boxand thenormal
cap site, was displaced 5' by -32 bp into the inserted
sequence. In addition, pl32/-18-specific mRNA levels
ap-peared somewhatlower than thoseencodedbypBC12/HIV/
IL-2 in both the presence and absence of tat (Table 2).
Nevertheless, these results clearly demonstrated that
shift-ing the site of transcription initiation within the HIV-I LTR
does not necessarily significantly affect trans-activation by
the viral tatgeneproduct (Table 1; Fig. 3).
Effect oftatonreportergenemRNAtranslational efficiency.
Ithas beenproposed that the reported enhancement in the
[image:4.612.316.555.89.280.2]translational efficiency of HIV-I-specific mRNAs in the presence of tat is due to relief of a specific translational
TABLE 2. Effect ofHIV-I LTR mutations on indicator gene mRNAandprotein levels
IL-2
IL-2 mRNA IL-2protein protein!
Clonetransfecteda U(%)b
U/mIL
(%) mRNAratio' -tat
pBC12/HIV/IL-2 203 (5.4) 48 (0.4) 0.072
pD+46/+77 360(9.6) 64 (0.6) 0.054
pD+45/+77 250(6.7) 48 (0.4) 0.059
pD+42/+77 232(6.2) 96(0.8) 0.126
pD+9/+77 237(6.3) 48 (0.4) 0.062
p132/-18 127 (3.4) 128 (1.0) 0.31
pBC12/CMV/IL-2 3,749 (100) 12,286 (100) 1.00 +tat
pBC12/HIV/IL-2 2,581 (56) 3,072 (25) 0.45 pD+46/+77 2,491 (54) 1,536 (13) 0.24 pD+45/+77 1,849(40) 768 (6.3) 0.16
pD+42/+77 794(17) 192 (1.6) 0.09
pD+9/+77 512(11) 48 (0.4) 0.04
p132/-18 521(11) 1,536 (13) 1.11
pBC12/CMV/IL-2 4,629 (100) 12,286 (100) 1.00
aEquimolar levels ofthe referenceplasmid pBC12AIwere also
cotrans-fectedineachculture.Inaddition, the negative control plasmidpXF3/oriwas
cotransfected inthe -tatcultures,andpBC12/CMV/t2wascotransfected in
the +tat cultures. A culturetransfectedwithpBC12/CMV/t2 alone yieldedno
detectableIL-2 mRNA(Fig. 2)orprotein (<2U/mI).
bIL-2 mRNAlevelsweredetermined by scanning of the autoradiographs
shown in Fig. 2 and 3 with an LKB soft-laser scanner. The values are
corrected forslightdifferences observed intheintensity ofthereference RNA signal. Activity is giveninarbitrary units.
' SecretedIL-2protein levelsweredetermined by bioassay (29).
dTheratio ofIL-2 proteintoIL-2 mRNAdetermined in each culture is expressed relativetopBC12/CMV/IL-2, which is arbitrarilyset at1.00.
1 2 3 4 5 6
147- 123-
110-
90-76 -
67-FIG. 3. Analysis of the HIV-I mRNA transcription startsitesin
transfectedcells.RNA from transfectedCOScellswasharvestedat
72 hposttransfection, and 10-p.g samples wereused in the
quanti-tativeS1 nuclease protectionassay visualizedhere. The size of the
rescued probe fragment wasdetermined with reference to
MspI-cleavedpBR322DNA size markers andmeasuresthe distance of the
transcriptionstartsite from the site oflabelingatposition+82(see
Materials and Methods fordetails). Lanes: 1, 0.5 jigofpoly(A)+
mRNA from HIV-I-infected H9 cells(a giftof M. Dukovich and W.
Greene); 2,pBC12/CMV/IL-2-transfectedCOS cells(negative
con-trol); 3,pBC12/HIV/IL-2+pXF3/ori; 4, pBC12/HIV/IL-2+pBC12/ CMV/t2; 5, p132/-18 + pXF3/ori; 6, p132/-18 + pBC12/CMV/t2. Thenumbersonthe leftindicatemolecular size in nucleotides.
1 2 3 4 5 6 7 8 9
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.360.516.435.604.2]inhibition (5, 8).Toexaminethisquestion,wecomparedthe ratiosofIL-2
protein
to IL-2 mRNA levels produced after transfection of cultures with several of the HIV-I LTR constructs(Table2). This ratio isameasureof theefficiency of translational utilization of each mRNA molecule by thecell (5).
Aspreviously described (5),theparental pBC12/HIV/IL-2
constructionyieldedanIL-2 mRNAwhich,inthe absence of
tat, wastranslated much lessefficiently (-10-to25-fold)by
the cell thanwas the IL-2 mRNAencodedby the
cytomeg-alovirus immediate-early vector. All of the deletion (pD)
mutantsyielded similar low translational efficiencies, includ-ing the extensively deletedmutant pD+9/+77. In the
pres-enceoftat, the threetat-responsive clones
(pBC12/HIV/IL-2, pD+46/+77, and pD+45/+77) all showed significant increases in mRNAtranslationalefficiency. In contrast, the translational efficiencies ofthe RNAs encoded by the two essentially nonresponsive clones pD+42/+77 and pD+9/ +77wereunaffectedbytat.This suggeststhat the transcrip-tionalandposttranscriptionaleffectsoftat aretightlylinked. The mutant
p132/-18
producedaninteresting phenotype.In the absence of tat, the mRNA encoded by pI32/-18 waspresent at a slightly lower steady-state level (Fig. 3) yet
encoded a higher level ofIL-2 protein than did the other
LTR mutants tested (Table 2). In the presence of tat, the
level of this mRNA increased only modestly, to a level
essentially
identical to that ofthe tat-nonresponsive clonepD+9/+77.
The level ofIL-2 proteinencoded by p132/-18mRNA was,however, -32-foldhigherthan that encodedby
pD+9/+77
mRNA, thussuggesting
a large differencebe-tween the translational efficiencies of these two similar mRNAmolecules.
Does tat affect the location of DNase I-hypersensitive sites
within the HIV-I LTR? The trans-acting gene product tat
enhancesHIV-I-specificgeneexpressionin partby
increas-ing the steady-state level ofHIV-I-specific mRNAs (5, 22, 25, 39).Thisincrease, which is dueto anincrease in therate
of HIV-I mRNAtranscription (12), mustbemediatedbythe
sequences located within the LTR TAR sequence. A
prop-erty which can be associated with shifts in the level of
transcription ofa gene is a shift in the pattern of DNase I hypersensitivity (7, 13).This isbelievedtobe duetochanges
in the componentsofthechromatin which enclosethe gene.
We therefore examined the effect oftat onthe
hypersensi-tivity profile
ofthe HIV-I LTRwithintransfected cells(Fig.
4).
COS cells were transfected withpBC12/HIV/IL-2
inthe presence or absence of the tat
expression
vectorpBC12/CMV/t2,
andpermeabilized
nuclei were preparedat72h aftertransfection (9, 10). The nuclei weretreatedwith
various levels of DNase I, and the nuclear DNA was then
isolated and cleaved with BamHI. This restriction enzyme
cutsthe4.7-kilobase
pBC12/HIV/IL-2
vector at auniquesitelocated atposition+765 relativetotheHIV-I LTR cap site.
The isolated DNA was subjected to electrophoresis on a
1.1% agarose gel and transferred to a nitrocellulose filter.
The
hypersensitive
sites were visualized with a probepre-pared against
a261-bp
XbaI-to-BamHIfragment
derived fromthe IL-2 indicator gene coding region.Two
hypersensitive
siteswereobserved within the HIV-I LTRby thisprocedure. The location and intensity of thesesites were not markedly affected by tat coexpression,
al-though the faster-migrating band did appear slightly more
diffuse in the presence of tat protein (Fig. 4, lane e). No
specific
bands were seen after probing of DNase I-treatednuclei derived from mock-transfected COS cells or when
naked pBC12/HIV/IL-2 DNA was used (data not shown).
-tat
j
7M
MA4l4 .
+tat
k b
-4.4 -- 2.3
--2.0
--
1.35--0.S7- - - TATA TAR
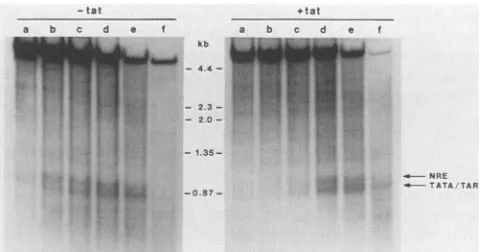
FIG. 4. DNase I-hypersensitive chromatin sites in the HIV-I LTR. Theautoradiograph visualizes DNase I-hypersensitive sites located within the HIV-1 LTR in COS cells transfected with pBC12/HIV/IL-2 with (+tat) and without (-tat)the tatexpression
vector pBC12/CMV/t2. Tatcoexpression resulted in a64-fold in-crease in IL-2 expression. The slower-migrating band maps to
position -175 to -125 in the HIV-I LTR, the site of a possible negative regulatory element (NRE).Thefaster-migrating bandmaps toHIV-I LTRposition -45 to +25, the siteof theTATAboxand the TAR element(Fig. 1). Lanes:a, no DNase I; b,c,d,e,andf, 128, 256, 512, 1,024, and 2,048 U of DNase I, respectively. kb, Kilobases.
The location of the two DNase I-hypersensitive sites was
carefully mappedin relationto DNA size markers
(HindIII-cleaved phage A DNA and HaeIII-cleaved XX174 DNA)
which were visualized by reprobing of the blot with the appropriate nick-translated phage DNA probes. This mea-surement indicated that thelargerbandwas910 + 25 bp in size,whereasthe smaller bandwas775 ± 35bpin size. This
correlates with the HIV-I LTRlocations -175 to -125, the
possible site ofa negative regulatoryelement(11, 34a),and
to -45 to +25 bp, which coincides with the HIV-I LTR
TATAbox and part of the TARelement.
DISCUSSION
In this study, we used site-directed mutagenesis ofthe
HIV-I LTR, combined witha transient-expression assay in
the HIV-I-permissive (8, 18) monkey cell line COS, to
examine the roles of specific LTR sequences in
trans-activationby theviraltatgeneproduct. Because this
trans-activationevent occurs atleast in partatthetranscriptional
level (12) and repeat structures have been shown to be
important in the regulationofexpression from several tran-scriptioncontrolregions (19),wefocusedparticularlyonthe
functionalsignificanceof inverted and direct repeats present
within the core TAR sequence (Fig. 1). Initially, we used
processive deletion mutagenesis to define the 3' border of
the core TAR element at position +44 relative to the cap
site. These dataconfirmpreviousresultsshowingthat the 3'
borderofTARlaybetween +31 and +54(22).Subsequently,
we used deletion andinsertion mutagenesisto examine the
roles of different areas within the TAR sequence between
-18 and +44 (Table 1). These studies revealed that most
mutations of this region dramatically reduced the
trans-activation response. Two minor mutations at about +10,
pBC12/HIV-Xho andpI4/+7, which disrupt the first of the
twodirect repeat elements noted in Fig. 1, were, however,
found to have little or no effect. Also of interest is
pD+9/+20, which is the largest internal TAR deletion
mu-tation to permit some level of residual trans-activation.
None of the TAR mutations, including extensive deletions
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.318.558.70.196.2]such aspD-18/+20 andpD+9/+77, were found to affect the
basal activity of theHIV-I LTR promoter significantly. This
observation argues against the hypothesis that the TAR element is a cis-acting negative regulator of HIV-I-specific
gene expression.
The results presented heredefineasmall, -60-bp element
which is critically important for tat-mediated trans-activa-tion of the HIV-I LTR and which retains a number of interestingstructural features. These include the two 14-bp imperfect direct repeats noted in Fig. 1, which are in turn
separated by a 10-bp palindrome (5'-TAGACCAGAT-3').
Only part ofthe previously noted (22, 24) imperfect 24-bp inverted repeat wasfound tobe important fortat-mediated trans-activation of HIV-I gene expression; however, this
could still form the basis fora 9-bp hairpin loop structure
(Fig. 1B). The limited mutational analysis described here
appears to focus attention on the second ofthe two direct
repeats (Fig. 1), since all deletions involving this element,
including pD+35/+38 and pD+44/+77, which affect this
element onlyslightly, resultin almost totalabrogationof the
trans-activationresponse. Itwill beofinterest to determine
whether syntheticcopies of this element canconfer a
trans-activation response on heterologous promoters when
pres-entin cis.
Ithaspreviously been shown that thefunction ofthe TAR
element isbothposition andorientation dependent (22, 25),
thus distinguishing TAR from inducible enhancer elements
which have been described for other systems (19). One possible function ofTAR, byanalogy toothertranscription
control regions(3, 15, 34),isas abindingsite foravirus-or
host-specifictranscription factor.OccupationofTARbythis
protein would, in turn, stabilize binding of transcription
factors to LTR U3 region sequences. In simian virus 40
late-gene trans-activation by simian virus 40 T antigen, stable binding of transcription factorsto Tantigen-binding
site II and the simian virus 4072-bp repeatelement occurs
only when the two binding sites are correctly and closely
spaced (3). Insertionofas little as 42 bp totally abrogated functional binding, and even a 4-bp insertion produced a significant effect(3). To testwhether the proximity ofTAR tothebodyoftheHIV-ILTRtranscriptioncontrol
region
inthe LTR U3 regionwascritical forfunction,wechangedthis
distance by insertion of8 or32 bp between the TATAbox
and the core TAR region (pl8/-18 and
p132/-18).
Thesemutations resulted in only a modest decrease in
trans-activation and therefore suggest that the position ofTAR
within theHIV-I LTR is flexible to atleast somedegree.
Because trans-activation appears to involve
transcrip-tional activation of the HIV-I LTR (12), we examined the
DNase I hypersensitivity profile of the LTR and asked
whetherthis was affected by tat coexpression. Our results
(Fig. 4) revealed two areas of
hypersensitivity
within theHIV-I LTRwhich mappedto -45to +25 bp (3'
site)
andto-175 to -125bp (5' site) butwere notmarkedlyaffectedby tat coexpression. The location of the 3' site extends from
-15 bp 5'to theTATAbox into theTAR region sequences
and the HIV-I LTR R region. The 5' site does not accord
with any knownimportant structurewithin theHIV-I LTR;
however, deletion of this sequence has been associated with
elevated expression from the HIV-I LTR, and this may,
therefore, be a negative regulatory element (11, 34a). No signal was noted atthe site ofthe HIV-I LTR Spl
binding
sites (14) (about -50 to -80) or at the location of the
inducibleHIV-Ienhancer element(23)(about -80to-105).
However,becauseonlyasubfraction of the introducedDNA maybefully transcriptionally activein this
transient-expres-sion assay, it ispossiblethat this assaycannotdetectaweak
but
significant signal
at these sites.Itis of interesttocomparethe in vivo DNase I
hypersen-sitivity analysis presented
here with a recentstudy
on the locationof DNaseI-protected
sites(footprints)
in theHIV-ILTR after in vitro incubation with a HeLa cell nuclear
extract (11). This report identified three
protected sites,
of which the most 5',extending
from -173 to-159,
appearscoincident with the 5' site noted in
Fig.
4. A secondweakly
protected site,
locatedat -97 to-78,
wasnot notedin ourdata. Of
particular
interest isathirdprotected
site,
extend-ing
from -42to +28 in theHIV-ILTRcoding
strand andasfaras +52 in the
noncoding
strand(11).
Thisprotection
ofthe TAR sequence
closely
mirrors both thelocation oftheDNase-hypersensitive
3' site noted inFig.
4(-45
to+25)
and the location of the sequences
required
for tattrans-activationasdefined
by
the above mutationalanalysis.
Theclosecorrelation between these in vivo dataand the
previ-ously
published
in vitroDNase Ifootprint analysis strongly
suggests the existence ofa cellular TAR
sequence-binding
protein
in cells that do not express the tat geneproduct.
Recent reports indicate that a number ofviral and cellular
trans-acting transcriptional
activators, including
thetrans-activator (tatl or
p40x)
encodedby
the human retrovirushumanT-cell
lymphotropic
virus typeI,
mayactvia cellularsequence-specific DNA-binding proteins (2, 13a, 17, 37).
Theevidence for a constitutive cellular TAR
sequence-binding
protein
may suggest a similar mechanism of actionfor theHIV-I tatgene
product.
ACKNOWLEDGMENTS
WethankAnn Perkinsfor technical assistance and LisaNieves andSharon Goodwinforsecretarialsupport. Wealso thankKlaus JantzenandHans-PeterFritton forhelpfuldiscussions.
LITERATURE CITED
1. Arya, S. K., C. Guo, S. F. Josephs,and F. Wong-Staal. 1985. Trans-activator gene ofhumanT-lymphotropic virus type III
(HTLV-III). Science229:69-73.
2. Berk,A. J. 1986. Adenoviruspromoters and ElA transactiva-tion. Annu. Rev.Genet.20:45-79.
3. Brady, J., M. R. Loeken, and G. Khoury. 1985. Interaction
between two transcriptional control sequences required for tumor-antigen-mediated simian virus 40 late gene expression.
Proc.Natl. Acad. Sci. USA 82:7299-7303.
4. Chen,I.S. Y.1986.Regulationof AIDS virusexpression.Cell
47:1-2.
5. Cullen, B. R. 1986. Trans-activation of human
immunodefi-ciencyvirusoccursviaabimodalmechanism.Cell 46:973-982.
6. Dayton, A. I.,J. G. Sodroski, C. A. Rosen, W. C. Goh, and
W.A.Haseltine. 1986.Thetrans-activatorgeneofthe human T celllymphotropicvirustypeIIIisrequiredfor
replication.
Cell 44:941-947.7. Elgin,S. C. R. 1981. DNaseI-hypersensitivesites ofchromatin.
Cell 27:413-415.
8. Feinberg,M.B.,R. F.Jarrett,A.Aldovini,R.C.Gallo,andF.
Wong-Staal.1986.HTLV-IIIexpressionand
production
involvecomplex regulationat the levels ofsplicingand translation of
viral RNA. Cell46:807-817.
9. Fritton,H.P.,T. Igo-Kemenes,J.Nowock, U.Strech-Jurk,M. Theisen, and A. E. Sippel. 1984. Alternative sets of DNase
I-hypersensitivesites characterizethevariousfunctionalstates ofthechickenlysozymegene. Nature(London)311:163-165.
10. Fritton, H.P.,A. E. Sippel,and T.Igo-Kemenes. 1983.
Nucle-ase-hypersensitivesites in thechromatin domainof thechicken
lysozymegene. Nucleic AcidsRes.11:3467-3485.
11. Garcia,J.A.,F. K.Wu,R.Mitsuyasu,andR.B.Gaynor.1987.
Interactionsof cellularproteins involved in the
transcriptional
regulation of the human immunodeficiency virus. EMBO J.
on November 10, 2019 by guest
http://jvi.asm.org/
6:3761-3770.
12. Hauber, J., A. Perkins, E. P. Heimer, and B. R. Cullen. 1987. Trans-activation ofhumanimmunodeficiency virusgene expres-sionismediatedby nuclear events. Proc. Natl. Acad. Sci. USA 84:6364-6368.
13. Igo-Kemenes, T., W.Horz,and H. G. Zachau. 1982. Chromatin. Annu. Rev. Biochem. 51:89-121.
13a.Jeang, K.-T., J. Brady, M. Radanovich, J. Duvall, and G. Khoury. 1987. p4Ox trans-activation oftheHTLV-I promoter. UCLA Symp. Mol. Cell. Biol. New Ser. 67:181-189.
14. Jones, K. A., J. T. Kadonaga, P. A. Luciw, and R. Tjian. 1986. Activation of the AIDS retrovirus promoter by the cellular transcription factor,Spl. Science232:755-759.
15. Jones, K. A., K. R. Yamamoto, and R. Tjian. 1985. Twodistinct transcription factors bind to the HSV thymidine kinase pro-moter in vitro. Cell42:559-579.
16. Knight, D. M., F. A. Flomerfelt, and J. Ghrayeb.1987. Expres-sion of the art/trs protein ofHIVand study of its role in viral envelope synthesis. Science236:837-840.
17. Kovesdi, I., R. Reichel, and J. R. Nevins. 1986.Identification of acellulartranscription factor involved in ElA trans-activation. Cell 45:219-228.
18. Levy, J. A., C. Cheng-Mayer, D. Dina, and P. Luciw. AIDS retrovirus (ARV-2) clone replicates in transfected human and animal fibroblasts. Science232:998-1001.
19. Maniatis, T., S. Goodbourn, and J. A. Fischer. 1987.Regulation of inducible and tissue-specific gene expression. Science 236:1237-1245.
20. Messing, J. 1983. New M13 vectors for cloning. Methods Enzymol. 101:20-78.
21. Morinaga, Y., T. Franceschini, S. Inouye, and M. Inouye. 1984. Improvementof oligonucleotide-directed site-specific mutagen-esis using double-stranded plasmid DNA. Biotechnology 2:636-639.
22. Muesing, M. A., D. H. Smith, and D. J. Capon.1987. Regulation of mRNA accumulation by ahuman immunodeficiency virus trans-activatorprotein. Cell 48:691-701.
23. Nable, G., and D. Baltimore. 1987. An inducible transcription factor activatesexpression of human immunodeficiency virus in Tcells. Nature(London)326:711-713.
24. Okamoto,T., and F.Wong-Staal. 1986.Demonstration of virus-specific transcriptional activator(s)incells infectedwith HTLV-IIIbyanin vitrocell-free system. Cell47:29-35.
25. Peterlin, B. M., P. A. Luciw, P. J. Barr, and M. D. Walker. 1986. Elevated levels of mRNA can account for the trans-activation ofhumanimmunodeficiency virus.Proc.Natl. Acad. Sci. USA 83:9734-9738.
26. Popovic, M., M. G. Sarngadharan, E. Read, and R. C.Gallo. 1984. Detection, isolation, andcontinuousproduction of cyto-pathic retroviruses (HTLV-III) from patients with AIDS and pre-AIDS. Science 224:497-500.
27. Ratner, L., A. Fisher, L. L. Jagodzinski, H. Mitsuya, R.-S. Liou,
R. C. Gallo, and F. Wong-Staal. 1987. Complete nucleotide sequences of functional clones of the AIDS virus. AIDS Res. 3:57-69.
28. Ratner, L., B. Starcich, S. F. Josephs, B. H. Hahn, E. P. Reddy, K. J. Livak,S. R. Petteway, Jr., M. L. Pearson, W. A. Haseltine, S. K. Arya, and F. Wong-Staal. 1985. Polymorphism of the 3' open reading frame of the virus associated with the acquired immune deficiency syndrome, human T-lymphotropic virus typeIII. NucleicAcids Res. 13:8219-8229.
29. Robb, R. J. 1985. Human interleukin 2. Methods Enzymol. 116:493-525.
30. Rosen, C. A., J. G. Sodroski, W. C. Goh, A. I. Dayton, J. Lippke, and W. A. Haseltine. 1986.Post-transcriptional regula-tion accounts for the trans-activaregula-tion of the human T-lympho-tropic virus type III. Nature (London) 319:555-559.
31. Rosen, C. A., J. G. Sodroski, and W. A. Haseltine. 1985. The locationof cis-acting regulatory sequencesinthehuman Tceli lymphotropic virus type III (HTLV-III/LAV) long terminal repeat. Cell 41:813-823.
32. Sanchez-Pescador, R., M. D. P-ower, P. J. Barr, K. S. Steimer, M. M.Stempien, S. L. Brown-Shimer, W. W. Gee, A. Renard, A. Randolph, J. A. Levy, D. Dina, and P. A. Luciw. 1985. Nucle-otidesequenceandexpression ofanAIDS-associatedretrovirus (ARV-2). Science227:484-492.
33. Sanger,F.,S.Nicklen, and A. R. Coulson. 1977. DNA sequenc-ing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA74:5463-5467.
34. Sawadogo, M.,and R. G. Roeder. 1985. Interaction ofa gene-specific transcription factor with the adenovirus major late promoter upstream ofthe TATAboxregion. Cell 43:167-175. 34a.Siekevitz, M., S. F. Josephs, M. Dukovich, N. Peffer, F.
Wong-Staal,and W. C. Greene. 1987.Activation of the HIV-ILTRby T cell mitogens and the trans-activator protein of HTLV-I. Science 238:1575-1578.
35. Sodroski, J., W.C. Goh, C.Rosen, A. Dayton,E.Terwilliger, and W. Haseltine. 1986. A second post-transcriptional trans-activatorgenerequired forHTLV-IIIreplication. Nature (Lon-don)321:412-417.
36. Tinoco, I., P. Borer, B. Dengler, M. Levine, 0. Uhlenbeck, D. Crothers, andJ. Gralla. 1973. Improved estimation of second-ary structure inribonucleic acids. Nature(London)NewBiol. 246:40-41.
37. Tsai, S. Y., I. Sagami, H. Wang, M.-J. Tsai, and B. W. O'Malley. 1987.Interactions betweenaDNA-binding
transcrip-tionfactor (COUP) and a non-DNA binding factor (S300-II).
Cell50:701-709.
38. Wain-Hobson,S.,P. Sonigo, 0. Danos, S. Cole, and M. Alizon. 1985. Nucleotide sequence of the AIDS virus, LAV. Cell 40:9-17.
39. Wright, C. M., B. K. Felber, H. Paskalis, and G. N.Pavlakis. 1986. Expression and characterization of the trans-activator of HTLV-III/LAVvirus. Science 234:988-992.